Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway



Abstract
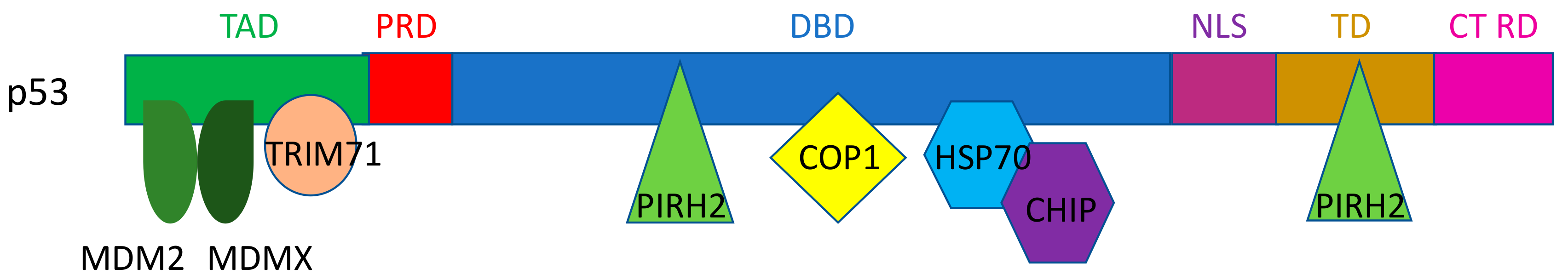
:1. Introduction
2. E3 Ligases for p53
2.1. MDM2/MDMX
2.2. Pirh2
2.3. COP1
2.4. CHIP
2.5. HUWE1
2.6. TRIM Proteins (TRIM24, TRIM28, TRIM29, TRIM39, TRIM69, and TRIM71)
2.7. RING1
2.8. FBW7α
3. E3 Ligases for p63 and p73
3.1. NEDD4
3.2. ITCH
3.3. WWP1
3.4. Pirh2
3.5. MDM2/MDMX
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TAD | Transactivation domain |
| MDM2 | Mouse double minute 2 |
| Pirh2 | p53 induced with a RING-H2 domain protein |
| CHIP | Carboxyl terminus of Hsc70 interacting protein |
| HUWE1 | HECT, UBA, and WWE domain-containing E3 ubiquitin ligase 1 |
| KO | Knockout |
| RING1 | Ring finger protein 1 |
| FBW7 | F-box and WD repeat domain-containing protein 7 |
| Itch/AIP4 | Itchy E3 ubiquitin protein ligase/atrophin-1 interacting protein 4 |
| WWP1 | WW domain-containing E3 ubiquitin protein ligase 1 |
References
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Woods, D.B.; Vousden, K.H. Regulation of p53 function. Exp. Cell Res. 2001, 264, 56–66. [Google Scholar] [CrossRef]
- Moll, U.M.; Petrenko, O. The MDM2-p53 interaction. Mol. Cancer Res. 2003, 1, 1001–1008. [Google Scholar]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar] [CrossRef]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef]
- Yamamoto, S.; Iwakuma, T. Regulators of oncogenic mutant TP53 gain of function. Cancers 2018, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Michael, D.; Oren, M. The p53–Mdm2 module and the ubiquitin system. Semin. Cancer Biol. 2003, 13, 49–58. [Google Scholar] [CrossRef]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Frum, R.A.; Grossman, S.R. Mechanisms of mutant p53 stabilization in cancer. In Mutant p53 and MDM2 in Cancer; Deb, S.P., Deb, S., Eds.; Springer: Dordrecht, The Netherlands, 2014; pp. 187–197. ISBN 978-94-017-9211-0. [Google Scholar]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumor suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Vousden, K.H.; Prives, C. Blinded by the Light: The growing complexity of p53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [Green Version]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a license to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [Green Version]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef]
- Kaghad, M.; Bonnet, H.; Yang, A.; Creancier, L.; Biscan, J.C.; Valent, A.; Minty, A.; Chalon, P.; Lelias, J.M.; Dumont, X.; et al. Monoallelically Expressed Gene Related to p53 at 1p36, a Region frequently deleted in neuroblastoma and other human cancers. Cell 1997, 90, 809–819. [Google Scholar] [CrossRef] [Green Version]
- Schmale, H.; Bamberger, C. A novel protein with strong homology to the tumor suppressor p53. Oncogene 1997, 15, 1363–1367. [Google Scholar] [CrossRef] [Green Version]
- Jost, C.A.; Marin, M.C.; Kaelin, W.G., Jr. p73 is a human p53-related protein that can induce apoptosis. Nature 1997, 389, 191–194. [Google Scholar] [CrossRef]
- Trink, B.; Okami, K.; Wu, L.; Sriuranpong, V.; Jen, J.; Sidransky, D. A new human p53 homologue. Nat. Med. 1998, 4, 747–748. [Google Scholar] [CrossRef]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dötsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 Homolog at 3q27–29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Laurenzi, V.D.; Melino, G. Evolution of functions within the p53/p63/p73 family. Ann. N. Y. Acad. Sci. 2000, 926, 90–100. [Google Scholar] [CrossRef]
- Melino, G.; Lu, X.; Gasco, M.; Crook, T.; Knight, R.A. Functional regulation of p73 and p63: Development and cancer. Trends Biochem. Sci. 2003, 28, 663–670. [Google Scholar] [CrossRef]
- Ishimoto, O.; Kawahara, C.; Enjo, K.; Obinata, M.; Nukiwa, T.; Ikawa, S. Possible oncogenic potential of ΔNp73: A newly identified isoform of human p73. Cancer Res. 2002, 62, 636–641. [Google Scholar]
- Moll, U.M.; Slade, N. p63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar]
- Dötsch, V.; Bernassola, F.; Coutandin, D.; Candi, E.; Melino, G. p63 and p73, the ancestors of p53. Cold Spring Harb. Perspect. Biol. 2010, 2, a004887. [Google Scholar] [CrossRef]
- Murray-Zmijewski, F.; Lane, D.P.; Bourdon, J.C. p53/p63/p73 isoforms: An orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ. 2006, 13, 962–972. [Google Scholar] [CrossRef]
- Vanbokhoven, H.; Melino, G.; Candi, E.; Declercq, W. p63, a Story of mice and men. J. Investig. Dermatol. 2011, 131, 1196–1207. [Google Scholar] [CrossRef]
- Rufini, A.; Agostini, M.; Grespi, F.; Tomasini, R.; Sayan, B.S.; Niklison-Chirou, M.V.; Conforti, F.; Velletri, T.; Mastino, A.; Mak, T.W.; et al. p73 in cancer. Genes Cancer 2011, 2, 491–502. [Google Scholar] [CrossRef]
- Mills, A.A.; Zheng, B.; Wang, X.-J.; Vogel, H.; Roop, D.R.; Bradley, A. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 1999, 398, 708–713. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Yang, A.; McKeon, F. p63 and p73: p53 mimics, menaces and more. Nat. Rev. Mol. Cell Biol. 2000, 1, 199–207. [Google Scholar] [CrossRef]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [CrossRef]
- Keyes, W.M.; Wu, Y.; Vogel, H.; Guo, X.; Lowe, S.W.; Mills, A.A. p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev. 2005, 19, 1986–1999. [Google Scholar] [CrossRef] [Green Version]
- Flores, E.R.; Sengupta, S.; Miller, J.B.; Newman, J.J.; Bronson, R.; Crowley, D.; Yang, A.; McKeon, F.; Jacks, T. Tumor predisposition in mice mutant for p63 and p73: Evidence for broader tumor suppressor functions for the p53 family. Cancer Cell 2005, 7, 363–373. [Google Scholar] [CrossRef] [Green Version]
- Tomasini, R.; Tsuchihara, K.; Wilhelm, M.; Fujitani, M.; Rufini, A.; Cheung, C.C.; Khan, F.; Itie-Youten, A.; Wakeham, A.; Tsao, M.; et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev. 2008, 22, 2677–2691. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, L.M.M.; Durell, S.R.; Mazur, S.J.; Appella, E. p53 N-terminal phosphorylation: A defining layer of complex regulation. Carcinogenesis 2012, 33, 1441–1449. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.L.; Veprintsev, D.B.; Fersht, A.R. Cooperative binding of tetrameric p53 to DNA. J. Mol. Biol. 2004, 341, 1145–1159. [Google Scholar] [CrossRef]
- Joerger, A.C.; Fersht, A.R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008, 77, 557–582. [Google Scholar] [CrossRef]
- Chène, P. The role of tetramerization in p53 function. Oncogene 2001, 20, 2611–2617. [Google Scholar] [CrossRef] [Green Version]
- Maltzman, W.; Czyzyk, L. UV irradiation stimulates levels of p53 cellular tumor antigen in nontransformed mouse cells. Mol. Cell. Biol. 1984, 4, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Price, B.D.; Calderwood, S.K. Increased sequence-specific p53-DNA binding activity after DNA damage is attenuated by phorbol esters. Oncogene 1993, 8, 3055–3062. [Google Scholar]
- Maki, C.G.; Howley, P.M. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol. Cell. Biol. 1997, 17, 355–363. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.N.; Roe, A.E.; Donehower, L.A.; Bradley, A. Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 1995, 378, 206–208. [Google Scholar] [CrossRef]
- Montes de Oca Luna, R.; Wagner, D.S.; Lozano, G. Rescue of early embryonic lethality in mdm2 -deficient mice by deletion of p53. Nature 1995, 378, 203–206. [Google Scholar] [CrossRef]
- Oliner, J.D.; Kinzler, K.W.; Meltzer, P.S.; George, D.L.; Vogelstein, B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature 1992, 358, 80–83. [Google Scholar] [CrossRef]
- Leach, F.S.; Tokino, T.; Meltzer, P.; Burrell, M.; Oliner, J.D.; Smith, S.; Hill, D.E.; Sidransky, D.; Kinzler, K.W.; Vogelstein, B. p53 Mutation and MDM2 amplification in human soft tissue sarcomas. Cancer Res. 1993, 53, 2231–2234. [Google Scholar]
