Overexpression of Hepatocyte Chemerin-156 Lowers Tumor Burden in a Murine Model of Diethylnitrosamine-Induced Hepatocellular Carcinoma



Abstract
1. Introduction
2. Results
2.1. Body Composition of Chemerin-156-Expressing Mice
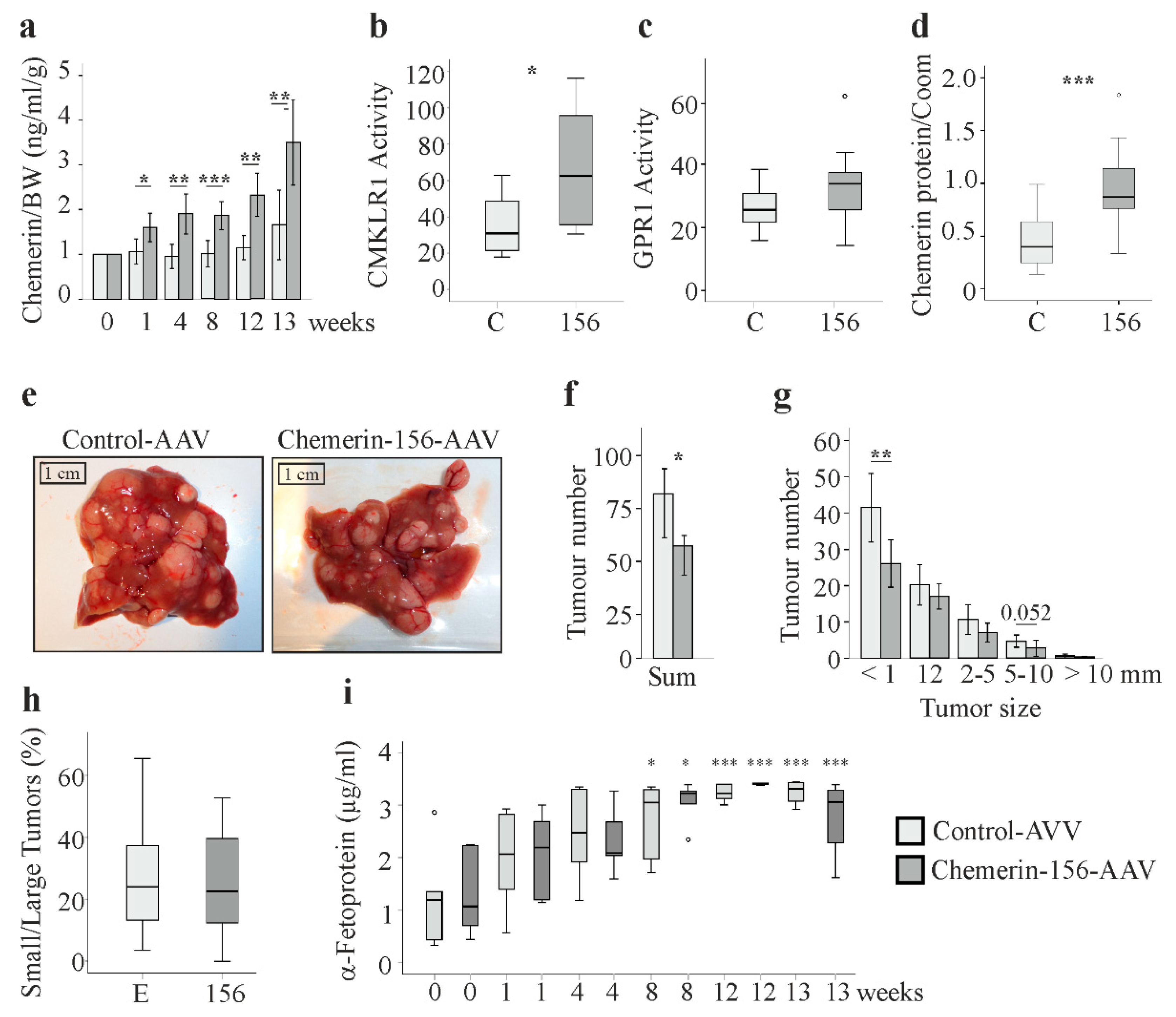
2.2. Serum and Hepatic Chemerin Protein and Activity of Serum Chemerin
2.3. Tumor Number and Size
2.4. Markers of Liver Injury
2.5. Genes and Proteins Already Described to Be Differentially Expressed in Cancer
2.6. Analysis of Genes Highly Expressed by Macrophages and Natural Killer Cells
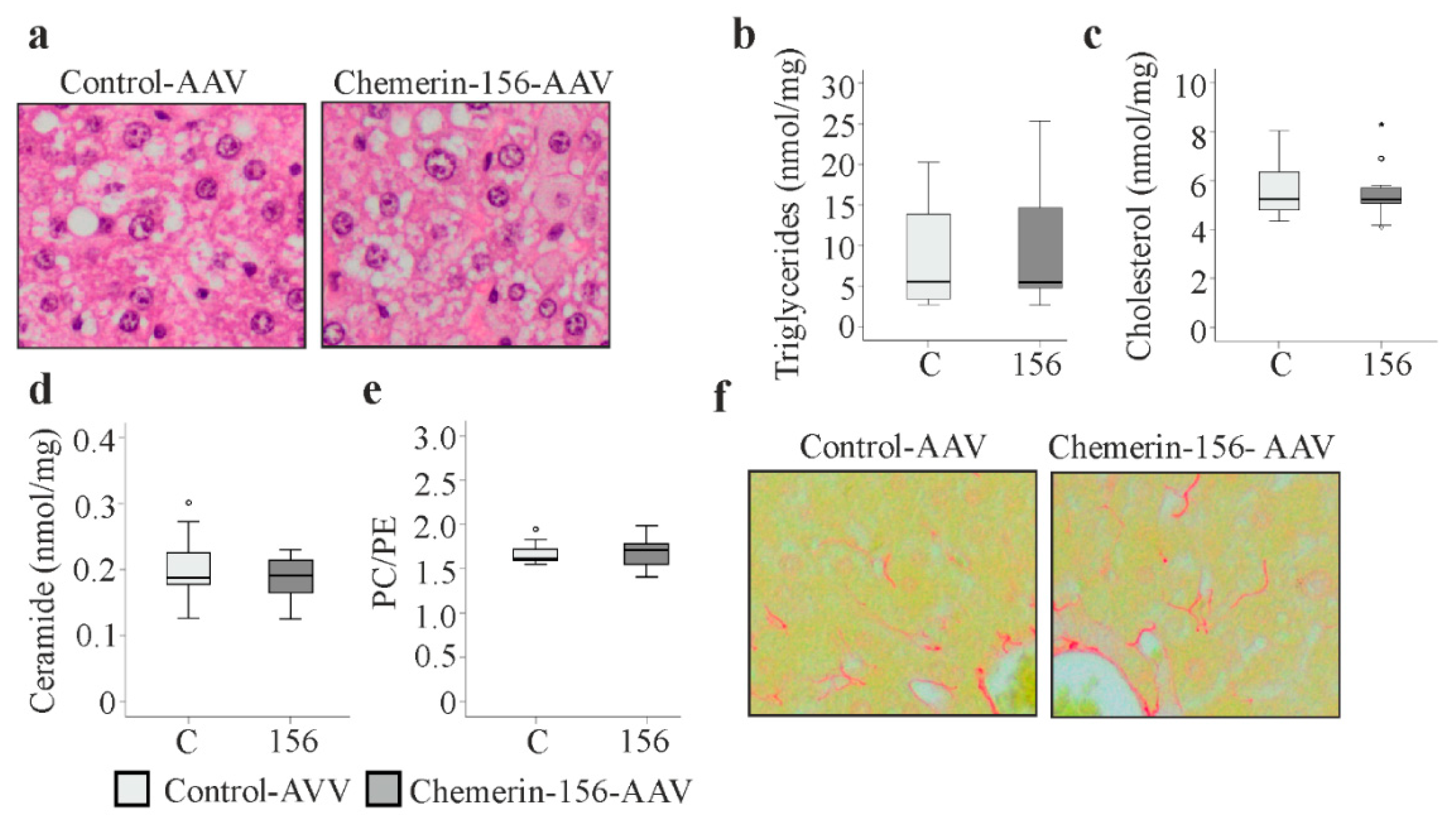
2.7. Lipid Metabolism in Tumorous and Non-Tumorous Liver Tissue
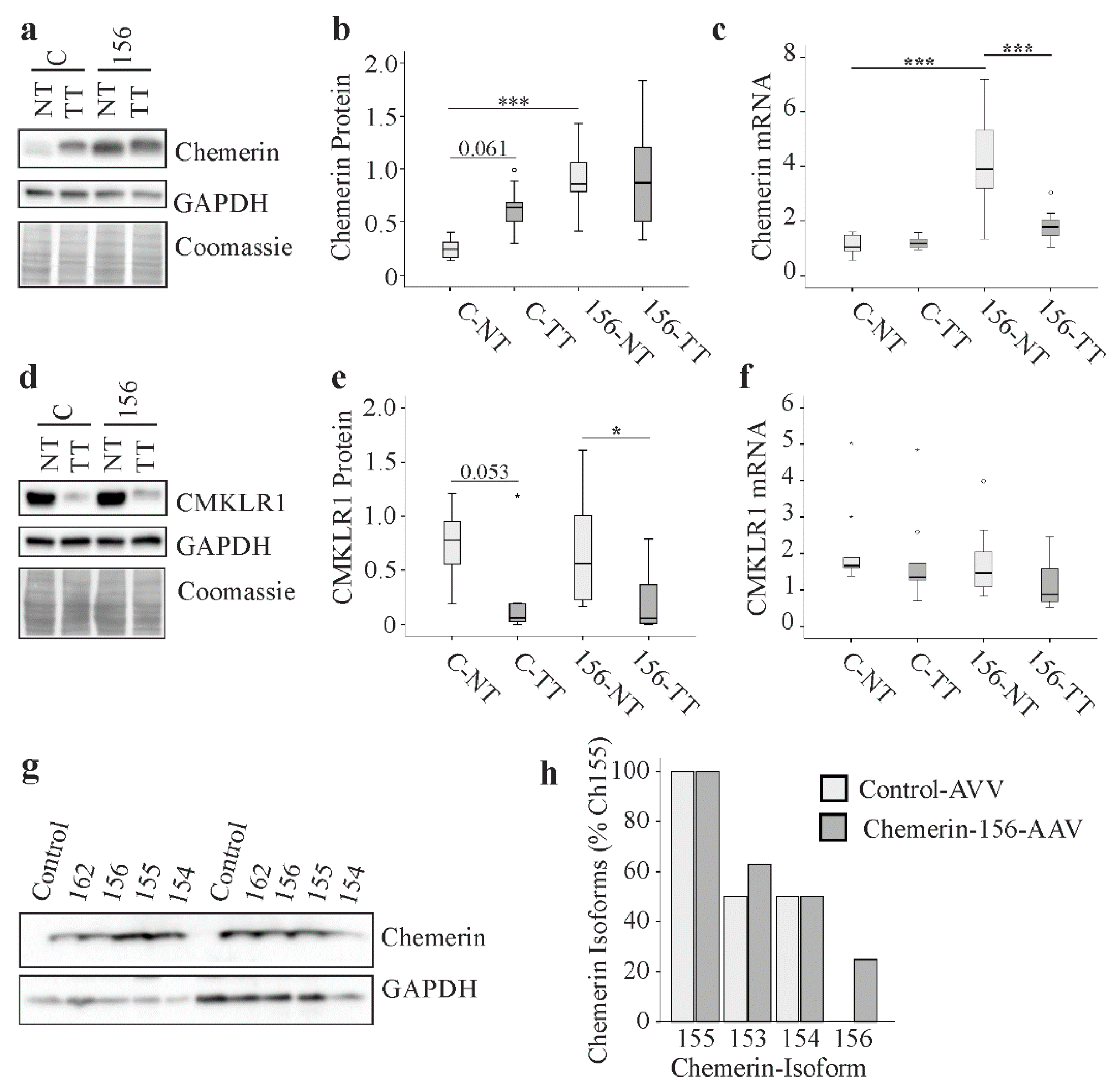
2.8. Chemerin and CMKLR1 in Murine HCC
2.9. Chemerin Isoforms in Murine Liver Tumors
3. Discussion
4. Materials and Methods
4.1. Adenoassociated Virus 8 (AAV8)
4.2. Animals
4.3. Sirius Red and Hematoxylin-Eosin Staining.
4.4. ELISAs
4.5. Measurement of CMKLR1 and GPR1 Activity in Mouse Serum
4.6. Mass Spectrometry of Chemerin Protein
4.7. Lipid Analysis
4.8. Immunoblot
4.9. Semiquantitative Real-Time RT-PCR
4.10. GeneChip Analysis
4.11. Recombinant Expression of Chemerin Isoforms in Hepa1–6 cells
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Badvie, S. Hepatocellular carcinoma. Postgrad. Med. J. 2000, 76, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Wan, S.; Kuo, N.; Kryczek, I.; Zou, W.; Welling, T.H. Myeloid cells in hepatocellular carcinoma. Hepatology 2015, 62, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Rourke, J.L.; Dranse, H.J.; Sinal, C.J. Towards an integrative approach to understanding the role of chemerin in human health and disease. Obes. Rev. 2013, 14, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Buechler, C. Chemerin in Liver Diseases. Endocrinol. Metab. Syndr. 2014, 3, 2–6. [Google Scholar] [CrossRef]
- Buechler, C.; Feder, S.; Haberl, E.M.; Aslanidis, C. Chemerin Isoforms and Activity in Obesity. Int. J. Mol. Sci. 2019, 20, 1128. [Google Scholar] [CrossRef]
- Shin, W.J.; Zabel, B.A.; Pachynski, R.K. Mechanisms and Functions of Chemerin in Cancer: Potential Roles in Therapeutic Intervention. Front. Immunol. 2018, 9, 2772. [Google Scholar] [CrossRef]
