DNA Damage Response and Oxidative Stress in Systemic Autoimmunity

 , ,
, , 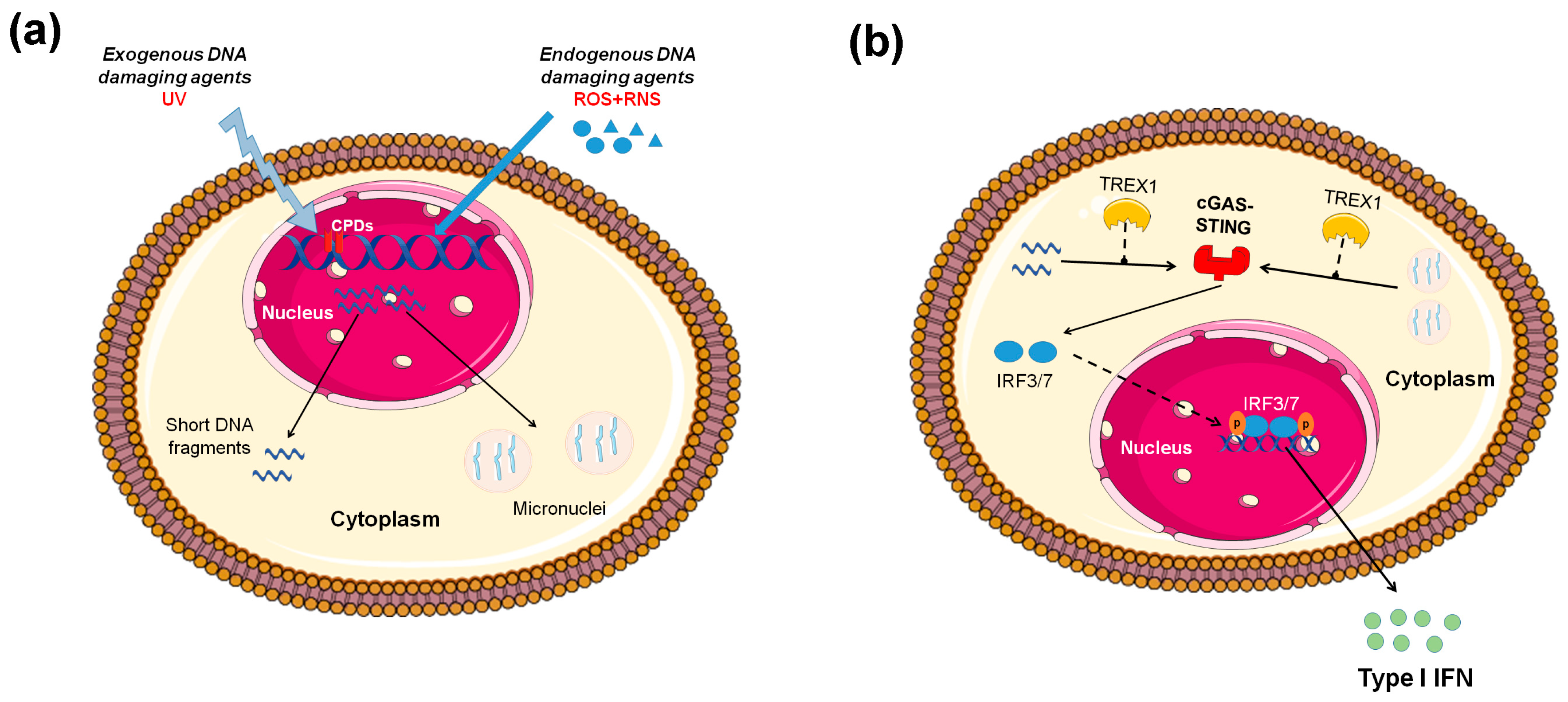{kind=link}
{kind=link}
Abstract
1. Introduction
2. Normal DNA Repair Pathways
2.1. Nucleotide Excision Repair (NER)
2.2. Base Excision Repair (BER)
2.3. Mismatch Repair (MMR)
2.4. Double-Strand Breaks (DSBs) Repair
2.4.1. Homologous Recombination Repair (HRR)
2.4.2. Canonical Non-Homologous End Joining (c-NHEJ)
2.4.3. Alternative Non-Homologous End Joining (alt-NHEJ)
2.4.4. Single-Strand Annealing (SSA)
2.5. Interstrand Cross-Link (ICL) Repair
2.6. Direct Repair Pathway
3. The Interplay between the DDR/R Network and the Immune Response: The Role of Oxidative Stress
3.1. DNA Double-Strand Breaks Per Se Induce Innate Immune Activation
3.2. Defective DNA Repair and Chronic Low-Level DNA Damage “Prime” the Innate Immune Response
3.3. Micronuclei: Connecting Nuclear DNA Damage and Cytosolic Innate Immune Receptors
3.4. Oxidative Stress Causes DNA Damage That Activates the Immune System
3.5. Oxidative Stress and Immune Senescence
3.6. Defects in Degradation of Endogenous DNA and Immune Activation
4. The DDR/R Network in Systemic Autoimmune Diseases
4.1. Systemic Lupus Erythematosus
4.1.1. Gene Polymorphisms Associated with Impaired DNA Repair Machinery and SLE
4.1.2. Increased Endogenous DNA Damage in SLE: Defective Repair or Increased Formation?
4.1.3. Autoantibodies against DNA Repair Enzymes
4.1.4. Defects in Apoptosis and SLE Pathogenesis
4.2. Systemic Sclerosis
4.3. Rheumatoid Arthritis
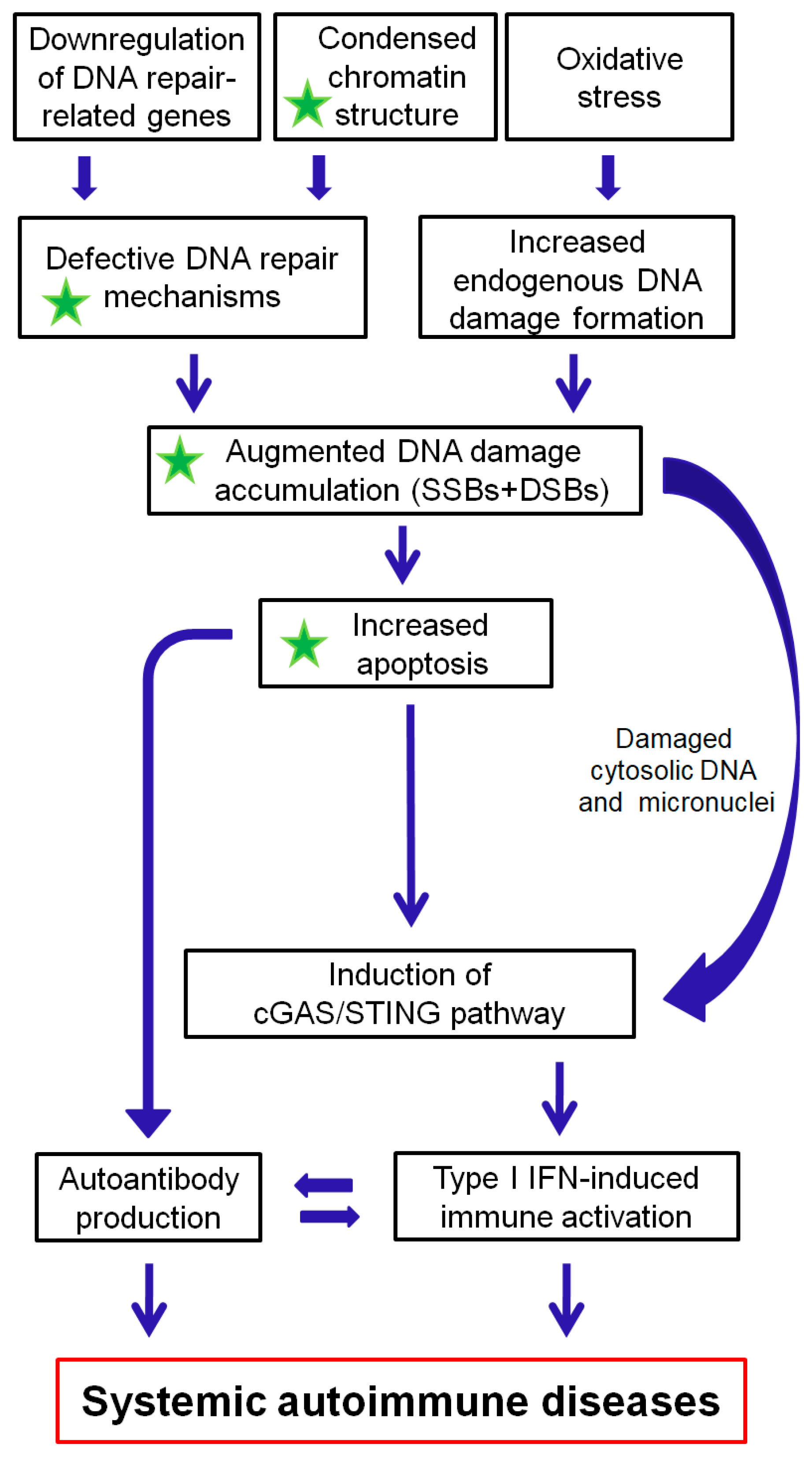
5. Conclusion and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Pateras, I.S.; Havaki, S.; Nikitopoulou, X.; Vougas, K.; Townsend, P.A.; Panayiotidis, M.I.; Georgakilas, A.G.; Gorgoulis, V.G. The DNA damage response and immune signaling alliance: Is it good or bad? Nature decides when and where. Pharmacol. Ther. 2015, 154, 36–56. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Sfikakis, P.P. Increased DNA double-strand breaks and enhanced apoptosis in patients with lupus nephritis. Lupus 2015, 24, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Vougas, K.; Gorgoulis, V.G.; Sfikakis, P.P. Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus. Arthritis Res. Ther. 2016, 18, 182. [Google Scholar] [CrossRef] [PubMed]
- Brzostek-Racine, S.; Gordon, C.; Van Scoy, S.; Reich, N.C. The DNA damage response induces IFN. J. Immunol. 2011, 187, 5336–5345. [Google Scholar] [CrossRef]
- Härtlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kröger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Shen, Y.J.; Le Bert, N.; Chitre, A.A.; Koo, C.X.; Nga, X.H.; Ho, S.S.W.; Khatoo, M.; Tan, N.Y.; Ishii, K.J.; Gasser, S. Genome-derived cytosolic DNA mediates type I interferon-dependent rejection of B cell lymphoma cells. Cell Rep. 2015, 11, 460–473. [Google Scholar] [CrossRef]
- Nakad, R.; Schumacher, B. DNA Damage Response and Immune Defense: Links and Mechanisms. Front. Genet. 2016, 7, 147. [Google Scholar] [CrossRef]
- Erdal, E.; Haider, S.; Rehwinkel, J.; Harris, A.L.; McHugh, P.J. A prosurvival DNA damage-induced cytoplasmic interferon response is mediated by end resection factors and is limited by Trex1. Genes Dev. 2017, 31, 353–369. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Lettice, L.; Tarnauskaitė, Ž.; Reddy, K.; Dix, F.; Revuelta, A.; Abbondati, E.; Rigby, R.E.; Rabe, B.; et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 2016, 35, 831–844. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, K.; Knittler, K.; Borowski, C.; Rudnik, S.; Damme, M.; Aden, K.; Spehlmann, M.E.; Frey, N.; Saftig, P.; Chalaris, A.; et al. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum. Mol. Genet. 2017, 26, 3960–3972. [Google Scholar] [CrossRef]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.-W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest. 2008, 118, 2516–2525. [Google Scholar] [CrossRef]