- Watanabe, T.; Hotta, T.; Ichikawa, A.; Kinoshita, T.; Nagai, H.; Uchida, T.; Murate, T.; Saito, H. The MDM2 oncogene overexpression in chronic lymphocytic leukemia and low-grade lymphoma of B-cell origin. Blood 1994, 84, 3158–3165. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Ichikawa, A.; Saito, H.; Hotta, T. Overexpression of the MDM2 oncogene in leukemia and lymphoma. Leuk. Lymphoma 1996, 21, 391–397. [Google Scholar] [CrossRef]
- Korkolopoulou, P.; Christodoulou, P.; Kouzelis, K.; Hadjiyannakis, M.; Priftis, A.; Stamoulis, G.; Seretis, A.; Thomas-Tsagli, E. MDM2 and p53 expression in gliomas: A multivariate survival analysis including proliferation markers and epidermal growth factor receptor. Br. J. Cancer 1997, 75, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Tos, A.P.D.; Doglioni, C.; Piccinin, S.; Sciot, R.; Furlanetto, A.; Boiocchi, M.; Cin, P.D.; Maestro, R.; Fletcher, C.D.M.; Tallini, G. Coordinated expression and amplification of the MDM2, CDK4, and HMGI-C genes in atypical lipomatous tumours. J. Pathol. 2000, 190, 531–536. [Google Scholar] [CrossRef]
- Rayburn, E.; Zhang, R.; He, J.; Wang, H. MDM2 and human malignancies: Expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr. Cancer Drug Targets 2005, 5, 27–41. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.N.; Hancock, A.R.; Vogel, H.; Donehower, L.A.; Bradley, A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 15608–15612. [Google Scholar] [CrossRef] [Green Version]
- Senturk, E.; Manfredi, J.J. Mdm2 and tumorigenesis: Evolving theories and unsolved mysteries. Genes Cancer 2012, 3, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Chen, J.; Marechal, V.; Levine, A.J. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell. Biol. 1993, 13, 4107–4114. [Google Scholar] [CrossRef]
- Parant, J.; Chavez-Reyes, A.; Little, N.A.; Yan, W.; Reinke, V.; Jochemsen, A.G.; Lozano, G. Rescue of embryonic lethality in Mdm4 -null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet. 2001, 29, 92–95. [Google Scholar] [CrossRef]
- Finch, R.A.; Donoviel, D.B.; Potter, D.; Shi, M.; Fan, A.; Freed, D.D.; Wang, C.; Zambrowicz, B.P.; Ramirez-Solis, R.; Sands, A.T.; et al. Mdmx is a negative regulator of p53 activity in vivo. Cancer Res. 2002, 62, 3221–3225. [Google Scholar]
- Migliorini, D.; Denchi, E.L.; Danovi, D.; Jochemsen, A.; Capillo, M.; Gobbi, A.; Helin, K.; Pelicci, P.G.; Marine, J.C. Mdm4 (Mdmx) Regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol. Cell. Biol. 2002, 22, 5527–5538. [Google Scholar] [CrossRef] [Green Version]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The roles of MDM2 and MDMX in cancer. Annu. Rev. Pathol. 2016, 11, 617–644. [Google Scholar] [CrossRef]
- Marine, J.C.; Jochemsen, A.G. MDMX (MDM4), a promising target for p53 reactivation therapy and beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a026237. [Google Scholar] [CrossRef]
- Stad, R.; Ramos, Y.F.M.; Little, N.; Grivell, S.; Attema, J.; van Der Eb, A.J.; Jochemsen, A.G. Hdmx stabilizes Mdm2 and p53. J. Biol. Chem. 2000, 275, 28039–28044. [Google Scholar] [CrossRef] [Green Version]
- Hakem, A.; Bohgaki, M.; Lemmers, B.; Tai, E.; Salmena, L.; Matysiak-Zablocki, E.; Jung, Y.S.; Karaskova, J.; Kaustov, L.; Duan, S.; et al. Role of Pirh2 in mediating the regulation of p53 and c-Myc. PLoS Genet. 2011, 7, e1002360. [Google Scholar] [CrossRef]
- Duan, W.; Gao, L.; Druhan, L.J.; Zhu, W.G.; Morrison, C.; Otterson, G.A.; Villalona-Calero, M.A. Expression of Pirh2, a newly identified ubiquitin protein ligase, in lung cancer. J. Natl. Cancer Inst. 2004, 96, 1718–1721. [Google Scholar] [CrossRef] [Green Version]
- Duan, W.; Gao, L.; Wu, X.; Zhang, Y.; Otterson, G.A.; Villalona-Calero, M.A. Differential response between the p53 ubiquitin–protein ligases Pirh2 and MdM2 following DNA damage in human cancer cells. Exp. Cell Res. 2006, 312, 3370–3378. [Google Scholar] [CrossRef]
- Huang, X.; Qian, X.; Cheng, C.; He, S.; Sun, L.; Ke, Q.; Zhang, L.; Pan, X.; He, F.; Wang, Q.; et al. Expression of Pirh2, a p27Kip1 ubiquitin ligase, in hepatocellular carcinoma: Correlation with p27Kip1 and cell proliferation. Hum. Pathol. 2011, 42, 507–515. [Google Scholar] [CrossRef]
- Leng, R.P.; Lin, Y.; Ma, W.; Wu, H.; Lemmers, B.; Chung, S.; Parant, J.M.; Lozano, G.; Hakem, R.; Benchimol, S. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 2003, 112, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Migliorini, D.; Bogaerts, S.; Defever, D.; Vyas, R.; Denecker, G.; Radaelli, E.; Zwolinska, A.; Depaepe, V.; Hochepied, T.; Skarnes, W.C.; et al. Cop1 constitutively regulates c-Jun protein stability and functions as a tumor suppressor in mice. J. Clin. Investig. 2011, 121, 1329–1343. [Google Scholar] [CrossRef] [Green Version]
- Marine, J.C. Spotlight on the role of COP1 in tumorigenesis. Nat. Rev. Cancer 2012, 12, 455–464. [Google Scholar] [CrossRef]
- Li, Y.; Wang, D.; Zhao, B.; Wang, W.; Huang, C.; Chen, Y.; Zheng, Y.; Keshari, R.P.; Xia, J.; Zhou, Z. High level of COP1 expression is associated with poor prognosis in primary gastric cancer. Int. J. Biol. Sci. 2012, 8, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Wang, L.; Xiao, R.; Pan, Q.; Huang, H.; Kuang, R. High expression of constitutive photomorphogenic 1 (COP1) is associated with poor prognosis in bladder cancer. Tumour Biol. 2016, 37, 8917–8922. [Google Scholar] [CrossRef]
- Zou, S.; Zhu, Y.; Wang, B.; Qian, F.; Zhang, X.; Wang, L.; Fu, C.; Bao, H.; Xie, M.; Gao, S.; et al. The ubiquitin ligase COP1 promotes glioma cell proliferation by preferentially downregulating tumor suppressor p53. Mol. Neurobiol. 2017, 54, 5008–5016. [Google Scholar] [CrossRef]
- Wei, W.; Kaelin, W.G. Good COP1 or bad COP1? In vivo veritas. J. Clin. Investig. 2011, 121, 1263–1265. [Google Scholar] [CrossRef] [Green Version]
- Sane, S.; Rezvani, K. Essential roles of E3 ubiquitin ligases in p53 regulation. Int. J. Mol. Sci. 2017, 18, 442. [Google Scholar] [CrossRef]
- Dornan, D.; Wertz, I.; Shimizu, H.; Arnott, D.; Frantz, G.D.; Dowd, P.; O’Rourke, K.; Koeppen, H.; Dixit, V.M. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004, 429, 86–92. [Google Scholar] [CrossRef]
- Marshall, H.; Bhaumik, M.; Aviv, H.; Moore, D.; Yao, M.; Dutta, J.; Rahim, H.; Gounder, M.; Ganesan, S.; Saleem, A.; et al. Deficiency of the dual ubiquitin/SUMO ligase Topors results in genetic instability and an increased rate of malignancy in mice. BMC Mol. Biol. 2010, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Oyanagi, H.; Takenaka, K.; Ishikawa, S.; Kawano, Y.; Adachi, Y.; Ueda, K.; Wada, H.; Tanaka, F. Expression of LUN gene that encodes a novel RING finger protein is correlated with development and progression of non-small cell lung cancer. Lung Cancer 2004, 46, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Saleem, A.; Dutta, J.; Malegaonkar, D.; Rasheed, F.; Rasheed, Z.; Rajendra, R.; Marshall, H.; Luo, M.; Li, H.; Rubin, E.H. The topoisomerase I- and p53-binding protein Topors is differentially expressed in normal and malignant human tissues and may function as a tumor suppressor. Oncogene 2004, 23, 5293–5300. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Ozaki, T.; Takada, Y.; Kageyama, H.; Nakamura, Y.; Hata, A.; Zhang, J.H.; Simonds, W.F.; Nakagawara, A.; Koseki, H. topors, a p53 and topoisomerase I-binding RING finger protein, is a coactivator of p53 in growth suppression induced by DNA damage. Oncogene 2005, 24, 3385–3396. [Google Scholar] [CrossRef] [Green Version]