- Liu-Chittenden, Y.; Jain, M.; Gaskins, K.; Wang, S.; Merino, M.J.; Kotian, S.; Kumar Gara, S.; Davis, S.; Zhang, L.; Kebebew, E. RARRES2 functions as a tumor suppressor by promoting beta-catenin phosphorylation/degradation and inhibiting p38 phosphorylation in adrenocortical carcinoma. Oncogene 2017, 36, 3541–3552. [Google Scholar] [CrossRef]
- Pachynski, R.K.; Zabel, B.A.; Kohrt, H.E.; Tejeda, N.M.; Monnier, J.; Swanson, C.D.; Holzer, A.K.; Gentles, A.J.; Sperinde, G.V.; Edalati, A.; et al. The chemoattractant chemerin suppresses melanoma by recruiting natural killer cell antitumor defenses. J. Exp. Med. 2012, 209, 1427–1435. [Google Scholar] [CrossRef]
- Tummler, C.; Snapkov, I.; Wickstrom, M.; Moens, U.; Ljungblad, L.; Maria Elfman, L.H.; Winberg, J.O.; Kogner, P.; Johnsen, J.I.; Sveinbjornsson, B. Inhibition of chemerin/CMKLR1 axis in neuroblastoma cells reduces clonogenicity and cell viability in vitro and impairs tumor growth in vivo. Oncotarget 2017, 8, 95135–95151. [Google Scholar] [CrossRef]
- Wang, N.; Wang, Q.J.; Feng, Y.Y.; Shang, W.; Cai, M. Overexpression of chemerin was associated with tumor angiogenesis and poor clinical outcome in squamous cell carcinoma of the oral tongue. Clin. Oral Investig. 2014, 18, 997–1004. [Google Scholar] [CrossRef]
- Kumar, J.D.; Kandola, S.; Tiszlavicz, L.; Reisz, Z.; Dockray, G.J.; Varro, A. The role of chemerin and ChemR23 in stimulating the invasion of squamous oesophageal cancer cells. Br. J. Cancer 2016, 114, 1152–1159. [Google Scholar] [CrossRef]
- Wang, C.; Wu, W.K.; Liu, X.; To, K.F.; Chen, G.G.; Yu, J.; Ng, E.K. Increased serum chemerin level promotes cellular invasiveness in gastric cancer: A clinical and experimental study. Peptides 2014, 51, 131–138. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.L.; Jiang, L.; Chen, J.K. Reduced expression of chemerin is associated with a poor prognosis and a lowed infiltration of both dendritic cells and natural killer cells in human hepatocellular carcinoma. Clin. Lab. 2011, 57, 879–885. [Google Scholar]
- Lin, Y.; Yang, X.; Liu, W.; Li, B.; Yin, W.; Shi, Y.; He, R. Chemerin has a protective role in hepatocellular carcinoma by inhibiting the expression of IL-6 and GM-CSF and MDSC accumulation. Oncogene 2017, 36, 3599–3608. [Google Scholar] [CrossRef]