- Ohnishi, S.; Ma, N.; Thanan, R.; Pinlaor, S.; Hammam, O.; Murata, M.; Kawanishi, S. DNA Damage in Inflammation-Related Carcinogenesis and Cancer Stem Cells. Oxid. Med. Cell. Longev. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Pálmai-Pallag, T.; Bachrati, C.Z. Inflammation-induced DNA damage and damage-induced inflammation: A vicious cycle. Microbes Infect. 2014, 16, 822–832. [Google Scholar] [CrossRef]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.; Sweasy, J.B. Interplay between DNA repair and inflammation, and the link to cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef]
- Fontes, F.L.; Pinheiro, D.M.; Oliveira, A.H.; Oliveira, R.K.; Lajus, T.B.; Agnez-Lima, L.F. Role of DNA repair in host immune response and inflammation. Mutat. Res. Rev. Mutat. Res. 2015, 763, 246–257. [Google Scholar] [CrossRef]
- Kiraly, O.; Gong, G.; Olipitz, W.; Muthupalani, S.; Engelward, B.P. Inflammation-Induced Cell Proliferation Potentiates DNA Damage-Induced Mutations In Vivo. PLoS Genet. 2015, 11, e1004901. [Google Scholar] [CrossRef]
- Neves-Costa, A.; Moita, L.F. Modulation of inflammation and disease tolerance by DNA damage response pathways. FEBS J. 2017, 284, 680–698. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Palomino, G.M.; Bassi, C.L.; Wastowski, I.J.; Xavier, D.J.; Lucisano-Valim, Y.M.; Crispim, J.C.O.; Rassi, D.M.; Marques-Netom, J.F.; Sakamoto-Hojo, E.T.; Moreau, P.; et al. Patients with systemic sclerosis present increased DNA damage differentially associated with DNA repair gene polymorphisms. J. Rheumatol. 2014, 41, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Sfikakis, P.P. DNA damage accumulation, defective chromatin organization and deficient DNA repair capacity in patients with rheumatoid arthritis. Clin. Immunol. 2019, 203, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.-H.; Cohen, P.L. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, 202. [Google Scholar] [CrossRef] [PubMed]
- Shuck, S.C.; Short, E.A.; Turchi, J.J. Eukaryotic nucleotide excision repair: From understanding mechanisms to influencing biology. Cell Res. 2008, 18, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; He, Y.-Y. Molecular regulation of UV-induced DNA repair. Photochem. Photobiol. 2015, 91, 254–264. [Google Scholar] [CrossRef]
- Schärer, O.D. Nucleotide excision repair in eukaryotes. Cold Spring Harb. Perspect. Biol. 2013, 5, a012609. [Google Scholar] [CrossRef]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; He, H.; Kelley, M.R.; Georgiadis, M.M. Redox regulation of DNA repair: Implications for human health and cancer therapeutic development. Antioxid. Redox Signal. 2010, 12, 1247–1269. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic targeting of the DNA mismatch repair pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, M.; Allen, C.; Nickoloff, J.A.; Hromas, R. Synthetic lethality: Exploiting the addiction of cancer to DNA repair. Blood 2011, 117, 6074–6082. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Padilla, I.; Romero, N.; Amir, E.; Matias-Guiu, X.; Vilar, E.; Muggia, F.; Garcia-Donas, J. Mismatch repair status and clinical outcome in endometrial cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2013, 88, 154–167. [Google Scholar] [CrossRef]
- Li, Y.-H.; Wang, X.; Pan, Y.; Lee, D.-H.; Chowdhury, D.; Kimmelman, A.C. Inhibition of non-homologous end joining repair impairs pancreatic cancer growth and enhances radiation response. PLoS ONE 2012, 7, e39588. [Google Scholar] [CrossRef]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Scott, S.; Pandita, T.K. The cellular control of DNA double-strand breaks. J. Cell. Biochem. 2006, 99, 1463–1475. [Google Scholar] [CrossRef]