- Guan, B.; Pungaliya, P.; Li, X.; Uquillas, C.; Mutton, L.N.; Rubin, E.H.; Bieberich, C.J. Ubiquitination by TOPORS regulates the prostate tumor suppressor NKX3.1. J. Biol. Chem. 2008, 283, 4834–4840. [Google Scholar] [CrossRef] [Green Version]
- Rajendra, R.; Malegaonkar, D.; Pungaliya, P.; Marshall, H.; Rasheed, Z.; Brownell, J.; Liu, L.F.; Lutzker, S.; Saleem, A.; Rubin, E.H. Topors functions as an E3 ubiquitin ligase with specific E2 enzymes and ubiquitinates p53. J. Biol. Chem. 2004, 279, 36440–36444. [Google Scholar] [CrossRef] [Green Version]
- Palubinsky, A.M.; Stankowski, J.N.; Kale, A.C.; Codreanu, S.G.; Singer, R.J.; Liebler, D.C.; Stanwood, G.D.; McLaughlin, B. CHIP is an essential determinant of neuronal mitochondrial stress signaling. Antioxid. Redox Signal. 2015, 23, 535–549. [Google Scholar] [CrossRef] [Green Version]
- Dai, Q.; Zhang, C.; Wu, Y.; McDonough, H.; Whaley, R.A.; Godfrey, V.; Li, H.H.; Madamanchi, N.; Xu, W.; Neckers, L.; et al. CHIP activates HSF1 and confers protection against apoptosis and cellular stress. EMBO J. 2003, 22, 5446–5458. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Zang, J.; Dai, H.J.; Li, F.; Guo, F. Ubiquitin ligase CHIP functions as an oncogene and activates the AKT signaling pathway in prostate cancer. Int. J. Oncol. 2018, 53, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Elengoe, A.; Naser, M.A.; Hamdan, S. A novel protein interaction between nucleotide binding domain of Hsp70 and p53 motif. Int. J. Genom. 2015, 2015, 391293. [Google Scholar] [CrossRef]
- Esser, C.; Scheffner, M.; Höhfeld, J. The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 2005, 280, 27443–27448. [Google Scholar] [CrossRef] [Green Version]
- Kon, N.; Zhong, J.; Qiang, L.; Accili, D.; Gu, W. Inactivation of arf-bp1 induces p53 activation and diabetic phenotypes in mice. J. Biol. Chem. 2012, 287, 5102–5111. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Q.; Gao, W.; Du, F.; Wang, X. Mule/ARF-BP1, a BH3-only E3 ubiquitin ligase, catalyzes the polyubiquitination of Mcl-1 and regulates apoptosis. Cell 2005, 121, 1085–1095. [Google Scholar] [CrossRef] [Green Version]
- Inoue, S.; Hao, Z.; Elia, A.J.; Cescon, D.; Zhou, L.; Silvester, J.; Snow, B.; Harris, I.S.; Sasaki, M.; Li, W.Y.; et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Genes Dev. 2013, 27, 1101–1114. [Google Scholar] [CrossRef] [Green Version]
- Qu, H.; Liu, H.; Jin, Y.; Cui, Z.; Han, G. HUWE1 upregulation has tumor suppressive effect in human prostate cancer cell lines through c-Myc. Biomed. Pharmacother. 2018, 106, 309–315. [Google Scholar] [CrossRef]
- Qi, C.F.; Kim, Y.S.; Xiang, S.; Abdullaev, Z.; Torrey, T.A.; Janz, S.; Kovalchuk, A.L.; Sun, J.; Chen, D.; Cho, W.C.; et al. Characterization of ARF-BP1/HUWE1 interactions with CTCF, MYC, ARF and p53 in MYC-driven B cell neoplasms. Int. J. Mol. Sci. 2012, 13, 6204–6219. [Google Scholar] [CrossRef] [Green Version]
- Confalonieri, S.; Quarto, M.; Goisis, G.; Nuciforo, P.; Donzelli, M.; Jodice, G.; Pelosi, G.; Viale, G.; Pece, S.; Di Fiore, P.P. Alterations of ubiquitin ligases in human cancer and their association with the natural history of the tumor. Oncogene 2009, 28, 2959–2968. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Marinoni, F.; Hock, A.; Hulleman, E.; Popov, N.; Beier, R.; Bernard, S.; Quarto, M.; Capra, M.; Goettig, S.; et al. The ubiquitin ligase HectH9 regulates transcriptional activation by Myc and is essential for tumor cell proliferation. Cell 2005, 123, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.Y.; Lee, Y.; Kim, J.H.; Chung, A.S.; Joo, J.H.; Kim, C.N.; Kim, N.S.; Choe, I.S.; Kim, J.W. Over-expression of human UREB1 in colorectal cancer: HECT domain of human UREB1 inhibits the activity of tumor suppressor p53 protein. Biochem. Biophys. Res. Commun. 2005, 326, 7–17. [Google Scholar] [CrossRef]
- Chen, D.; Kon, N.; Li, M.; Zhang, W.; Qin, J.; Gu, W. ARF-BP1/Mule is a critical mediator of the ARF tumor suppressor. Cell 2005, 121, 1071–1083. [Google Scholar] [CrossRef] [Green Version]
- Cammas, F.; Mark, M.; Dollé, P.; Dierich, A.; Chambon, P.; Losson, R. Mice lacking the transcriptional corepressor TIF1β are defective in early postimplantation development. Development 2000, 127, 2955–2963. [Google Scholar]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef]
- Wang, C.; Ivanov, A.; Chen, L.; Fredericks, W.J.; Seto, E.; Rauscher, F.J.; Chen, J. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J. 2005, 24, 3279–3290. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.U.; Moulin, M.; Coumailleau, F.; Wong, W.W.; Miasari, M.; Carter, H.; Silke, J.; Cohen-Tannoudji, M.; Vince, J.E.; Vaux, D.L. CARP2 deficiency does not alter induction of NF-κB by TNFα. Curr. Biol. 2009, 19, R15–R17. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Rozan, L.M.; McDonald, E.R.; Navaraj, A.; Liu, J.J.; Matthew, E.M.; Wang, W.; Dicker, D.T.; El-Deiry, W.S. CARPs are ubiquitin ligases that promote MDM2-independent p53 and phospho-p53ser20 degradation. J. Biol. Chem. 2007, 282, 3273–3281. [Google Scholar] [CrossRef] [Green Version]
- Yagishita, N.; Ohneda, K.; Amano, T.; Yamasaki, S.; Sugiura, A.; Tsuchimochi, K.; Shin, H.; Kawahara, K.; Ohneda, O.; Ohta, T.; et al. Essential role of synoviolin in embryogenesis. J. Biol. Chem. 2005, 280, 7909–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamasaki, S.; Yagishita, N.; Sasaki, T.; Nakazawa, M.; Kato, Y.; Yamadera, T.; Bae, E.; Toriyama, S.; Ikeda, R.; Zhang, L.; et al. Cytoplasmic destruction of p53 by the endoplasmic reticulum-resident ubiquitin ligase ‘Synoviolin’. EMBO J. 2007, 26, 113–122. [Google Scholar] [CrossRef]
- Khetchoumian, K.; Teletin, M.; Tisserand, J.; Mark, M.; Herquel, B.; Ignat, M.; Zucman-Rossi, J.; Cammas, F.; Lerouge, T.; Thibault, C.; et al. Loss of Trim24 (Tif1α) gene function confers oncogenic activity to retinoic acid receptor α. Nat. Genet. 2007, 39, 1500–1506. [Google Scholar] [CrossRef]
- Tsai, W.W.; Wang, Z.; Yiu, T.T.; Akdemir, K.C.; Xia, W.; Winter, S.; Tsai, C.Y.; Shi, X.; Schwarzer, D.; Plunkett, W.; et al. TRIM24 links a non-canonical histone signature to breast cancer. Nature 2010, 468, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambon, M.; Orsetti, B.; Berthe, M.L.; Bascoul-Mollevi, C.; Rodriguez, C.; Duong, V.; Gleizes, M.; Thénot, S.; Bibeau, F.; Theillet, C.; et al. Prognostic significance of TRIM24/TIF-1α gene expression in breast cancer. Am. J. Pathol. 2011, 178, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Pathiraja, T.N.; Thakkar, K.N.; Jiang, S.; Stratton, S.; Liu, Z.; Gagea, M.; Xi, S.; Shah, P.K.; Phan, L.; Lee, M.H.; et al. TRIM24 links glucose metabolism with transformation of human mammary epithelial cells. Oncogene 2015, 34, 2836–2845. [Google Scholar] [CrossRef] [Green Version]
