Novel Loss-of-Function Variants in CDC14A are Associated with Recessive Sensorineural Hearing Loss in Iranian and Pakistani Patients

 , , , ,
, , , ,  ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. Clinical Presentation
2.2. Identification of Two Novel CDC14A Variants
2.3. Functional Characterization of the Splice Variant c.1421+2T>C
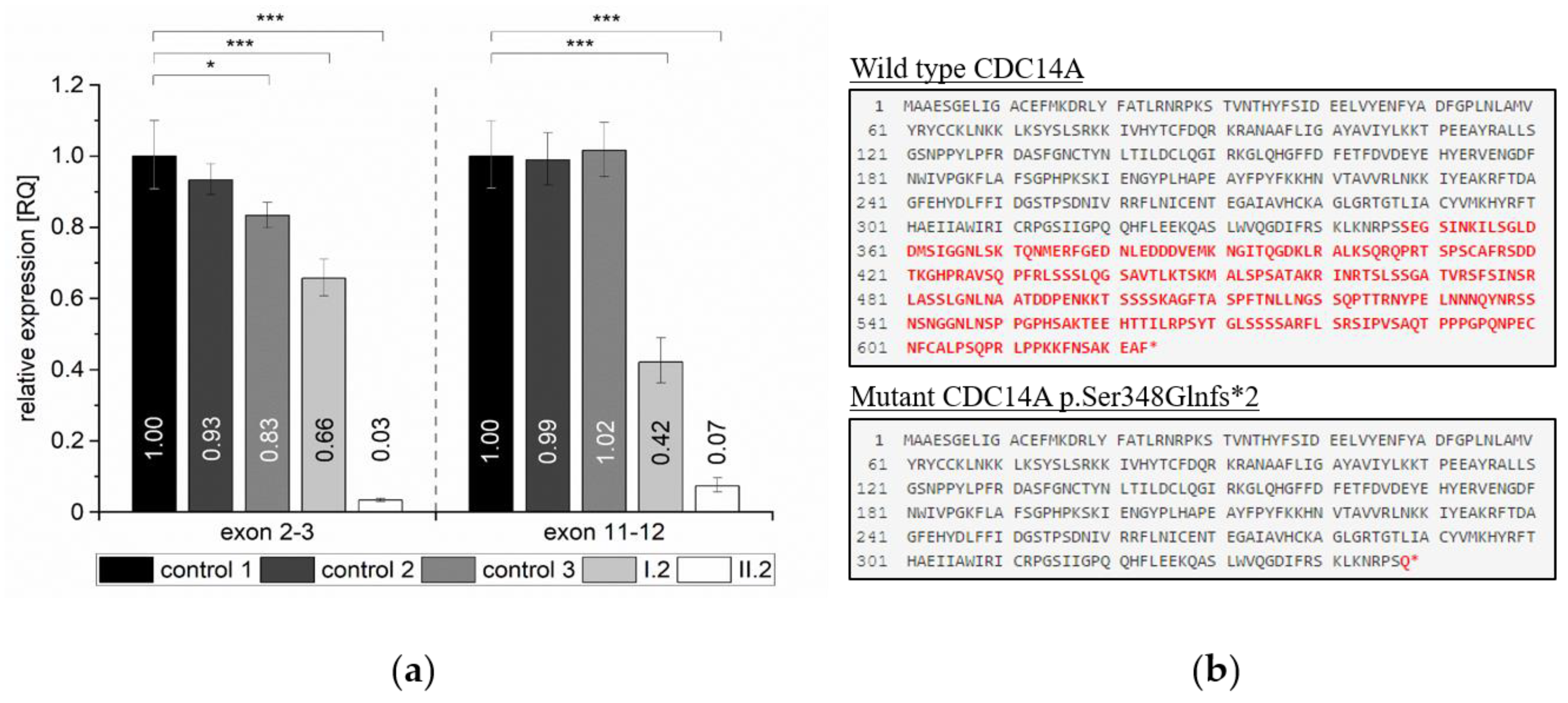
2.4. Quantification of Relative Expression Levels for CDC14A
3. Discussion
4. Materials and Methods
4.1. Patient Recruitment and Clinical Assessment
4.2. Exome Sequencing
4.3. In Silico Variant Analysis
4.4. Gene Mapping Approaches
4.5. Validation of The CDC14A Variants and Segregation Analysis
4.6. Minigene Assay
4.7. Expression Analysis Using Reverse Transcription Quantitative Real-Time PCR (RT-qPCR)
4.8. Statistical Data Collection
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ARNSHL | Autosomal Recessive Non-Syndromic Hearing Loss |
| CDC14A | Cell Division Cycle 14A |
| DFNB32 | Deafness B32 |
| DSPc/PTPc | Dual-specificity phosphatase domain |
| DSPn | Dual-specificity phosphatase domain |
| GJB2 | Gap Junction Protein Beta 2 |
| HIIMS | Hearing Impairment and Infertile Male Syndrome |
| HL | Hearing Loss |
| LD | Linkage Disequilibrium |
| LOD | Logarithm of the Odds |
| NMD | Nonsense-Mediated mRNA Decay |
| PTC | Premature Termination Codon |
| QC | Quality Control |
| RT-qPCR | Reverse Transcription Quantitative Real-Time Polymerase Chain Reaction |
| RQ | Relative Quantification |
| SNP | Single Nucleotide Polymorphism |
| WT | Wild Type |
References
- Vona, B.; Nanda, I.; Hofrichter, M.A.; Shehata-Dieler, W.; Haaf, T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol. Cell. Probes 2015, 29, 260–270. [Google Scholar] [CrossRef]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org/ (accessed on 10 December 2019).
- Delmaghani, S.; Aghaie, A.; Bouyacoub, Y.; El Hachmi, H.; Bonnet, C.; Riahi, Z.; Chardenoux, S.; Perfettini, I.; Hardelin, J.P.; Houmeida, A.; et al. Mutations in CDC14A, Encoding a Protein Phosphatase Involved in Hair Cell Ciliogenesis, Cause Autosomal-Recessive Severe to Profound Deafness. Am. J. Hum. Genet. 2016, 98, 1266–1270. [Google Scholar] [CrossRef] [PubMed]
- Imtiaz, A.; Belyantseva, I.A.; Beirl, A.J.; Fenollar-Ferrer, C.; Bashir, R.; Bukhari, I.; Bouzid, A.; Shaukat, U.; Azaiez, H.; Booth, K.T.; et al. CDC14A phosphatase is essential for hearing and male fertility in mouse and human. Hum. Mol. Genet. 2018, 27, 780–798. [Google Scholar] [CrossRef]