- Li, J.J.; Yin, H.K.; Guan, D.X.; Zhao, J.S.; Feng, Y.X.; Deng, Y.Z.; Wang, X.; Li, N.; Wang, X.F.; Cheng, S.Q.; et al. Chemerin suppresses hepatocellular carcinoma metastasis through CMKLR1-PTEN-Akt axis. Br. J. Cancer 2018, 118, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Heindryckx, F.; Colle, I.; Van Vlierberghe, H. Experimental mouse models for hepatocellular carcinoma research. Int. J. Exp. Pathol. 2009, 90, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Chu, I.S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Tolba, R.; Kraus, T.; Liedtke, C.; Schwarz, M.; Weiskirchen, R. Diethylnitrosamine (DEN)-induced carcinogenic liver injury in mice. Lab. Anim. 2015, 49, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Tong, M.; Neusner, A.; Longato, L.; Lawton, M.; Wands, J.R.; de la Monte, S.M. Nitrosamine exposure causes insulin resistance diseases: Relevance to type 2 diabetes mellitus, non-alcoholic steatohepatitis, and Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 827–844. [Google Scholar] [PubMed]
- Buchmann, A.; Bauer-Hofmann, R.; Mahr, J.; Drinkwater, N.R.; Luz, A.; Schwarz, M. Mutational activation of the c-Ha-ras gene in liver tumors of different rodent strains: Correlation with susceptibility to hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 1991, 88, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Diwan, B.A.; Rice, J.M.; Ohshima, M.; Ward, J.M. Interstrain differences in susceptibility to liver carcinogenesis initiated by N-nitrosodiethylamine and its promotion by phenobarbital in C57BL/6NCr, C3H/HeNCrMTV- and DBA/2NCr mice. Carcinogenesis 1986, 7, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.H.; Nomura, K.; Kanda, H.; Kusakabe, M.; Yoshiki, A.; Sakakura, T.; Kitagawa, T. Strain specific sensitivity to diethylnitrosamine-induced carcinogenesis is maintained in hepatocytes of C3H/HeN in equilibrium with C57BL/6N chimeric mice. Cancer Res. 1991, 51, 3257–3260. [Google Scholar] [PubMed]
- Schneider, C.; Teufel, A.; Yevsa, T.; Staib, F.; Hohmeyer, A.; Walenda, G.; Zimmermann, H.W.; Vucur, M.; Huss, S.; Gassler, N.; et al. Adaptive immunity suppresses formation and progression of diethylnitrosamine-induced liver cancer. Gut 2012, 61, 1733–1743. [Google Scholar] [CrossRef]
- Bursch, W.; Chabicovsky, M.; Wastl, U.; Grasl-Kraupp, B.; Bukowska, K.; Taper, H.; Schulte-Hermann, R. Apoptosis in stages of mouse hepatocarcinogenesis: Failure to counterbalance cell proliferation and to account for strain differences in tumor susceptibility. Toxicol. Sci. 2005, 85, 515–529. [Google Scholar] [CrossRef][Green Version]