- Allton, K.; Jain, A.K.; Herz, H.M.; Tsai, W.W.; Jung, S.Y.; Qin, J.; Bergmann, A.; Johnson, R.L.; Barton, M.C. Trim24 targets endogenous p53 for degradation. Proc. Natl. Acad. Sci. USA 2009, 106, 11612–11616. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; He, L.; Li, Z.; Han, X.; Liang, J.; Si, W.; Chen, Z.; Li, L.; Xie, G.; Li, W.; et al. SCFJFK is a bona fide E3 ligase for ING4 and a potent promoter of the angiogenesis and metastasis of breast cancer. Genes Dev. 2015, 29, 672–685. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Shi, L.; Li, W.; Yu, W.; Liang, J.; Zhang, H.; Yang, X.; Wang, Y.; Li, R.; Yao, X.; et al. JFK, a Kelch domain-containing F-box protein, links the SCF complex to p53 regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 10195–10200. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Shin, J.Y.; Seo, J.; Lee, K.D.; Lee, E.W.; Lee, M.S.; Lee, H.W.; Choi, I.J.; Jeong, J.S.; Chun, K.H.; et al. Acceleration of gastric tumorigenesis through MKRN1-mediated posttranslational regulation of p14ARF. J. Natl. Cancer Inst. 2012, 104, 1660–1672. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.W.; Lee, M.S.; Camus, S.; Ghim, J.; Yang, M.R.; Oh, W.; Ha, N.C.; Lane, D.P.; Song, J. Differential regulation of p53 and p21 by MKRN1 E3 ligase controls cell cycle arrest and apoptosis. EMBO J. 2009, 28, 2100–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Huang, N.J.; Chen, C.; Tang, W.; Kornbluth, S. Ubiquitylation of p53 by the APC/C inhibitor Trim39. Proc. Natl. Acad. Sci. USA 2012, 109, 20931–20936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulman, B.R.M.; Liang, X.; Stahlhut, C.; DelConte, C.; Stefani, G.; Slack, F.J. The let-7 microRNA target gene, Mlin41/Trim71 is required for mouse embryonic survival and neural tube closure. Cell Cycle 2008, 7, 3935–3942. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lai, F.; Niswander, L. The ubiquitin ligase mLin41 temporally promotes neural progenitor cell maintenance through FGF signaling. Genes Dev. 2012, 26, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Hao, Q.; Wang, J.; Li, J.; Huang, C.; Zhang, Y.; Wu, X.; Lu, H.; Zhou, X. Ubiquitin ligase TRIM71 suppresses ovarian tumorigenesis by degrading mutant p53. Cell Death Dis. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.T.T.; Richter, D.; Michel, G.; Mitschka, S.; Kolanus, W.; Cuevas, E.; Wulczyn, F.G. The ubiquitin ligase LIN41/TRIM71 targets p53 to antagonize cell death and differentiation pathways during stem cell differentiation. Cell Death Differ. 2017, 24, 1063–1078. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Li, P.; Shao, X.; Yang, Y.; Liu, X.; Feng, M.; Yu, Q.; Hu, R.; Wang, Z. The E3 ligase RING1 targets p53 for degradation and promotes cancer cell proliferation and survival. Cancer Res. 2018, 78, 359–371. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.H.; Perez-Losada, J.; Wu, D.; Delrosario, R.; Tsunematsu, R.; Nakayama, K.I.; Brown, K.; Bryson, S.; Balmain, A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature 2004, 432, 775–779. [Google Scholar] [CrossRef]
- Tripathi, V.; Kaur, E.; Kharat, S.S.; Hussain, M.; Damodaran, A.P.; Kulshrestha, S.; Sengupta, S. Abrogation of FBW7α-dependent p53 degradation enhances p53’s function as a tumor suppressor. J. Biol. Chem. 2019. [Google Scholar] [CrossRef]
- Kimura, T.; Gotoh, M.; Nakamura, Y.; Arakawa, H. hCDC4b, a regulator of cyclin E, as a direct transcriptional target of p53. Cancer Sci. 2003, 94, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Perez-Losada, J.; Mao, J.H.; Balmain, A. Control of genomic instability and epithelial tumor development by the p53-Fbxw7/Cdc4 pathway. Cancer Res. 2005, 65, 6488–6492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Sun, Y.; Chen, X.; Squires, J.; Nowroozizadeh, B.; Liang, C.; Huang, J. p53 mutation directs AURKA overexpression via miR-25 and FBXW7 in prostatic small cell neuroendocrine carcinoma. Mol. Cancer Res. 2015, 13, 584–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcagno, D.Q.; Freitas, V.M.; Leal, M.F.; de Souza, C.R.T.; Demachki, S.; Montenegro, R.; Assumpção, P.P.; Khayat, A.S.; Smith, M.C.; dos Santos, A.K.R.; et al. MYC, FBXW7 and TP53 copy number variation and expression in gastric cancer. BMC Gastroenterol. 2013, 13, 141. [Google Scholar] [CrossRef] [PubMed]
- Grim, J.E.; Knoblaugh, S.E.; Guthrie, K.A.; Hagar, A.; Swanger, J.; Hespelt, J.; Delrow, J.J.; Small, T.; Grady, W.M.; Nakayama, K.I.; et al. Fbw7 and p53 cooperatively suppress advanced and chromosomally unstable intestinal cancer. Mol. Cell. Biol. 2012, 32, 2160–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokobori, T.; Mimori, K.; Iwatsuki, M.; Ishii, H.; Onoyama, I.; Fukagawa, T.; Kuwano, H.; Nakayama, K.I.; Mori, M. p53-altered FBXW7 expression determines poor prognosis in gastric cancer cases. Cancer Res. 2009, 69, 3788–3794. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Inuzuka, H.; Zhong, J.; Wan, L.; Fukushima, H.; Sarkar, F.H.; Wei, W. Tumor suppressor functions of FBW7 in cancer development and progression. FEBS Lett. 2012, 586, 1409–1418. [Google Scholar] [CrossRef] [Green Version]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Li, L.J.; Zheng, J.C.; Kang, R.; Yan, J.Q. Targeting Trim69 alleviates high fat diet (HFD)-induced hippocampal injury in mice by inhibiting apoptosis and inflammation through ASK1 inactivation. Biochem. Biophys. Res. Commun. 2019, 515, 658–664. [Google Scholar] [CrossRef]
- Rong, X.; Rao, J.; Li, D.; Jing, Q.; Lu, Y.; Ji, Y. TRIM69 inhibits cataractogenesis by negatively regulating p53. Redox Biol. 2019, 22, 101157. [Google Scholar] [CrossRef]
- Khan, O.Y.; Fu, G.; Ismail, A.; Srinivasan, S.; Cao, X.; Tu, Y.; Lu, S.; Nawaz, Z. Multifunction steroid receptor coactivator, E6-associated protein, is involved in development of the prostate gland. Mol. Endocrinol. 2006, 20, 544–559. [Google Scholar] [CrossRef] [Green Version]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef]
- Yao, T.P.; Oh, S.P.; Fuchs, M.; Zhou, N.D.; Ch’ng, L.E.; Newsome, D.; Bronson, R.T.; Li, E.; Livingston, D.M.; Eckner, R. Gene dosage–dependent embryonic development and proliferation defects in mice lacking the transcriptional integrator p300. Cell 1998, 93, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.G.; Özdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [Green Version]
- Grossman, S.R.; Perez, M.; Kung, A.L.; Joseph, M.; Mansur, C.; Xiao, Z.X.; Kumar, S.; Howley, P.M.; Livingston, D.M. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol. Cell 1998, 2, 405–415. [Google Scholar] [CrossRef]
- Querido, E.; Morisson, M.R.; Chu-Pham-Dang, H.; Thirlwell, S.W.L.; Boivin, D.; Branton, P.E. Identification of three functions of the adenovirus E4orf6 protein that mediate p53 degradation by the E4orf6-E1B55K complex. J. Virol. 2001, 75, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Boutell, C.; Everett, R.D. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and ubiquitinates p53. J. Biol. Chem. 2003, 278, 36596–36602. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, T.; Matsuzawa, S.; Kress, C.L.; Bruey, J.M.; Krajewska, M.; Lefebvre, S.; Zapata, J.M.; Ronai, Z.; Reed, J.C. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc. Natl. Acad. Sci. USA 2007, 104, 6371–6376. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, W.; Font-Burgada, J.; Palmer, T.; Hamil, A.S.; Biswas, S.K.; Poidinger, M.; Borcherding, N.; Xie, Q.; Ellies, L.G.; et al. Ubiquitin-conjugating enzyme Ubc13 controls breast cancer metastasis through a TAK1-p38 MAP kinase cascade. Proc. Natl. Acad. Sci. USA 2014, 111, 13870–13875. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Shen, S.; Zhang, Z.; Zhang, W.; Xiao, W. Ubiquitin-conjugating enzyme complex Uev1A-Ubc13 promotes breast cancer metastasis through nuclear factor-κB mediated matrix metalloproteinase-1 gene regulation. Breast Cancer Res. 2014, 16, R75. [Google Scholar] [CrossRef] [Green Version]
- Laine, A.; Topisirovic, I.; Zhai, D.; Reed, J.C.; Borden, K.L.B.; Ronai, Z. Regulation of p53 localization and activity by Ubc13. Mol. Cell. Biol. 2006, 26, 8901–8913. [Google Scholar] [CrossRef] [Green Version]
- Cam, L.L.; Lacroix, M.; Ciemerych, M.A.; Sardet, C.; Sicinski, P. The E4F protein is required for mitotic progression during embryonic cell cycles. Mol. Cell. Biol. 2004, 24, 6467–6475. [Google Scholar] [CrossRef] [Green Version]
- Hatchi, E.; Rodier, G.; Lacroix, M.; Caramel, J.; Kirsh, O.; Jacquet, C.; Schrepfer, E.; Lagarrigue, S.; Linares, L.K.; Lledo, G.; et al. E4F1 deficiency results in oxidative stress–mediated cell death of leukemic cells. J. Exp. Med. 2011, 208, 1403–1417. [Google Scholar] [CrossRef]