- Mills, K.D.; Ferguson, D.O.; Alt, F.W. The role of DNA breaks in genomic instability and tumorigenesis. Immunol. Rev. 2003, 194, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. Homologous recombination in cancer development, treatment and development of drug resistance. Carcinogenesis 2010, 31, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Shah Punatar, R.; Martin, M.J.; Wyatt, H.D.; Chan, Y.W.; West, S.C. Resolution of single and double Holliday junction recombination intermediates by GEN1. Proc. Natl. Acad. Sci. USA 2017, 114, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Pandita, T.K.; Hazra, T.K. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016, 7, 13049. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar]
- Bunting, S.F.; Nussenzweig, A. End-joining, translocations and cancer. Nat. Rev. Cancer 2013, 13, 443–454. [Google Scholar] [CrossRef]
- Zha, S.; Guo, C.; Boboila, C.; Oksenych, V.; Cheng, H.-L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2011, 469, 250–254. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Yu, A.M.; McVey, M. Synthesis-dependent microhomology-mediated end joining accounts for multiple types of repair junctions. Nucleic Acids Res. 2010, 38, 5706–5717. [Google Scholar] [CrossRef]
- Simsek, D.; Jasin, M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 2010, 17, 410–416. [Google Scholar] [CrossRef]
- Deriano, L.; Roth, D.B. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef] [PubMed]
- Hartlerode, J.; Scully, R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 2009, 423, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. 2016, 32, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Anai, H.; Hanada, K. Mechanisms of interstrand DNA crosslink repair and human disorders. Genes Environ. 2016, 38, 9. [Google Scholar] [CrossRef]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Daly, M.; Arends, M.J.; Patel, K.J. Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 2012, 489, 571–575. [Google Scholar] [CrossRef]
- Wang, Y.; Leung, J.W.; Jiang, Y.; Lowery, M.G.; Do, H.; Vasquez, K.M.; Chen, J.; Wang, W.; Li, L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol. Cell 2013, 49, 997–1009. [Google Scholar] [CrossRef]
- Clauson, C.; Schärer, O.D.; Niedernhofer, L. Advances in understanding the complex mechanisms of DNA interstrand cross-link repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012732. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M.; Naumann, S.; Roos, W.P. MGMT: Key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents. DNA Repair (Amst.) 2007, 6, 1079–1099. [Google Scholar] [CrossRef]
- Hiddinga, B.I.; Pauwels, P.; Janssens, A.; van Meerbeeck, J.P. O6-Methylguanine-DNA methyltransferase (MGMT): A drugable target in lung cancer? Lung Cancer (Amst. Neth.) 2017, 107, 91–99. [Google Scholar] [CrossRef]
- Plummer, R. Perspective on the pipeline of drugs being developed with modulation of DNA damage as a target. Clin. Cancer Res. 2010, 16, 4527–4531. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.H.; Liu, W.L.; Cao, H.; Wen, C.; Chen, L.; Jiang, G. O(6)-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis. 2013, 4, e876. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Leitch, A.; Hayward, B.E.; Garner, A.; Parmar, R.; Griffith, E.; Ali, M.; Semple, C.; Aicardi, J.; Babul-Hirji, R.; et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet. 2006, 38, 910–916. [Google Scholar] [CrossRef] [PubMed]
- Pizzi, S.; Sertic, S.; Orcesi, S.; Cereda, C.; Bianchi, M.; Jackson, A.P.; Lazzaro, F.; Plevani, P.; Muzi-Falconi, M. Reduction of hRNase H2 activity in Aicardi-Goutières syndrome cells leads to replication stress and genome instability. Hum. Mol. Genet. 2015, 24, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Hiller, B.; Achleitner, M.; Glage, S.; Naumann, R.; Behrendt, R.; Roers, A. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med. 2012, 209, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Crow, Y.J.; Hayward, B.E.; Parmar, R.; Robins, P.; Leitch, A.; Ali, M.; Black, D.N.; van Bokhoven, H.; Brunner, H.G.; Hamel, B.C.; et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet. 2006, 38, 917–920. [Google Scholar] [CrossRef]