- Schmid, A.; Rignall, B.; Pichler, B.J.; Schwarz, M. Quantitative analysis of the growth kinetics of chemically induced mouse liver tumors by magnetic resonance imaging. Toxicol. Sci. 2012, 126, 52–59. [Google Scholar] [CrossRef]
- Zhao, L.; Yamaguchi, Y.; Shen, W.J.; Morser, J.; Leung, L.L.K. Dynamic and tissue-specific proteolytic processing of chemerin in obese mice. PLoS ONE 2018, 13, e0202780. [Google Scholar] [CrossRef]
- Galle, P.R.; Foerster, F.; Kudo, M.; Chan, S.L.; Llovet, J.M.; Qin, S.; Schelman, W.R.; Chintharlapalli, S.; Abada, P.B.; Sherman, M.; et al. Biology and significance of alpha-fetoprotein in hepatocellular carcinoma. Liver Int. 2019. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Bioactive Lipid Species and Metabolic Pathways in Progression and Resolution of Nonalcoholic Steatohepatitis. Gastroenterology 2018, 155, 282–302.e8. [Google Scholar] [CrossRef]
- Van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef]
- Henderson, J.M.; Polak, N.; Chen, J.; Roediger, B.; Weninger, W.; Kench, J.G.; McCaughan, G.W.; Zhang, H.E.; Gorrell, M.D. Multiple liver insults synergize to accelerate experimental hepatocellular carcinoma. Sci. Rep. 2018, 8, 10283. [Google Scholar] [CrossRef] [PubMed]
- Magee, N.; Zhang, Y. Role of early growth response 1 in liver metabolism and liver cancer. Hepatoma Res. 2017, 3, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Lukk, M.; Kutter, C.; Davies, S.; Alexander, G.; Odom, D.T. Global gene expression profiling reveals SPINK1 as a potential hepatocellular carcinoma marker. PLoS ONE 2013, 8, e59459. [Google Scholar] [CrossRef]
- Teng, F.; Guo, M.; Liu, F.; Wang, C.; Dong, J.; Zhang, L.; Zou, Y.; Chen, R.; Sun, K.; Fu, H.; et al. Treatment with an SLC12A1 antagonist inhibits tumorigenesis in a subset of hepatocellular carcinomas. Oncotarget 2016, 7, 53571–53582. [Google Scholar] [CrossRef]
- Wang, B.; Hsu, S.H.; Frankel, W.; Ghoshal, K.; Jacob, S.T. Stat3-mediated activation of microRNA-23a suppresses gluconeogenesis in hepatocellular carcinoma by down-regulating glucose-6-phosphatase and peroxisome proliferator-activated receptor gamma, coactivator 1 alpha. Hepatology 2012, 56, 186–197. [Google Scholar] [CrossRef]