- Rodier, G.; Kirsh, O.; Baraibar, M.; Houlès, T.; Lacroix, M.; Delpech, H.; Hatchi, E.; Arnould, S.; Severac, D.; Dubois, E.; et al. The Transcription Factor E4F1 Coordinates CHK1-dependent checkpoint and mitochondrial functions. Cell Rep. 2015, 11, 220–233. [Google Scholar] [CrossRef]
- Le Cam, L.; Linares, L.K.; Paul, C.; Julien, E.; Lacroix, M.; Hatchi, E.; Triboulet, R.; Bossis, G.; Shmueli, A.; Rodriguez, M.S.; et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 2006, 127, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Weng, L.; Yuan, B.; Wang, Z.; Jia, L.; Jin, R.; Lu, H.; Li, X.C.; Liu, Y.J.; Zhang, Z. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat. Immunol. 2016, 17, 1373–1380. [Google Scholar] [CrossRef]
- Hatakeyama, S. TRIM proteins and cancer. Nat. Rev. Cancer 2011, 11, 792–804. [Google Scholar] [CrossRef]
- Hatakeyama, S. Early evidence for the role of TRIM29 in multiple cancer models. Expert Opin. Ther. Targets 2016, 20, 767–770. [Google Scholar] [CrossRef] [Green Version]
- Kosaka, Y.; Inoue, H.; Ohmachi, T.; Yokoe, T.; Matsumoto, T.; Mimori, K.; Tanaka, F.; Watanabe, M.; Mori, M. Tripartite motif-containing 29 (TRIM29) is a novel marker for lymph node metastasis in gastric cancer. Ann. Surg. Oncol. 2007, 14, 2543–2549. [Google Scholar] [CrossRef]
- Ai, L.; Kim, W.J.; Alpay, M.; Tang, M.; Pardo, C.E.; Hatakeyama, S.; May, W.S.; Kladde, M.P.; Heldermon, C.D.; Siegel, E.M.; et al. TRIM29 suppresses TWIST1 and invasive breast cancer behavior. Cancer Res. 2014, 74, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Yanagi, T.; Watanabe, M.; Hata, H.; Kitamura, S.; Imafuku, K.; Yanagi, H.; Homma, A.; Wang, L.; Takahashi, H.; Shimizu, H.; et al. Loss of TRIM29 alters keratin distribution to promote cell invasion in squamous cell carcinoma. Cancer Res. 2018, 78, 6795–6806. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Villagra, A.; Peng, L.; Coppola, D.; Glozak, M.; Sotomayor, E.M.; Chen, J.; Lane, W.S.; Seto, E. The ATDC (TRIM29) protein binds p53 and antagonizes p53-mediated functions. Mol. Cell. Biol. 2010, 30, 3004–3015. [Google Scholar] [CrossRef] [Green Version]
- Kaneko-Oshikawa, C.; Nakagawa, T.; Yamada, M.; Yoshikawa, H.; Matsumoto, M.; Yada, M.; Hatakeyama, S.; Nakayama, K.; Nakayama, K.I. Mammalian E4 is required for cardiac development and maintenance of the nervous system. Mol. Cell. Biol. 2005, 25, 10953–10964. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lv, Y.; Zhang, Y.; Gao, H. Regulation of p53 level by UBE4B in breast cancer. PLoS ONE 2014, 9, e90154. [Google Scholar] [CrossRef]
- Carén, H.; Ejeskär, K.; Fransson, S.; Hesson, L.; Latif, F.; Sjöberg, R.M.; Krona, C.; Martinsson, T. A cluster of genes located in 1p36 are down-regulated in neuroblastomas with poor prognosis, but not due to CpG island methylation. Mol. Cancer 2005, 4, 10. [Google Scholar] [CrossRef] [Green Version]
- Zage, P.E.; Sirisaengtaksin, N.; Liu, Y.; Gireud, M.; Brown, B.S.; Palla, S.; Richards, K.N.; Hughes, D.P.M.; Bean, A.J. UBE4B Levels are correlated with clinical outcomes in neuroblastoma patients and with altered neuroblastoma cell proliferation and sensitivity to EGFR inhibitors. Cancer 2013, 119, 915. [Google Scholar] [CrossRef]
- Wu, H.; Leng, R.P. UBE4B, a ubiquitin chain assembly factor, is required for MDM2-mediated p53 polyubiquitination and degradation. Cell Cycle 2011, 10, 1912–1915. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Pomeroy, S.L.; Ferreira, M.; Teider, N.; Mariani, J.; Nakayama, K.I.; Hatakeyama, S.; Tron, V.A.; Saltibus, L.F.; Spyracopoulos, L.; et al. UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 2011, 17, 347–355. [Google Scholar] [CrossRef]
- Cahilly-Snyder, L.; Yang-Feng, T.; Francke, U.; George, D.L. Molecular analysis and chromosomal mapping of amplified genes isolated from a transformed mouse 3T3 cell line. Somat. Cell Mol. Genet. 1987, 13, 235–244. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Honda, R.; Tanaka, H.; Yasuda, H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997, 420, 25–27. [Google Scholar] [CrossRef] [Green Version]
- Honda, R.; Yasuda, H. Activity of MDM2, a ubiquitin ligase, toward p53 or itself is dependent on the RING finger domain of the ligase. Oncogene 2000, 19, 1473–1476. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Brooks, C.L.; Wu-Baer, F.; Chen, D.; Baer, R.; Gu, W. Mono-versus polyubiquitination: Differential control of p53 fate by Mdm2. Science 2003, 302, 1972–1975. [Google Scholar] [CrossRef] [Green Version]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [CrossRef]
- Perry, M.E.; Piette, J.; Zawadzki, J.A.; Harvey, D.; Levine, A.J. The mdm-2 gene is induced in response to UV light in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1993, 90, 11623–11627. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Bayle, J.H.; Olson, D.; Levine, A.J. The p53-mdm-2 autoregulatory feedback loop. Genes Dev. 1993, 7, 1126–1132. [Google Scholar] [CrossRef] [Green Version]
- Shvarts, A.; Steegenga, W.T.; Riteco, N.; van Laar, T.; Dekker, P.; Bazuine, M.; van Ham, R.C.; van der Houven van Oordt, W.; Hateboer, G.; van der Eb, A.J.; et al. MDMX: A novel p53-binding protein with some functional properties of MDM2. EMBO J. 1996, 15, 5349–5357. [Google Scholar] [CrossRef]
- Stad, R.; Little, N.A.; Xirodimas, D.P.; Frenk, R.; van der Eb, A.J.; Lane, D.P.; Saville, M.K.; Jochemsen, A.G. Mdmx stabilizes p53 and Mdm2 via two distinct mechanisms. EMBO Rep. 2001, 2, 1029–1034. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Iyappan, S.; Scheffner, M. Hetero-oligomerization with MdmX rescues the ubiquitin/Nedd8 ligase activity of RING finger mutants of Mdm2. J. Biol. Chem. 2007, 282, 10901–10907. [Google Scholar] [CrossRef] [Green Version]
- Itahana, K.; Mao, H.; Jin, A.; Itahana, Y.; Clegg, H.V.; Lindström, M.S.; Bhat, K.P.; Godfrey, V.L.; Evan, G.I.; Zhang, Y. Targeted inactivation of Mdm2 RING finger E3 ubiquitin ligase activity in the mouse reveals mechanistic insights into p53 regulation. Cancer Cell 2007, 12, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef] [Green Version]
- Pant, V.; Xiong, S.; Iwakuma, T.; Quintás-Cardama, A.; Lozano, G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl. Acad. Sci. USA 2011, 108, 11995–12000. [Google Scholar] [CrossRef] [Green Version]
- Tollini, L.A.; Jin, A.; Park, J.; Zhang, Y. Regulation of p53 by Mdm2 E3 Ligase function is dispensable in embryogenesis and development, but essential in response to DNA damage. Cancer Cell 2014, 26, 235–247. [Google Scholar] [CrossRef] [Green Version]
- Stommel, J.M.; Wahl, G.M. Accelerated MDM2 auto-degradation induced by DNA-damage kinases is required for p53 activation. EMBO J. 2004, 23, 1547–1556. [Google Scholar] [CrossRef] [Green Version]
- Xiong, S.; Pant, V.; Suh, Y.A.; Van Pelt, C.S.; Wang, Y.; Valentin–Vega, Y.A.; Post, S.M.; Lozano, G. Spontaneous tumorigenesis in mice overexpressing the p53 negative regulator Mdm4. Cancer Res. 2010, 70, 7148–7154. [Google Scholar] [CrossRef] [Green Version]
- Clercq, S.D.; Gembarska, A.; Denecker, G.; Maetens, M.; Naessens, M.; Haigh, K.; Haigh, J.J.; Marine, J.C. Widespread overexpression of epitope-tagged Mdm4 does not accelerate rumor formation in vivo. Mol. Cell. Biol. 2010, 30, 5394–5405. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Qian, Y.; Chen, X. Pirh2 RING-finger E3 ubiquitin ligase: Its role in tumorigenesis and cancer therapy. FEBS Lett. 2012, 586, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Klijn, J.G.M.; Zhang, Y.; Sieuwerts, A.M.; Look, M.P.; Yang, F.; Talantov, D.; Timmermans, M.; Meijer-van Gelder, M.E.; Yu, J.; et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005, 365, 671–679. [Google Scholar] [CrossRef]