- Chowdhury, D.; Beresford, P.J.; Zhu, P.; Zhang, D.; Sung, J.S.; Demple, B.; Perrino, F.W.; Lieberman, J. The exonuclease TREX1 is in the SET complex and acts in concert with NM23–H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 2006, 23, 133–142. [Google Scholar] [CrossRef]
- Yang, Y.G.; Lindahl, T.; Barnes, D.E. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007, 131, 873–886. [Google Scholar] [CrossRef]
- Günther, C.; Kind, B.; Reijns, M.A.; Berndt, N.; Martinez-Bueno, M.; Wolf, C.; Tüngler, V.; Chara, O.; Lee, Y.A.; Hübner, N.; et al. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Invest. 2015, 125, 413–424. [Google Scholar] [CrossRef]
- Gekara, N.O. DNA damage-induced immune response: Micronuclei provide key platform. J. Cell. Biol. 2017, 216, 2999–3001. [Google Scholar] [CrossRef]
- Gao, D.; Li, T.; Li, X.-D.; Chen, X.; Li, Q.-Z.; Wight-Carter, M.; Chen, Z. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 2015, 112, 5699–5705. [Google Scholar] [CrossRef] [PubMed]
- Fucikova, J.; Moserova, I.; Urbanova, L.; Bezu, L.; Kepp, O.; Cremer, I.; Salek, C.; Strnad, P.; Kroemer, G.; Galluzzi, L.; et al. Prognostic and Predictive Value of DAMPs and DAMP-Associated Processes in Cancer. Front. Immunol. 2015, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2016, 17, 97–111. [Google Scholar] [CrossRef]
- Morita, M.; Stamp, G.; Robins, P.; Dulic, A.; Rosewell, I.; Hrivnak, G.; Daly, G.; Lindahl, T.; Barnes, D.E. Gene-targeted mice lacking the Trex1 (DNase III) 3′-->5′ DNA exonuclease develop inflammatory myocarditis. Mol. Cell. Biol. 2004, 24, 6719–6727. [Google Scholar] [CrossRef]
- Stetson, D.B.; Ko, J.S.; Heidmann, T.; Medzhitov, R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 2008, 134, 587–598. [Google Scholar] [CrossRef]
- Hasan, M.; Dobbs, N.; Khan, S.; White, M.A.; Wakeland, E.K.; Li, Q.Z.; Yan, N. Cutting edge: Inhibiting TBK1 by compound II ameliorates autoimmune disease in mice. J. Immunol. 2015, 195, 4573–4577. [Google Scholar] [CrossRef]
- Zuo, L.; Prather, E.R.; Stetskiv, M.; Garrison, D.E.; Meade, J.R.; Peace, T.I.; Zhou, T. Inflammaging and Oxidative Stress in Human Diseases: From Molecular Mechanisms to Novel Treatments. Int. J. Mol. Sci. 2019, 20, 4472. [Google Scholar] [CrossRef]
- Sedelnikova, O.A.; Redon, C.E.; Dickey, J.S.; Nakamura, A.J.; Georgakilas, A.G.; Bonner, W.M. Role of oxidatively induced DNA lesions in human pathogenesis. Mutat. Res. 2010, 704, 152–159. [Google Scholar] [CrossRef]
- Efremov, Y.R.; Proskurina, A.S.; Potter, E.A.; Dolgova, E.V.; Efremova, O.V.; Taranov, O.S.; Ostanin, A.A.; Chernykh, E.R.; Kolchanov, N.A.; Bogachev, S.S. Cancer Stem Cells: Emergent Nature of Tumor Emergency. Front. Genet. 2018, 9, 544. [Google Scholar] [CrossRef]
- Altieri, F.; Grillo, C.; Maceroni, M.; Chichiarelli, S. DNA damage and repair: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 891–930. [Google Scholar] [CrossRef] [PubMed]
- Kryston, T.B.; Georgiev, A.B.; Pissis, P.; Georgakilas, A.G. Role of oxidative stress and DNA damage in human carcinogenesis. Mutat. Res. 2011, 711, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Čolak, E.; Ignjatović, S.; Radosavljević, A.; Žorić, L. The association of enzymatic and non-enzymatic antioxidant defense parameters with inflammatory markers in patients with exudative form of age-related macular degeneration. J. Clin. Biochem. Nutr. 2017, 60, 100–107. [Google Scholar] [CrossRef]
- Bokhari, B.; Sharma, S. Stress Marks on the Genome: Use or Lose? Int. J. Mol. Sci. 2019, 20, 364. [Google Scholar] [CrossRef]
- Perl, A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2013, 9, 674–686. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Doridot, L.; Jeljeli, M.; Chêne, C.; Batteux, F. Implication of oxidative stress in the pathogenesis of systemic sclerosis via inflammation, autoimmunity and fibrosis. Redox Biol. 2019, 101122. [Google Scholar] [CrossRef]
- Vona, R.; Giovannetti, A.; Gambardella, L.; Malorni, W.; Pietraforte, D.; Straface, E. Oxidative stress in the pathogenesis of systemic scleroderma: An overview. J. Cell. Mol. Med. 2018, 22, 3308–3314. [Google Scholar] [CrossRef]