- Stegmeier, F.; Amon, A. Closing mitosis: The functions of the Cdc14 phosphatase and its regulation. Annu. Rev. Genet. 2004, 38, 203–232. [Google Scholar] [CrossRef] [PubMed]
- Mocciaro, A.; Schiebel, E. Cdc14: A highly conserved family of phosphatases with non-conserved functions? J. Cell Sci. 2010, 123, 2867–2876. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef]
- Hilgert, N.; Smith, R.J.; Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: Which ones should be analyzed in DNA diagnostics? Mutat. Res. 2009, 681, 189–196. [Google Scholar] [CrossRef]
- Wang, G.S.; Cooper, T.A. Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat. Rev. Genet. 2007, 8, 749–761. [Google Scholar] [CrossRef]
- Lopez-Bigas, N.; Audit, B.; Ouzounis, C.; Parra, G.; Guigo, R. Are splicing mutations the most frequent cause of hereditary disease? FEBS Lett. 2005, 579, 1900–1903. [Google Scholar] [CrossRef]
- Kapustin, Y.; Chan, E.; Sarkar, R.; Wong, F.; Vorechovsky, I.; Winston, R.M.; Tatusova, T.; Dibb, N.J. Cryptic splice sites and split genes. Nucleic Acids Res. 2011, 39, 5837–5844. [Google Scholar] [CrossRef]
- Balagopal, V.; Fluch, L.; Nissan, T. Ways and means of eukaryotic mRNA decay. Biochim. Biophys. Acta 2012, 1819, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Coyle, S.M.; Doudna, J.A. Coupled 5′ nucleotide recognition and processivity in Xrn1-mediated mRNA decay. Mol. Cell 2011, 41, 600–608. [Google Scholar] [CrossRef] [PubMed]
- Meola, N.; Domanski, M.; Karadoulama, E.; Chen, Y.; Gentil, C.; Pultz, D.; Vitting-Seerup, K.; Lykke-Andersen, S.; Andersen, J.S.; Sandelin, A.; et al. Identification of a Nuclear Exosome Decay Pathway for Processed Transcripts. Mol. Cell 2016, 64, 520–533. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef]
- Maquat, L.E. Nonsense-mediated mRNA decay: Splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol. 2004, 5, 89–99. [Google Scholar] [CrossRef]
- Mazzoli, M.G.; Van Camp, G.U.; Newton, V.; Giarbini, N.; Declau, F.; Parving, A. Recommendations for the description of genetic and audiological data for families with nonsyndromic hereditary hearing impairment. Audiol. Med. 2003, 1, 148–150. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Fattahi, Z.; Beheshtian, M.; Mohseni, M.; Poustchi, H.; Sellars, E.; Nezhadi, S.H.; Amini, A.; Arzhangi, S.; Jalalvand, K.; Jamali, P.; et al. Iranome: A catalog of genomic variations in the Iranian population. Hum. Mutat. 2019, 40, 1968–1984. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRXiv 2019. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T.; et al. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef]