- Chin, K.; DeVries, S.; Fridlyand, J.; Spellman, P.T.; Roydasgupta, R.; Kuo, W.L.; Lapuk, A.; Neve, R.M.; Qian, Z.; Ryder, T.; et al. Genomic and transcriptional aberrations linked to breast cancer pathophysiologies. Cancer Cell 2006, 10, 529–541. [Google Scholar] [CrossRef] [Green Version]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef]
- Berchuck, A.; Iversen, E.S.; Lancaster, J.M.; Pittman, J.; Luo, J.; Lee, P.; Murphy, S.; Dressman, H.K.; Febbo, P.G.; West, M.; et al. Patterns of gene expression that characterize long-term survival in advanced stage serous ovarian cancers. Clin. Cancer Res. 2005, 11, 3686–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raponi, M.; Zhang, Y.; Yu, J.; Chen, G.; Lee, G.; Taylor, J.M.G.; Macdonald, J.; Thomas, D.; Moskaluk, C.; Wang, Y.; et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66, 7466–7472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, E.; Denti, S.; Catena, R.; Rossetti, G.; Polo, S.; Gasparian, S.; Putignano, S.; Rogge, L.; Pardi, R. Characterization of human constitutive photomorphogenesis protein 1, a RING finger ubiquitin ligase that interacts with Jun transcription factors and modulates their transcriptional activity. J. Biol. Chem. 2003, 278, 19682–19690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Andersen, J.B.; Song, H.T.; Judge, A.D.; Seo, D.; Ishikawa, T.; Marquardt, J.U.; Kitade, M.; Durkin, M.E.; Raggi, C.; et al. Definition of ubiquitination modulator COP1 as a novel therapeutic target in human hepatocellular carcinoma. Cancer Res. 2010, 70, 8264–8269. [Google Scholar] [CrossRef] [Green Version]
- Muller, P.; Hrstka, R.; Coomber, D.; Lane, D.P.; Vojtesek, B. Chaperone-dependent stabilization and degradation of p53 mutants. Oncogene 2008, 27, 3371–3383. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Luk, C.T.; Schroer, S.A.; Smith, A.M.; Li, X.; Cai, E.P.; Gaisano, H.; MacDonald, P.E.; Hao, Z.; Mak, T.W.; et al. Dichotomous role of pancreatic HUWE1/MULE/ARF-BP1 in modulating β cell apoptosis in mice under physiological and genotoxic conditions. Diabetologia 2014, 57, 1889–1898. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.J.; Xu, W.M.; Yang, M.; Wang, K.; Chen, Y.; Huang, X.J.; Ma, Q.H. HUWE1 plays important role in mouse preimplantation embryo development and the dysregulation is associated with poor embryo development in humans. Sci. Rep. 2016, 6, 37928. [Google Scholar] [CrossRef]
- King, B.; Boccalatte, F.; Moran-Crusio, K.; Wolf, E.; Wang, J.; Kayembe, C.; Lazaris, C.; Yu, X.; Aranda-Orgilles, B.; Lasorella, A.; et al. The ubiquitin ligase Huwe1 regulates the maintenance and lymphoid commitment of hematopoietic stem cells. Nat. Immunol. 2016, 17, 1312–1321. [Google Scholar] [CrossRef] [Green Version]
- Fok, K.L.; Bose, R.; Sheng, K.; Chang, C.W.; Katz-Egorov, M.; Culty, M.; Su, S.; Yang, M.; Ruan, Y.C.; Chan, H.C.; et al. Huwe1 regulates the establishment and maintenance of spermatogonia by suppressing DNA damage response. Endocrinology 2017, 158, 4000–4016. [Google Scholar] [CrossRef]
- Ozato, K.; Shin, D.M.; Chang, T.H.; Morse, H.C. TRIM family proteins and their emerging roles in innate immunity. Nat. Rev. Immunol. 2008, 8, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, S. TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 2017, 42, 297–311. [Google Scholar] [CrossRef]
- Okamoto, K.; Kitabayashi, I.; Taya, Y. KAP1 dictates p53 response induced by chemotherapeutic agents via Mdm2 interaction. Biochem. Biophys. Res. Commun. 2006, 351, 216–222. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, S.; Li, Q.; Hu, S.; Tashakori, M.; Van Pelt, C.; You, M.J.; Pageon, L.; Lozano, G. Tissue specific and age dependent effects of global Mdm2 loss. J. Pathol. 2014, 233, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Rousseaux, M.W.; Revelli, J.P.; Vázquez-Vélez, G.E.; Kim, J.Y.; Craigen, E.; Gonzales, K.; Beckinghausen, J.; Zoghbi, H.Y. Depleting Trim28 in adult mice is well tolerated and reduces levels of α-synuclein and tau. Elife 2018, 7, e36768. [Google Scholar] [CrossRef]
- Sho, T.; Tsukiyama, T.; Sato, T.; Kondo, T.; Cheng, J.; Saku, T.; Asaka, M.; Hatakeyama, S. TRIM29 negatively regulates p53 via inhibition of Tip60. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 1245–1253. [Google Scholar] [CrossRef]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.J.; Welcker, M.; Clurman, B.E. Tumor suppression by the Fbw7 ubiquitin ligase: Mechanisms and opportunities. Cancer Cell 2014, 26, 455–464. [Google Scholar] [CrossRef] [Green Version]
- Maisse, C.; Guerrieri, P.; Melino, G. p73 and p63 protein stability: The way to regulate function? Biochem. Pharmacol. 2003, 66, 1555–1561. [Google Scholar] [CrossRef]
- Shin, J.S.; Ha, J.H.; Lee, D.H.; Ryu, K.S.; Bae, K.H.; Park, B.C.; Park, S.G.; Yi, G.S.; Chi, S.W. Structural convergence of unstructured p53 family transactivation domains in MDM2 recognition. Cell Cycle 2015, 14, 533–543. [Google Scholar] [CrossRef] [Green Version]
- Fouladkou, F.; Lu, C.; Jiang, C.; Zhou, L.; She, Y.; Walls, J.R.; Kawabe, H.; Brose, N.; Henkelman, R.M.; Huang, A.; et al. The ubiquitin ligase Nedd4-1 is required for heart development and is a suppressor of thrombospondin-1. J. Biol. Chem. 2010, 285, 6770–6780. [Google Scholar] [CrossRef] [Green Version]
- Boase, N.A.; Kumar, S. NEDD4: The founding member of a family of ubiquitin-protein ligases. Gene 2015, 557, 113–122. [Google Scholar] [CrossRef]
- Bakkers, J.; Camacho-Carvajal, M.; Nowak, M.; Kramer, C.; Danger, B.; Hammerschmidt, M. Destabilization of ΔNp63α by Nedd4-mediated ubiquitination and Ubc9-mediated sumoylation, and its implications on dorsoventral patterning of the zebrafish embryo. Cell Cycle 2005, 4, 790–800. [Google Scholar] [CrossRef] [Green Version]
- Giamboi-Miraglia, A.; Cianfarani, F.; Cattani, C.; Lena, A.M.; Serra, V.; Campione, E.; Terrinoni, A.; Zambruno, G.; Odorisio, T.; Daniele, N.D.; et al. The E3 ligase Itch knockout mice show hyperproliferation and wound healing alteration. FEBS J. 2015, 282, 4435–4449. [Google Scholar] [CrossRef]
- Li, P.F.; Zhang, Q.G. Inhibition of ITCH suppresses proliferation and induces apoptosis of lung cancer cells. Cell. Physiol. Biochem. 2018, 48, 1703–1709. [Google Scholar] [CrossRef]
- Rossi, M.; Aqeilan, R.I.; Neale, M.; Candi, E.; Salomoni, P.; Knight, R.A.; Croce, C.M.; Melino, G. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc. Natl. Acad. Sci. USA 2006, 103, 12753–12758. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Simone, M.D.; Pollice, A.; Santoro, R.; Mantia, G.L.; Guerrini, L.; Calabrò, V. Itch/AIP4 associates with and promotes p63 protein degradation. Cell Cycle 2006, 5, 1816–1822. [Google Scholar] [CrossRef]
- Shu, L.; Zhang, H.; Boyce, B.; Xing, L. Ubiquitin E3 ligase Wwp1 negatively regulates osteoblast function by inhibiting osteoblast differentiation and migration. J. Bone Miner. Res. 2013, 28, 1925–1935. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zan, P.; Li, S.; Liu, J.; Wang, J.; Chen, D.; Wang, H.; Qian, Y.; Luo, L.; Huang, X. Knockdown of WWP1 inhibits growth and invasion, but induces apoptosis of osteosarcoma cells. Int. J. Clin. Exp. Pathol. 2015, 8, 7869–7877. [Google Scholar]
- Nguyen Huu, N.S.; Ryder, W.D.J.; Zeps, N.; Flasza, M.; Chiu, M.; Hanby, A.M.; Poulsom, R.; Clarke, R.B.; Baron, M. Tumour-promoting activity of altered WWP1 expression in breast cancer and its utility as a prognostic indicator. J. Pathol. 2008, 216, 93–102. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, Z.; Sheehan, C.E.; Slodkowska, E.; Sheehan, C.B.; Boguniewicz, A.; Ross, J.S. Overexpression of WWP1 is associated with the estrogen receptor and insulin-like growth factor receptor 1 in breast carcinoma. Int. J. Cancer 2009, 124, 2829–2836. [Google Scholar] [CrossRef]