- Altindag, O.; Karakoc, M.; Kocyigit, A.; Celik, H.; Soran, N. Increased DNA damage and oxidative stress in patients with rheumatoid arthritis. Clin. Biochem. 2007, 40, 167–171. [Google Scholar] [CrossRef]
- Gehrke, N.; Mertens, C.; Zillinger, T.; Wenzel, J.; Bald, T.; Zahn, S.; Tüting, T.; Hartmann, G.; Barchet, W. Oxidative Damage of DNA Confers Resistance to Cytosolic Nuclease TREX1 Degradation and Potentiates STING-Dependent Immune Sensing. Immunity 2013, 39, 482–495. [Google Scholar] [CrossRef]
- Kemp, M.G.; Lindsey-Boltz, L.A.; Sancar, A. UV Light Potentiates STING (Stimulator of Interferon Genes)-dependent Innate Immune Signaling through Deregulation of ULK1 (Unc51-like Kinase 1). J. Biol. Chem. 2015, 290, 12184–12194. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Rai, P.; Onder, T.T.; Young, J.J.; McFaline, J.L.; Pang, B.; Dedon, P.C.; Weinberg, R.A. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc. Natl. Acad. Sci. USA 2009, 106, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; Wang, Y.; Vanhoutte, P.M. Senescence of cultured porcine coronary arterial endothelial cells is associated with accelerated oxidative stress and activation of NFkB. J. Vasc. Res. 2010, 47, 287–298. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Li, G.; Luna, C.; Qiu, J.; Epstein, D.L.; Gonzalez, P. Alterations in microRNA expression in stress-induced cellular senescence. Mech. Ageing Dev. 2009, 130, 731–741. [Google Scholar] [CrossRef]
- De Cecco, M.; Ito, T.; Petrashen, A.P.; Elias, A.E.; Skvir, N.J.; Criscione, S.W.; Caligiana, A.; Brocculi, G.; Adney, E.M.; Boeke, J.D.; et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 2019, 566, 73–78. [Google Scholar] [CrossRef]
- Watad, A.; Bragazzi, N.L.; Adawi, M.; Amital, H.; Toubi, E.; Porat, B.S.; Shoenfeld, Y. Autoimmunity in the Elderly: Insights from Basic Science and Clinics—A Mini-Review. Gerontology 2017, 63, 515–523. [Google Scholar] [CrossRef]
- Agrawal, A.; Sridharan, A.; Prakash, S.; Agrawal, H. Dendritic cells and aging: Consequences for autoimmunity. Expert Rev. Clin. Immunol. 2012, 8, 73–80. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Morsiani, C.; Conte, M.; Santoro, A.; Grignolio, A.; Monti, D.; Capri, M.; Salvioli, S. The Continuum of Aging and Age-Related Diseases: Common Mechanisms but Different Rates. Front. Med. (Lausanne) 2018, 5, 61. [Google Scholar] [CrossRef]
- Corrales-Medina, V.F.; Alvarez, K.N.; Weissfeld, L.A.; Angus, D.C.; Chirinos, J.A.; Chang, C.C.; Newman, A.; Loehr, L.; Folsom, A.R.; Elkind, M.S.; et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA 2015, 313, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.N.; Ovsyannikova, I.G.; Poland, G.A.; Kennedy, R.B. Immunosenescence and human vaccine immune responses. Immun. Ageing 2019, 16, 25. [Google Scholar] [CrossRef] [PubMed]
- Kawane, K.; Ohtani, M.; Miwa, K.; Kizawa, T.; Kanbara, Y.; Yoshioka, Y.; Yoshikawa, H.; Nagata, S. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 2006, 443, 998–1002. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Systemic lupus erythematosus. N. Engl. J. Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef]
- Baccala, R.; Hoebe, K.; Kono, D.H.; Beutler, B.; Theofilopoulos, A.N. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat. Med. 2007, 13, 543–551. [Google Scholar] [CrossRef]
- Grieves, J.L.; Fye, J.M.; Harvey, S.; Grayson, J.M.; Hollis, T.; Perrino, F.W. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. USA 2015, 112, 5117–5122. [Google Scholar] [CrossRef]
- Noble, P.W.; Bernatsky, S.; Clarke, A.E.; Isenberg, D.A.; Ramsey-Goldman, R.; Hansen, J.E. DNA-damaging autoantibodies and cancer: The lupus butterfly theory. Nat. Rev. Rheumatol. 2016, 12, 429–434. [Google Scholar] [CrossRef]