- Wang, W.; Smits, R.; Hao, H.; He, C. Wnt/beta-Catenin Signaling in Liver Cancers. Cancers 2019, 11, 926. [Google Scholar] [CrossRef]
- He, X.C.; Yin, T.; Grindley, J.C.; Tian, Q.; Sato, T.; Tao, W.A.; Dirisina, R.; Porter-Westpfahl, K.S.; Hembree, M.; Johnson, T.; et al. PTEN-deficient intestinal stem cells initiate intestinal polyposis. Nat. Genet. 2007, 39, 189–198. [Google Scholar] [CrossRef]
- Min, L.; He, B.; Hui, L. Mitogen-activated protein kinases in hepatocellular carcinoma development. Semin. Cancer Biol. 2011, 21, 10–20. [Google Scholar] [CrossRef]
- Nakagawa, H.; Hirata, Y.; Takeda, K.; Hayakawa, Y.; Sato, T.; Kinoshita, H.; Sakamoto, K.; Nakata, W.; Hikiba, Y.; Omata, M.; et al. Apoptosis signal-regulating kinase 1 inhibits hepatocarcinogenesis by controlling the tumor-suppressing function of stress-activated mitogen-activated protein kinase. Hepatology 2011, 54, 185–195. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Ju, Q.; Li, G.C. Tumor markers for hepatocellular carcinoma. Mol. Clin. Oncol. 2013, 1, 593–598. [Google Scholar] [CrossRef]
- Hindupur, S.K.; Colombi, M.; Fuhs, S.R.; Matter, M.S.; Guri, Y.; Adam, K.; Cornu, M.; Piscuoglio, S.; Ng, C.K.Y.; Betz, C.; et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature 2018, 555, 678–682. [Google Scholar] [CrossRef]
- Hu, D.G.; Marri, S.; McKinnon, R.A.; Mackenzie, P.I.; Meech, R. Deregulation of the Genes that Are Involved in Drug Absorption, Distribution, Metabolism, and Excretion in Hepatocellular Carcinoma. J. Pharmacol. Exp. Ther. 2019, 368, 363–381. [Google Scholar] [CrossRef] [PubMed]
- Saelee, P.; Wongkham, S.; Puapairoj, A.; Khuntikeo, N.; Petmitr, S.; Chariyalertsak, S.; Sumethchotimaytha, W.; Karalak, A. Novel PNLIPRP3 and DOCK8 gene expression and prognostic implications of DNA loss on chromosome 10q25.3 in hepatocellular carcinoma. Asian Pac. J. Cancer Prev. APJCP 2009, 10, 501–506. [Google Scholar]
- Morales, A.; Mari, M.; Garcia-Ruiz, C.; Colell, A.; Fernandez-Checa, J.C. Hepatocarcinogenesis and ceramide/cholesterol metabolism. Anti-Cancer Agents Med. Chem. 2012, 12, 364–375. [Google Scholar] [CrossRef][Green Version]
- Jenkins, C.M.; Mancuso, D.J.; Yan, W.; Sims, H.F.; Gibson, B.; Gross, R.W. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 2004, 279, 48968–48975. [Google Scholar] [CrossRef]