- Chen, C.; Sun, X.; Guo, P.; Dong, X.Y.; Sethi, P.; Zhou, W.; Zhou, Z.; Petros, J.; Frierson, H.F.; Vessella, R.L.; et al. Ubiquitin E3 ligase WWP1 as an oncogenic factor in human prostate cancer. Oncogene 2007, 26, 2386–2394. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Z.; Chen, C. WW domain-containing E3 ubiquitin protein ligase 1 targets p63 transcription factor for ubiquitin-mediated proteasomal degradation and regulates apoptosis. Cell Death Differ. 2008, 15, 1941–1951. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Scialpi, F.; Bernassola, F.; El Sherbini, E.S.; Melino, G. The E3 ubiquitin ligase WWP1 regulates ΔNp63-dependent transcription through Lys63 linkages. Biochem. Biophys. Res. Commun. 2010, 402, 425–430. [Google Scholar] [CrossRef]
- Tetzlaff, M.T.; Yu, W.; Li, M.; Zhang, P.; Finegold, M.; Mahon, K.; Harper, J.W.; Schwartz, R.J.; Elledge, S.J. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc. Natl. Acad. Sci. USA 2004, 101, 3338–3345. [Google Scholar] [CrossRef] [Green Version]
- Tsunematsu, R.; Nakayama, K.; Oike, Y.; Nishiyama, M.; Ishida, N.; Hatakeyama, S.; Bessho, Y.; Kageyama, R.; Suda, T.; Nakayama, K.I. Mouse Fbw7/Sel-10/Cdc4 is required for Notch degradation during vascular development. J. Biol. Chem. 2004, 279, 9417–9423. [Google Scholar] [CrossRef] [Green Version]
- Galli, F.; Rossi, M.; D’Alessandra, Y.; De Simone, M.; Lopardo, T.; Haupt, Y.; Alsheich-Bartok, O.; Anzi, S.; Shaulian, E.; Calabrò, V.; et al. MDM2 and Fbw7 cooperate to induce p63 protein degradation following DNA damage and cell differentiation. J. Cell. Sci. 2010, 123, 2423–2433. [Google Scholar] [CrossRef] [Green Version]
- Jung, Y.S.; Qian, Y.; Yan, W.; Chen, X. Pirh2 E3 ubiquitin ligase modulates keratinocyte differentiation through p63. J. Investig. Dermatol. 2013, 133, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; De Laurenzi, V.; Munarriz, E.; Green, D.R.; Liu, Y.C.; Vousden, K.H.; Cesareni, G.; Melino, G. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005, 24, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Kubo, N.; Okoshi, R.; Nakashima, K.; Shimozato, O.; Nakagawara, A.; Ozaki, T. MDM2 promotes the proteasomal degradation of p73 through the interaction with Itch in HeLa cells. Biochem. Biophys. Res. Commun. 2010, 403, 405–411. [Google Scholar] [CrossRef]
- Aki, D.; Zhang, W.; Liu, Y.C. The E3 ligase Itch in immune regulation and beyond. Immunol. Rev. 2015, 266, 6–26. [Google Scholar] [CrossRef]
- Neira, J.L.; Díaz-García, C.; Prieto, M.; Coutinho, A. The C-terminal SAM domain of p73 binds to the N terminus of MDM2. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 760–770. [Google Scholar] [CrossRef]
- Peschiaroli, A.; Scialpi, F.; Bernassola, F.; Pagano, M.; Melino, G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene 2009, 28, 3157–3166. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Eldakhakhny, S.; Conforti, F.; Crosbie, E.J.; Melino, G.; Sayan, B.S. Pir2/Rnf144b is a potential endometrial cancer biomarker that promotes cell proliferation. Cell Death Dis. 2018, 9, 504. [Google Scholar] [CrossRef] [Green Version]
- Sayan, B.S.; Yang, A.L.; Conforti, F.; Tucci, P.; Piro, M.C.; Browne, G.J.; Agostini, M.; Bernardini, S.; Knight, R.A.; Mak, T.W.; et al. Differential control of TAp73 and ΔNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc. Natl. Acad. Sci. USA 2010, 107, 12877–12882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudryashova, E.; Wu, J.; Havton, L.A.; Spencer, M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009, 18, 1353–1367. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Z.; De La Torre, R.; Barling, A.; Tsujikawa, T.; Hornick, N.; Hanifin, J.; Simpson, E.; Wang, Y.; Swanzey, E.; et al. Trim32 deficiency enhances Th2 immunity and predisposes to features of atopic dermatitis. J. Investig. Dermatol. 2017, 137, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Kano, S.; Miyajima, N.; Fukuda, S.; Hatakeyama, S. Tripartite motif protein 32 facilitates cell growth and migration via degradation of Abl-interactor 2. Cancer Res. 2008, 68, 5572–5580. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Cano, L.; Hillje, A.L.; Fuertes-Alvarez, S.; Marques, M.M.; Blanch, A.; Ian, R.W.; Irwin, M.S.; Schwamborn, J.C.; Marín, M.C. Regulatory feedback loop between TP73 and TRIM32. Cell Death Dis. 2013, 4, e704. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, N.; Maddika, S. WWP2-WWP1 ubiquitin ligase complex coordinated by PPM1G maintains the balance between cellular p73 and ΔNp73 levels. Mol. Cell. Biol. 2014, 34, 3754–3764. [Google Scholar] [CrossRef] [Green Version]
- Min, B.; Ryu, J.; Chi, S.W.; Yi, G.S. Ubiquitination-dependent degradation of p73 by the mitochondrial E3 ubiquitin ligase Hades. Biochem. Biophys. Res. Commun. 2015, 467, 316–321. [Google Scholar] [CrossRef]
- Stindt, M.H.; Muller, P.A.; Ludwig, R.L.; Kehrloesser, S.; Dötsch, V.; Vousden, K.H. Functional interplay between MDM2, p63/p73 and mutant p53. Oncogene 2015, 34, 4300–4310. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Zhou, Z.; Zhou, W.; Dong, J.T.; Seth, A. The WWP1 E3 ubiquitin ligase: A potential molecular target for breast cancer. Cancer Res. 2007, 67, 353. [Google Scholar]
- Zhi, X.; Chen, C. WWP1: A versatile ubiquitin E3 ligase in signaling and diseases. Cell. Mol. Life Sci. 2012, 69, 1425–1434. [Google Scholar] [CrossRef] [PubMed]
- Halaby, M.; Hakem, R.; Hakem, A. Pirh2: An E3 ligase with central roles in the regulation of cell cycle, DNA damage response, and differentiation. Cell Cycle 2013, 12, 2733–2737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.S.; Qian, Y.; Chen, X. The p73 tumor suppressor is targeted by Pirh2 RING finger E3 ubiquitin ligase for the proteasome-dependent degradation. J. Biol. Chem. 2011, 286, 35388–35395. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Abou, Z.R.; Flores, E.R.; Leng, R.P. Pirh2, a ubiquitin E3 ligase, inhibits p73 transcriptional activity by promoting its ubiquitination. Mol. Cancer Res. 2011, 9, 1780–1790. [Google Scholar] [CrossRef] [Green Version]
- Zawacka-Pankau, J.; Kostecka, A.; Sznarkowska, A.; Hedström, E.; Kawiak, A. p73 tumor suppressor protein: A close relative of p53 not only in structure but also in anti-cancer approach? Cell Cycle 2010, 9, 720–728. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Wienzek, S.; König, C.; Roth, J. Inactivation of the p53-homologue p73 by the mdm2-oncoprotein. Oncogene 1999, 18, 2101–2106. [Google Scholar] [CrossRef] [Green Version]
- Bálint, E.; Bates, S.; Vousden, K.H. Mdm2 binds p73α without targeting degradation. Oncogene 1999, 18, 3923–3929. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Chen, L.; Jost, C.A.; Maya, R.; Keller, D.; Wang, X.; Kaelin, W.G.; Oren, M.; Chen, J.; Lu, H. MDM2 suppresses p73 function without promoting p73 degradation. Mol. Cell. Biol. 1999, 19, 3257–3266. [Google Scholar] [CrossRef] [Green Version]
- Little, N.A.; Jochemsen, A.G. Hdmx and Mdm2 can repress transcription activation by p53 but not by p63. Oncogene 2001, 20, 4576–4580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Arooz, T.; Siu, W.Y.; Chiu, C.H.S.; Lau, A.; Yamashita, K.; Poon, R.Y.C. MDM2 and MDMX can interact differently with ARF and members of the p53 family. FEBS Lett. 2001, 490, 202–208. [Google Scholar] [CrossRef]
- Kadakia, M.; Slader, C.; Berberich, S.J. Regulation of p63 function by Mdm2 and MdmX. DNA Cell Biol. 2001, 20, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Ongkeko, W.M.; Wang, X.Q.; Siu, W.Y.; Lau, A.W.; Yamashita, K.; Harris, A.L.; Cox, L.S.; Poon, R.Y. MDM2 and MDMX bind and stabilize the p53-related protein p73. Curr. Biol. 1999, 9, 829–832. [Google Scholar] [CrossRef] [Green Version]