- Senejani, A.G.; Liu, Y.; Kidane, D.; Maher, S.E.; Zeiss, C.J.; Park, H.J.; Kashgarian, M.; McNiff, J.M.; Zelterman, D.; Bothwell, A.L.; et al. Mutation of POLB causes lupus in mice. Cell Rep. 2014, 6, 1–8. [Google Scholar] [CrossRef]
- Liu, L.; Tran, E.; Zhao, Y.; Huang, Y.; Flavell, R.; Lu, B. Gadd45 beta and Gadd45 gamma are critical for regulating autoimmunity. J. Exp. Med. 2005, 202, 1341–1347. [Google Scholar] [CrossRef]
- Bassi, C.; Xavier, D.; Palomino, G.; Nicolucci, P.; Soares, C.; Sakamoto-Hojo, E.; Donadi, E. Efficiency of the DNA repair and polymorphisms of the XRCC1, XRCC3 and XRCC4 DNA repair genes in systemic lupus erythematosus. Lupus 2008, 17, 988–995. [Google Scholar] [CrossRef]
- Lin, Y.J.; Wan, L.; Huang, C.; Chen, S.; Huang, Y.; Lai, C.; Lin, W.; Liu, H.P.; Wu, Y.S.; Chen, C.M.; et al. Polymorphisms in the DNA repair gene XRCC1 and associations with systemic lupus erythematosus risk in the Taiwanese Han Chinese population. Lupus 2009, 18, 1246–1251. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.J.; Gao, J.P.; Li, J.; Han, J.W.; Xu, Q.; Hu, W.; Pan, T.; Cheng, Y.; Yu, Z.Y.; Ni, C.; et al. Follow-up study identifies two novel susceptibility loci PRKCB and 8p11.21 for systemic lupus erythematosus. Rheumatology (Oxford) 2011, 50, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Zheng, H.F.; Cui, Y.; Sun, L.; Ye, D.Q.; Hu, Z.; Xu, J.H.; Cai, Z.M.; Huang, W.; Zhao, G.P.; et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Tsao, B.P.; Cantor, R.M.; Grossman, J.M.; Shen, N.; Teophilov, N.T.; Wallace, D.J.; Arnett, F.C.; Hartung, K.; Goldstein, R.; Kalunian, K.C. PARP alleles within the linked chromosomal region are associated with systemic lupus erythematosus. J. Clin. Invest. 1999, 103, 1135–1140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mireles-Canales, M.P.; González-Chávez, S.A.; Quiñonez-Flores, C.M.; León-López, E.A.; Pacheco-Tena, C. DNA Damage and Deficiencies in the Mechanisms of Its Repair: Implications in the Pathogenesis of Systemic Lupus Erythematosus. J. Immunol. Res. 2018, 2018, 8214379. [Google Scholar] [CrossRef]
- Ballestar, E.; Esteller, M.; Richardson, B.C. The epigenetic face of systemic lupus erythematosus. J. Immunol. 2006, 176, 7143–7147. [Google Scholar] [CrossRef]
- Davies, R.C.; Pettijohn, K.; Fike, F.; Wang, J.; Nahas, S.A.; Tunuguntla, R.; Hu, H.; Gatti, R.A.; McCurdy, D. Defective DNA double-strand break repair in pediatric systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 568–578. [Google Scholar] [CrossRef]
- McConnell, J.R.; Crockard, A.D.; Cairns, A.P.; Bell, A.L. Neutrophils from systemic lupus erythematosus patients demonstrate increased nuclear DNA damage. Clin. Exp. Rheumatol. 2002, 20, 653–660. [Google Scholar]
- Kaplan, M. Neutrophils in the pathogenesis and manifestations of SLE. Nat. Rev. Rheumatol. 2011, 7, 691–699. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef]
- Lee, H.M.; Sugino, H.; Aoki, C.; Nishimoto, N. Underexpression of mitochondrial-DNA encoded ATP synthesis-related genes and DNA repair genes in systemic lupus erythematosus. Arthritis Res. Ther. 2011, 13, R63. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Vega, A.M.; Martínez-Bueno, M.; Oparina, N.Y.; López Herráez, D.; Kristjansdottir, H.; Steinsson, K.; Kozyrev, S.V.; Alarcón-Riquelme, M.E. Whole Exome Sequencing of Patients from Multicase Families with Systemic Lupus Erythematosus Identifies Multiple Rare Variants. Sci. Rep. 2018, 8, 8775. [Google Scholar] [CrossRef] [PubMed]
- Cerboni, B.; Morozzi, G.; Galeazzi, M.; Bellisai, F.; Micheli, V.; Pompucci, G.; Sestini, S. Poly(ADP-ribose) polymerase activity in systemic lupus erythematosus and systemic sclerosis. Hum. Immunol. 2009, 70, 487–491. [Google Scholar] [CrossRef]
- Lee, K.J.; Dong, X.; Wang, J.; Takeda, Y.; Dynan, W.S. Identification of human autoantibodies to the DNA ligase IV/XRCC4 complex and mapping of an autoimmune epitope to a potential regulatory region. J. Immunol. 2002, 169, 3413–3421. [Google Scholar] [CrossRef]