- Fromer, M.; Purcell, S.M. Using XHMM Software to Detect Copy Number Variation in Whole-Exome Sequencing Data. Curr. Protoc. Hum. Genet. 2014, 81, 7–23. [Google Scholar] [CrossRef]
- Shapiro, M.B.; Senapathy, P. RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987, 15, 7155–7174. [Google Scholar] [CrossRef]
- Yeo, G.; Burge, C.B. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J. Comput. Biol. 2004, 11, 377–394. [Google Scholar] [CrossRef]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved splice site detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Lin, X.; Salzberg, S.L. GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Res. 2001, 29, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Seelow, D.; Schuelke, M.; Hildebrandt, F.; Nurnberg, P. HomozygosityMapper—An interactive approach to homozygosity mapping. Nucleic Acids Res. 2009, 37, W593–W599. [Google Scholar] [CrossRef]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef]
- Ruschendorf, F.; Nurnberg, P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics 2005, 21, 2123–2125. [Google Scholar] [CrossRef]
- Abecasis, G.R.; Cherny, S.S.; Cookson, W.O.; Cardon, L.R. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002, 30, 97–101. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Tompson, S.W.; Young, T.L. Assaying the Effects of Splice Site Variants by Exon Trapping in a Mammalian Cell Line. Bio Protoc 2017, 7. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family ID | Variant | Sex | HL Onset | HL Severity | Male Infertility | Ethnicity | Reference |
|---|---|---|---|---|---|---|---|
| HLRB11 | c.376delT p.Tyr126Ilefs*64 | Male Female | Congenital | moderate-to-profound, progressive | Yes | Pakistani | Imtiaz et al. [4] |
| HLAI24 | c.417C>G p.Tyr139Ter | Male Female | Congenital | moderate-to-profound, progressive | Yes | Pakistani | Imtiaz et al. [4] |
| FT1 | c.935G>A p.Arg312Gln | Male Female | Congenital | moderate-to-severe, progressive | n.a. | Tunisian | Imtiaz et al. [4] |
| HPK1 | c.839-3C>G p.? | Male | Congenital | moderate-to-profound, progressive | Yes | Pakistani | Imtiaz et al. [4] |
| MORL1 | c.934C>G p.Arg312Gly | Male Female | Congenital | severe-to-profound, progressive | Yes | Iranian | Imtiaz et al. [4] |
| PKDF539 | c.959A>C p.Gln320Pro | Male | Congenital | severe-to-profound, progressive | Yes | Pakistani | Imtiaz et al. [4] |
| Mauritanian family | c.1015C>T p.Arg339Ter | Male | Congenital | Profound | No | Mauritanian | Delmaghani et al. [3] |