- Calabrò, V.; Mansueto, G.; Parisi, T.; Vivo, M.; Calogero, R.A.; La Mantia, G. The human MDM2 oncoprotein increases the transcriptional activity and the protein level of the p53 homolog p63. J. Biol. Chem. 2002, 277, 2674–2681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zdzalik, M.; Pustelny, K.; Kedracka-Krok, S.; Huben, K.; Pecak, A.; Wladyka, B.; Jankowski, S.; Dubin, A.; Potempa, J.; Dubin, G. Interaction of regulators Mdm2 and Mdmx with transcription factors p53, p63 and p73. Cell Cycle 2010, 9, 4584–4591. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Leng, R.P. MDM2 mediates p73 ubiquitination: A new molecular mechanism for suppression of p73 function. Oncotarget 2015, 6, 21479–21492. [Google Scholar] [CrossRef] [Green Version]
- Riley, M.F.; You, M.J.; Multani, A.S.; Lozano, G. Mdm2 overexpression and p73 loss exacerbate genomic instability and dampen apoptosis resulting in B-cell lymphoma. Oncogene 2016, 35, 358–365. [Google Scholar] [CrossRef] [Green Version]
- Feeley, K.P.; Adams, C.M.; Mitra, R.; Eischen, C.M. Mdm2 is required for survival and growth of p53-deficient cancer cells. Cancer Res. 2017, 77, 3823–3833. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| E3 Ligase | Phenotype of KO Mice | General Role in Cancer | p53 Interaction Site | Year # |
|---|---|---|---|---|
| MDM2 | Embryonically lethal [44,45] | p53 dependent and independent oncogenesis [46,47,48,49,50,51,52,53,54,55] | 1–51 aa and C-terminus [56,57] | 1997 [8] |
| MDMX | Embryonically lethal [58,59,60] | Oncogene [54,55,61,62] | 2000 [63] | |
| PIRH2 | Predisposed to tumorigenesis [64] | Oncogene [65,66,67] Tumor Suppressor [64] | 82–292 aa and the tetramerization domain [68] | 2003 [68] |
| COP1 | Embryonically lethal [69] | Oncogene [70,71,72,73] Tumor Suppressor [69,74] | Regions within the DNA-binding domain [75] | 2004 [76] |
| TOPORS | Viable but high perinatal mortality, genomic instability [77] | Tumor Suppressor [77,78,79,80] Oncogene [81] | 2004 [82] | |
| CHIP | Sterile, high levels of protein oxidation and lipid peroxidation, reduced antioxidant defense system and energetic status [83], sensitive to heat stress [84] | Oncogene [85] | DNA binding domain in p53, Hsp70 and CHIP complex [86] | 2005 [87] |
| HUWE1 (ARF-BP1, MULE) | Embryonically lethal [88] | Tumor suppressor [89,90,91] Oncogene [92,93,94,95] | 2005 [96] | |
| TRIM28 | Embryonically lethal [97] | Oncogene [98] | 2005 [99] | |
| CARP1/2 | CARP2 KO showed no abnormality [100] | 2007 [101] | ||
| SYNOVIOLIN | Embryonically lethal [102] | 2007 [103] | ||
| TRIM 24 | Metastatic HCC [104] | Liver specific Tumor Suppressor [104] Oncogene in breast cancer [105,106,107] | 2009 [108] | |
| JFK/SKP1-CUL1-F-box | Oncogene in breast cancer [109] | 2009 [110] | ||
| MKRN1 | Senescence and tumorigenesis in gastric cancer [111] | 2009 [112] | ||
| TRIM 39 | 2012 [113] | |||
| TRIM 71 | Embryonically lethal [114,115] | Tumor suppressor by degrading mutant p53 [116] | TA domain of mutant p53 [116] | 2017 [117] |
| RING1 | Oncogene [118] | 2018 [118] | ||
| FBW7α | Tumorigenic [119] | Oncogene [120] Tumor Suppressor [119,121,122,123,124,125,126,127,128] | 2019 [120] | |
| TRIM 69 | Increased metabolic disorder after high fat diet treatment [129] | 2019 [130] |
| Name | Type | Phenotype of KO Mice | General Role in Cancer | Year * |
|---|---|---|---|---|
| E6 HPV oncoprotein-E6AP complex | Only in HPV infected cells | Viable but small prostate gland, increased p53 protein levels in prostrate [131] | 1990 [132] | |
| P300/MDM2 | E4 ligase | Embryonic lethal [133] | Oncogene and tumor suppressor [134] | 1998 [135] |
| E4ORF6 and E1B55K | 2001 [136] | |||
| ICP0 | HSV1 Viral Oncoprotein targeting p53 for translocation | 2003 [137] | ||
| UBC13 | E2 conjugating enzyme causing proteasome independent degradation- cytosolic localization, tetramerization inhibition | Embryonic lethal [138] | Oncogene [139,140] | 2006 [141] |
| E4F1 | An atypical E3 ligase (lacking HECT/RING), does not cause degradation or nuclear transport but carry out localization to chromatin | Embryonic lethal [142] | Oncogene [143,144] | 2006 [145] |
| TRIM 29 | TRIM protein lacking RING finger | Increased macrophage production [146] | Oncogene [147,148,149] Tumor Suppressor [150,151] | 2010 [152] |
| UBE4b | E4 Ligase | Embryonic lethal [153] | Oncogene in breast cancer [154] Tumor suppressor [155,156] | 2011 [157,158] |
| E3 Ligase | Isoform Specificity | Phenotype of KO Mice | General Role in Cancer | p63 Interaction Site | Year # |
|---|---|---|---|---|---|
| MDM2 | TAp63α * TAp63γ * | Embryonically lethal [44,45] | Oncogene [46,47,48,49,50,51,52,53,54,55] | TA Domain [200] | 2001 [175] * |
| MDMX | TAp63α * TAp63γ * | Embryonically lethal [58,59,60] | Oncogene [54,55,61,62] | 2001 [175] * | |
| NEDD4 | ΔNp63α | Embryonic lethality at mid gestation with heart defects [201] | Both [202] | PPPY motif in SAM domain [203] | 2005 [203] |
| ITCH | ΔNp63α ΔNp63α | High rate of proliferation and improved wound healing [204] | Oncogene [205] | 109-120 aa of TAp63 and 15-26 aa of ΔNp63 [206,207] | 2006 [206,207] |
| WWP1 | TAp63α ΔNp63α * | Increased rate of bone formation rates [208] | Oncogene in osteosarcoma [209], breast cancer [210,211], and prostate cancer [212] | PPPY motif in SAM domain [213] | 2008 [213] 2010 [214] |
| FBW7-MDM2 | ΔNp63α | Embryo lethality at day 10.5 due to defects in cardiovascular development [215,216] | Oncogene [120] Tumor Suppressor [119,121,122,123,124,125,126,127,128] | Region surrounding S383 [217] | 2010 [217] |
| PIRH2 | TAp63α ΔNp63α | Oncogene [67,71,72] Tumor Suppressor [64] | 2013 [218] |
| E3 Ligase | Isoform Specificity | Phenotype of KO Mice | General Role in Cancer | p73 Interaction Site | Year # |
|---|---|---|---|---|---|
| MDM2 | TAp73α * TAp73β * ΔNp73α ** | Embryonically lethal [44,45] | Oncogene [46,47,48,49,50,51,52,53,54,55] | TA Domain [200] SAM Domain [222] | 1999 [170,171,172] * |
| MDMX | TAp73α * TAp73β * | Embryonically lethal [58,59,60] | Oncogene [54,55,61,62] | 2001 [174] * | |
| ITCH | TAp73α ΔNp73α | High rate of proliferation and improved wound healing [204] | Oncogene [205] | PY region just before the SAM domain of p73, and particularly the Y487 aa residue of TAp73 [219] | 2005 [219] |
| FBXO45 | TAp73α | SAM domain [223] | 2009 [223] | ||
| PIR2/RNF144B | ΔNp73α | Oncogene [224] | 2010 [225] | ||
| PIRH 2 | TAp73α * TAp73β | Predisposed to tumorigenesis [64] | Oncogene [66,67,71] Tumor Suppressor [64] | 2011 [166,167] * | |
| TRIM 32 | TAp73α | Myopathy and neurological deficiencies [226] AD- atopic dermatitis-like inflammatory skin condition [227] | Oncogene [228] | 2013 [229] | |
| WWP2-WWP1 Complex | ΔNp73α | 2014 [230] | |||
| HADES | 2015 [231] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bang, S.; Kaur, S.; Kurokawa, M. Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway. Int. J. Mol. Sci. 2020, 21, 261. https://doi.org/10.3390/ijms21010261
Bang S, Kaur S, Kurokawa M. Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway. International Journal of Molecular Sciences. 2020; 21(1):261. https://doi.org/10.3390/ijms21010261
Chicago/Turabian StyleBang, Scott, Sandeep Kaur, and Manabu Kurokawa. 2020. "Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway" International Journal of Molecular Sciences 21, no. 1: 261. https://doi.org/10.3390/ijms21010261
APA StyleBang, S., Kaur, S., & Kurokawa, M. (2020). Regulation of the p53 Family Proteins by the Ubiquitin Proteasomal Pathway. International Journal of Molecular Sciences, 21(1), 261. https://doi.org/10.3390/ijms21010261