- Fell, V.L.; Schild-Poulter, C. The Ku heterodimer: Function in DNA repair and beyond. Mutat. Res. Rev. Mutat. Res. 2015, 763, 15–29. [Google Scholar] [CrossRef]
- Luo, H.; Wang, L.; Bao, D.; Wang, L.; Zhao, H.; Lian, Y.; Yan, M.; Mohan, C.; Li, Q.Z. Novel Autoantibodies Related to Cell Death and DNA Repair Pathways in Systemic Lupus Erythematosus. Genom. Proteom. Bioinform. 2019, 17, 248–259. [Google Scholar] [CrossRef]
- Mahajan, A.; Herrmann, M.; Muñoz, L.E. Clearance Deficiency and Cell Death Pathways: A Model for the Pathogenesis of SLE. Front. Immunol. 2016, 7, 35. [Google Scholar] [CrossRef]
- Tsokos, G.C.; Lo, M.S.; Costa Reis, P.; Sullivan, K.E. New insights into the immunopathogenesis of systemic lupus erythematosus. Nat. Rev. Rheumatol. 2016, 12, 716–730. [Google Scholar] [CrossRef]
- Park, J.W.; Moon, S.Y.; Lee, J.H.; Park, J.K.; Lee, D.S.; Jung, K.C.; Song, Y.W.; Lee, E.B. Bone marrow analysis of immune cells and apoptosis in patients with systemic lupus erythematosus. Lupus 2014, 23, 975–985. [Google Scholar] [CrossRef]
- Cohen, P.L.; Caricchio, R.; Abraham, V.; Camenisch, T.D.; Jennette, J.C.; Roubey, R.A.; Earp, H.S.; Matsushima, G.; Reap, E.A. Delayed apoptotic cell clearance and lupus-like autoimmunity in mice lacking the c-mer membrane tyrosine kinase. J. Exp. Med. 2002, 196, 135–140. [Google Scholar] [CrossRef]
- Gabrielli, A.; Avvedimento, E.V.; Krieg, T. Scleroderma. N. Engl. J. Med. 2009, 360, 1989–2003. [Google Scholar] [CrossRef] [PubMed]
- Sambo, P.; Baroni, S.S.; Luchetti, M.; Paroncini, P.; Dusi, S.; Orlandini, G.; Gabrielli, A. Oxidative stress in scleroderma: Maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum. 2001, 44, 2653–2664. [Google Scholar] [CrossRef]
- Tsou, P.S.; Talia, N.N.; Pinney, A.J.; Kendzicky, A.; Piera-Velazquez, S.; Jimenez, S.A.; Seibold, J.R.; Phillips, K.; Koch, A.E. Effect of oxidative stress on protein tyrosine phosphatase 1B in scleroderma dermal fibroblasts. Arthritis Rheum. 2012, 64, 1978–1989. [Google Scholar] [CrossRef] [PubMed]
- Servettaz, A.; Goulvestre, C.; Kavian, N.; Nicco, C.; Guilpain, P.; Chéreau, C.; Vuiblet, V.; Guillevin, L.; Mouthon, L.; Weill, B.; et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J. Immunol. 2009, 182, 5855–5864. [Google Scholar] [CrossRef]
- Svegliati, S.; Marrone, G.; Pezone, A.; Spadoni, T.; Grieco, A.; Moroncini, G.; Grieco, D.; Vinciguerra, M.; Agnese, S.; Jüngel, A.; et al. Oxidative DNA damage induces the ATM-mediated transcriptional suppression of the Wnt inhibitor WIF-1 in systemic sclerosis and fibrosis. Sci. Signal. 2014, 7, ra84. [Google Scholar] [CrossRef]
- Mak, A.C.; Tang, P.L.; Cleveland, C.; Smith, M.H.; Kari Connolly, M.; Katsumoto, T.R.; Wolters, P.J.; Kwok, P.Y.; Criswell, L.A. Brief Report: Whole-Exome Sequencing for Identification of Potential Causal Variants for Diffuse Cutaneous Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 2257–2262. [Google Scholar] [CrossRef]
- Shao, L. DNA Damage Response Signals Transduce Stress From Rheumatoid Arthritis Risk Factors Into T Cell Dysfunction. Front. Immunol. 2018, 9, 3055. [Google Scholar] [CrossRef]
- Firestein, G.S.; Nguyen, K.; Aupperle, K.R.; Yeo, M.; Boyle, D.L.; Zvaifler, N.J. Apoptosis in rheumatoid arthritis: p53 overexpression in rheumatoid arthritis synovium. Am. J. Pathol. 1996, 149, 2143. [Google Scholar]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Simelyte, E. DNA mismatch repair enzyme expression in synovial tissue. Ann. Rheum. Dis. 2004, 63, 1695–1699. [Google Scholar] [CrossRef] [PubMed]
- Martelli-Palomino, G.; Paoliello-Paschoalato, A.B.; Crispim, J.C.; Rassi, D.M.; Oliveira, R.D.; Louzada, P.; Lucisano-Valim, Y.M.; Donadi, E.A. DNA damage increase in peripheral neutrophils from patients with rheumatoid arthritis is associated with the disease activity and the presence of shared epitope. Clin. Exp. Rheumatol. 2017, 35, 247–254. [Google Scholar] [PubMed]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular mitochondrial DNA and oxidatively damaged DNA in synovial fluid of patients with rheumatoid arthritis. Arthritis Res. Ther. 2003, 5, R234. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Fujii, H.; Colmegna, I.; Oishi, H.; Goronzy, J.J.; Weyand, C.M. Deficiency of the DNA repair enzyme ATM in rheumatoid arthritis. J. Exp. Med. 2009, 206, 1435–1449. [Google Scholar] [CrossRef]