- Selitsky, S.R.; Dinh, T.A.; Toth, C.L.; Kurtz, C.L.; Honda, M.; Struck, B.R.; Kaneko, S.; Vickers, K.C.; Lemon, S.M.; Sethupathy, P. Transcriptomic Analysis of Chronic Hepatitis B and C and Liver Cancer Reveals MicroRNA-Mediated Control of Cholesterol Synthesis Programs. mBio 2015, 6, e01500-15. [Google Scholar] [CrossRef] [PubMed]
- Haberl, E.M.; Pohl, R.; Rein-Fischboeck, L.; Feder, S.; Sinal, C.J.; Buechler, C. Chemerin in a Mouse Model of Non-alcoholic Steatohepatitis and Hepatocarcinogenesis. Anticancer Res. 2018, 38, 2649–2657. [Google Scholar] [CrossRef] [PubMed]
- Parikh, J.G.; Kulkarni, A.; Johns, C. alpha-smooth muscle actin-positive fibroblasts correlate with poor survival in hepatocellular carcinoma. Oncol. Lett. 2014, 7, 573–575. [Google Scholar] [CrossRef]
- Huang, J.; Diaz-Meco, M.T.; Moscat, J. The macroenviromental control of cancer metabolism by p62. Cell Cycle 2018, 17, 2110–2121. [Google Scholar] [CrossRef]
- Santos, N.P.; Colaco, A.A.; Oliveira, P.A. Animal models as a tool in hepatocellular carcinoma research: A Review. Tumour Biol. 2017, 39, 1010428317695923. [Google Scholar] [CrossRef]
- Gross, C.; Steiger, K.; Sayyed, S.; Heid, I.; Feuchtinger, A.; Walch, A.; Hess, J.; Unger, K.; Zitzelsberger, H.; Settles, M.; et al. Model Matters: Differences in Orthotopic Rat Hepatocellular Carcinoma Physiology Determine Therapy Response to Sorafenib. Clin. Cancer Res. 2015, 21, 4440–4450. [Google Scholar] [CrossRef]
- Guillabert, A.; Wittamer, V.; Bondue, B.; Godot, V.; Imbault, V.; Parmentier, M.; Communi, D. Role of neutrophil proteinase 3 and mast cell chymase in chemerin proteolytic regulation. J. Leukoc. Biol. 2008, 84, 1530–1538. [Google Scholar] [CrossRef] [PubMed]
- Sansoe, G.; Aragno, M.; Mastrocola, R.; Mengozzi, G.; Novo, E.; Parola, M. Role of Chymase in the Development of Liver Cirrhosis and Its Complications: Experimental and Human Data. PLoS ONE 2016, 11, e0162644. [Google Scholar] [CrossRef] [PubMed]
- Godin, C.; Dupont, S.; Ezzoukhry, Z.; Louandre, C.; Chatelain, D.; Henaut, L.; Sabbagh, C.; Regimbeau, J.M.; Maziere, J.C.; Barbare, J.C.; et al. Heterogeneous sensitivity of hepatocellular carcinoma to sorafenib revealed by the short-term culture of tumor fragments. Anticancer Res. 2013, 33, 1415–1420. [Google Scholar] [PubMed]
- The Human Protein Atlas. Available online: https://www.proteinatlas.org (accessed on 19 December 2019).