| PKSN10 | c.1033C>T p.Arg345Ter | Male Female | Congenital | moderate-to-profound, progressive | No | Pakistani | Imtiaz et al. [4] |
| MORL2 | c.1126C>T p.Arg376Ter | Male Female | Congenital | moderate-to-profound | No | Iranian | Imtiaz et al. [4] |
| Iranian family | c.1126C>T p.Arg376Ter | Male Female | Congenital | severe-to-profound | No | Iranian | Delmaghani et al. [3] |
| Family 1 | c.1421+2T>C p.Val472Leufs*20 | Female | Congenital | severe-to-profound | No | Iranian | Present study |
| Family 2 | c.1041dup p.Ser348Glnfs*2 | Male | Congenital | Profound | n.a. | Pakistani | Present study |
| Exon | 5′-3′ Primer Sequence (Forward) | 5′-3′ Primer Sequence (Reverse) |
|---|---|---|
| CDC14A Ex2_3 | CCCACTATTTCTCCATCGATGA | GTACACCATTGCCAAGTTCAG |
| CDC14A Ex11_12 | TGGCCTAGATGATATGTCTATTG | CTTCTAAGTTATCCTCTCCAAATC |
| GAPDH | TGCACCACCAACTGCTTAGC | GGCATGGACTGTGGTCATGAG |
| IPO8 | CGAGCTAGATCTTGCTGGGT | CGCTAATTCAACGGCATTTCTT |
| HPRT1 | TGACACTGGCAAAACAATGCA | GGTCCTTTTCACCAGCAAGCT |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doll, J.; Kolb, S.; Schnapp, L.; Rad, A.; Rüschendorf, F.; Khan, I.; Adli, A.; Hasanzadeh, A.; Liedtke, D.; Knaup, S.; et al. Novel Loss-of-Function Variants in CDC14A are Associated with Recessive Sensorineural Hearing Loss in Iranian and Pakistani Patients. Int. J. Mol. Sci. 2020, 21, 311. https://doi.org/10.3390/ijms21010311
Doll J, Kolb S, Schnapp L, Rad A, Rüschendorf F, Khan I, Adli A, Hasanzadeh A, Liedtke D, Knaup S, et al. Novel Loss-of-Function Variants in CDC14A are Associated with Recessive Sensorineural Hearing Loss in Iranian and Pakistani Patients. International Journal of Molecular Sciences. 2020; 21(1):311. https://doi.org/10.3390/ijms21010311
Chicago/Turabian StyleDoll, Julia, Susanne Kolb, Linda Schnapp, Aboulfazl Rad, Franz Rüschendorf, Imran Khan, Abolfazl Adli, Atefeh Hasanzadeh, Daniel Liedtke, Sabine Knaup, and et al. 2020. "Novel Loss-of-Function Variants in CDC14A are Associated with Recessive Sensorineural Hearing Loss in Iranian and Pakistani Patients" International Journal of Molecular Sciences 21, no. 1: 311. https://doi.org/10.3390/ijms21010311
APA StyleDoll, J., Kolb, S., Schnapp, L., Rad, A., Rüschendorf, F., Khan, I., Adli, A., Hasanzadeh, A., Liedtke, D., Knaup, S., Hofrichter, M. A., Müller, T., Dittrich, M., Kong, I.-K., Kim, H.-G., Haaf, T., & Vona, B. (2020). Novel Loss-of-Function Variants in CDC14A are Associated with Recessive Sensorineural Hearing Loss in Iranian and Pakistani Patients. International Journal of Molecular Sciences, 21(1), 311. https://doi.org/10.3390/ijms21010311