- Bashir, S.; Harris, G.; Denman, M.A.; Blake, D.R.; Winyard, P.G. Oxidative DNA damage and cellular sensitivity to oxidative stress in human autoimmune diseases. Ann. Rheum. Dis. 1993, 52, 659–666. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Y.; Jin, K.; Wen, Z.; Cao, W.; Wu, B.; Wen, R.; Tian, L.; Berry, G.J.; Goronzy, J.J.; et al. The DNA Repair Nuclease MRE11A Functions as a Mitochondrial Protector and Prevents T Cell Pyroptosis and Tissue Inflammation. Cell Metab. 2019, 30, 477–492. [Google Scholar] [CrossRef]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for combining immune checkpoint blockade with PARP inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef]
- McNally, J.P.; Millen, S.H.; Chaturvedi, V.; Lakes, N.; Terrell, C.E.; Elfers, E.E.; Carroll, K.R.; Hogan, S.P.; Andreassen, P.R.; Kanter, J.; et al. Manipulating DNA damage-response signaling for the treatment of immune-mediated diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E4782–E4791. [Google Scholar] [CrossRef]
- Mishra, N.; Reilly, C.M.; Brown, D.R.; Ruiz, P.; Gilkeson, G.S. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J. Clin. Invest. 2003, 111, 539–552. [Google Scholar] [CrossRef] [PubMed]
- Regna, N.L.; Vieson, M.D.; Luo, X.M.; Chafin, C.B.; Puthiyaveetil, A.G.; Hammond, S.E.; Caudell, D.L.; Jarpe, M.B.; Reilly, C.M. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin. Immunol. 2016, 162, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liao, X.; Vieson, M.D.; Chen, M.; Scott, R.; Kazmierczak, J.; Luo, X.M.; Reilly, C.M. Selective HDAC6 inhibition decreases early stage of lupus nephritis by down-regulating both innate and adaptive immune responses. Clin. Exp. Immunol. 2018, 191, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Waibel, M.; Christiansen, A.J.; Hibbs, M.L.; Shortt, J.; Jones, S.A.; Simpson, I.; Light, A.; O’Donnell, K.; Morand, E.F.; Tarlinton, D.M.; et al. Manipulation of B-cell responses with histone deacetylase inhibitors. Nat. Commun. 2015, 6, 6838. [Google Scholar] [CrossRef] [PubMed]
- Vojinovic, J.; Damjanov, N.; D’Urzo, C.; Furlan, A.; Susic, G.; Pasic, S.; Iagaru, N.; Stefan, M.; Dinarello, C.A. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 1452–1458. [Google Scholar] [CrossRef]
- Li, Y.; Shen, Y.; Hohensinner, P.; Ju, J.; Wen, Z.; Goodman, S.B.; Zhang, H.; Goronzy, J.J.; Weyand, C.M. Deficient Activity of the Nuclease MRE11A Induces T Cell Aging and Promotes Arthritogenic Effector Functions in Patients with Rheumatoid Arthritis. Immunity 2016, 45, 903–916. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souliotis, V.L.; Vlachogiannis, N.I.; Pappa, M.; Argyriou, A.; Ntouros, P.A.; Sfikakis, P.P. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. Int. J. Mol. Sci. 2020, 21, 55. https://doi.org/10.3390/ijms21010055
Souliotis VL, Vlachogiannis NI, Pappa M, Argyriou A, Ntouros PA, Sfikakis PP. DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. International Journal of Molecular Sciences. 2020; 21(1):55. https://doi.org/10.3390/ijms21010055
Chicago/Turabian StyleSouliotis, Vassilis L., Nikolaos I. Vlachogiannis, Maria Pappa, Alexandra Argyriou, Panagiotis A. Ntouros, and Petros P. Sfikakis. 2020. "DNA Damage Response and Oxidative Stress in Systemic Autoimmunity" International Journal of Molecular Sciences 21, no. 1: 55. https://doi.org/10.3390/ijms21010055
APA StyleSouliotis, V. L., Vlachogiannis, N. I., Pappa, M., Argyriou, A., Ntouros, P. A., & Sfikakis, P. P. (2020). DNA Damage Response and Oxidative Stress in Systemic Autoimmunity. International Journal of Molecular Sciences, 21(1), 55. https://doi.org/10.3390/ijms21010055