- Wanninger, J.; Bauer, S.; Eisinger, K.; Weiss, T.S.; Walter, R.; Hellerbrand, C.; Schaffler, A.; Higuchi, A.; Walsh, K.; Buechler, C. Adiponectin upregulates hepatocyte CMKLR1 which is reduced in human fatty liver. Mol. Cell. Endocrinol. 2012, 349, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Haberl, E.M.; Feder, S.; Pohl, R.; Rein-Fischboeck, L.; Wanninger, J.; Eichelberger, L.; Duerholz, K.; Weiss, T.S.; Buechler, C. Differential regulation of hepatic chemerin according to the etiology of hepatocellular carcinoma. In preparation.
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2015, 67, 103–117. [Google Scholar] [CrossRef]
- Horn, P.; von Loeffelholz, C.; Forkert, F.; Stengel, S.; Reuken, P.; Aschenbach, R.; Stallmach, A.; Bruns, T. Low circulating chemerin levels correlate with hepatic dysfunction and increased mortality in decompensated liver cirrhosis. Sci. Rep. 2018, 8, 9242. [Google Scholar] [CrossRef] [PubMed]
- Feder, S.; Kandulski, A.; Schacherer, D.; Weiss, T.S.; Buechler, C. Serum Chemerin Does Not Differentiate Colorectal Liver Metastases from Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 3919. [Google Scholar] [CrossRef]
- Chang, S.S.; Eisenberg, D.; Zhao, L.; Adams, C.; Leib, R.; Morser, J.; Leung, L. Chemerin activation in human obesity. Obesity (Silver Spring) 2016, 24, 1522–1529. [Google Scholar] [CrossRef]
- Carotenuto, P.; Fassan, M.; Pandolfo, R.; Lampis, A.; Vicentini, C.; Cascione, L.; Paulus-Hock, V.; Boulter, L.; Guest, R.; Quagliata, L.; et al. Wnt signalling modulates transcribed-ultraconserved regions in hepatobiliary cancers. Gut 2017, 66, 1268–1277. [Google Scholar] [CrossRef]
- Preziosi, M.; Poddar, M.; Singh, S.; Monga, S.P. Hepatocyte Wnts Are Dispensable During Diethylnitrosamine and Carbon Tetrachloride-Induced Injury and Hepatocellular Cancer. Gene Expr. 2018, 18, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Ohata, T.; Yokoo, H.; Kamiyama, T.; Fukai, M.; Aiyama, T.; Hatanaka, Y.; Hatanaka, K.; Wakayama, K.; Orimo, T.; Kakisaka, T.; et al. Fatty acid-binding protein 5 function in hepatocellular carcinoma through induction of epithelial-mesenchymal transition. Cancer Med. 2017, 6, 1049–1061. [Google Scholar] [CrossRef]
- Liu, X.; Liang, Y.; Song, R.; Yang, G.; Han, J.; Lan, Y.; Pan, S.; Zhu, M.; Liu, Y.; Wang, Y.; et al. Long non-coding RNA NEAT1-modulated abnormal lipolysis via ATGL drives hepatocellular carcinoma proliferation. Mol. Cancer 2018, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Bhat, M.; Skill, N.; Marcus, V.; Deschenes, M.; Tan, X.; Bouteaud, J.; Negi, S.; Awan, Z.; Aikin, R.; Kwan, J.; et al. Decreased PCSK9 expression in human hepatocellular carcinoma. BMC Gastroenterol. 2015, 15, 176. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Liu, Y.J.; Xia, L.M.; Yan, W.; Zhu, Q.; Tian, D.A. Very low density lipoprotein receptor subtype II silencing by RNA interference inhibits cell proliferation in hepatoma cell lines. Hepato-Gastroenterology 2010, 57, 882–890. [Google Scholar] [PubMed]
- Krautbauer, S.; Meier, E.M.; Rein-Fischboeck, L.; Pohl, R.; Weiss, T.S.; Sigruener, A.; Aslanidis, C.; Liebisch, G.; Buechler, C. Ceramide and polyunsaturated phospholipids are strongly reduced in human hepatocellular carcinoma. Biochim. Biophys. Acta 2016, 1861, 1767–1774. [Google Scholar] [CrossRef]
- Enriquez-Cortina, C.; Bello-Monroy, O.; Rosales-Cruz, P.; Souza, V.; Miranda, R.U.; Toledo-Perez, R.; Luna-Lopez, A.; Simoni-Nieves, A.; Hernandez-Pando, R.; Gutierrez-Ruiz, M.C.; et al. Cholesterol overload in the liver aggravates oxidative stress-mediated DNA damage and accelerates hepatocarcinogenesis. Oncotarget 2017, 8, 104136–104148. [Google Scholar] [CrossRef]
- Schmitz, G.; Langmann, T. Transcriptional regulatory networks in lipid metabolism control ABCA1 expression. Biochim. Biophys. Acta 2005, 1735, 1–19. [Google Scholar] [CrossRef]
- Ernst, M.C.; Issa, M.; Goralski, K.B.; Sinal, C.J. Chemerin exacerbates glucose intolerance in mouse models of obesity and diabetes. Endocrinology 2010, 151, 1998–2007. [Google Scholar] [CrossRef]
- Terada, T.; Matsunaga, Y. Increased mast cells in hepatocellular carcinoma and intrahepatic cholangiocarcinoma. J. Hepatol. 2000, 33, 961–966. [Google Scholar] [CrossRef]
- Sajic, T.; Ciuffa, R.; Lemos, V.; Xu, P.; Leone, V.; Li, C.; Williams, E.G.; Makris, G.; Banaei-Esfahani, A.; Heikenwalder, M.; et al. A new class of protein biomarkers based on subcellular distribution: Application to a mouse liver cancer model. Sci. Rep. 2019, 9, 6913. [Google Scholar] [CrossRef]
- Rourke, J.L.; Muruganandan, S.; Dranse, H.J.; McMullen, N.M.; Sinal, C.J. Gpr1 is an active chemerin receptor influencing glucose homeostasis in obese mice. J. Endocrinol. 2014, 222, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Toulany, J.; Parlee, S.D.; Sinal, C.J.; Slayter, K.; McNeil, S.; Goralski, K.B. CMKLR1 activation ex vivo does not increase proportionally to serum total chemerin in obese humans. Endocr. Connect. 2016, 5, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Horing, M.; Ejsing, C.S.; Hermansson, M.; Liebisch, G. Quantification of Cholesterol and Cholesteryl Ester by Direct Flow Injection High-Resolution Fourier Transform Mass Spectrometry Utilizing Species-Specific Response Factors. Anal. Chem. 2019, 91, 3459–3466. [Google Scholar] [CrossRef] [PubMed]
- Bauer, S.; Wanninger, J.; Schmidhofer, S.; Weigert, J.; Neumeier, M.; Dorn, C.; Hellerbrand, C.; Zimara, N.; Schaffler, A.; Aslanidis, C.; et al. Sterol regulatory element-binding protein 2 (SREBP2) activation after excess triglyceride storage induces Chemerin in hypertrophic adipocytes. Endocrinology 2011, 152, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Bohler(Dutta), A. Adding Automated Statistical Analysis and Biological Evaluation Modules to www.arrayanalysis.org. Master’s Thesis, Manipal University, Manipal, India, 2011. [Google Scholar]
- Eijssen, L.M.; Jaillard, M.; Adriaens, M.E.; Gaj, S.; de Groot, P.J.; Muller, M.; Evelo, C.T. User-friendly solutions for microarray quality control and pre-processing on ArrayAnalysis.org. Nucleic Acids Res. 2013, 41, W71–W76. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Animal Group | Tissue | Animal Number | Median and Range |
|---|---|---|---|---|
| F4/80 | Control-AAV | NT | 9 | 1.7 (1.1–6.1) |
| Control-AAV | TT | 9 | 2.1 (1.1–5.1) | |
| Chemerin-156-AAV | NT | 12 | 1.4 (0.6–5.8) | |
| Chemerin-156-AAV | TT | 12 | 1.2 (0.5–4.9) | |
| CCL3 | Control-AAV | NT | 9 | 0.6 (0.2–0.9) |
| Control-AAV | TT | 8 | 0.4 (0.2–0.9) | |
| Chemerin-156-AAV | NT | 12 | 0.5 (0.3–1.4) | |
| Chemerin-156-AAV | TT | 12 | 0.3 (0.2 – 0.9) | |
| CD38 | Control-AAV | NT | 9 | 1.5 (1.1–3.3) |
| Control-AAV | TT | 9 | 2.4 (1.8–4.1) | |
| Chemerin-156-AAV | NT | 12 | 1.7 (0.5–3.3) | |
| Chemerin-156-AAV | TT | 12 | 2.3 (2.0–3.1) | |
| IL-6 | Control-AAV | NT | 9 | 1.7 (0.3–2.6) |
| Control-AAV | TT | 8 | 0.5 (0.2–2.4) | |
| Chemerin-156-AAV | NT | 12 | 2.3 (0.2–7.2) | |
| Chemerin-156-AAV | TT | 12 | 1.3 (0.1–3.5) | |
| NCR1 | Control-AAV | NT | 9 | 1.6 (0.9–2.7) |
| Control-AAV | TT | 9 | 2.0 (1.3–3.4) | |
| Chemerin-156-AAV | NT | 12 | 2.0 (0.6–4.9) | |
| Chemerin-156-AAV | TT | 12 | 1.9 (0.9–5.1) | |
| Ly49c | Control-AAV | NT | 9 | 0.9 (0.6–1.6) |
| Control-AAV | TT | 9 | 0.7 (0.4–1.5) | |
| Chemerin-156-AAV | NT | 12 | 0.8 (0.4–1.3) | |
| Chemerin-156-AAV | TT | 12 | 0.5 (0.3–1.1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haberl, E.M.; Pohl, R.; Rein-Fischboeck, L.; Feder, S.; Sinal, C.J.; Bruckmann, A.; Hoering, M.; Krautbauer, S.; Liebisch, G.; Buechler, C. Overexpression of Hepatocyte Chemerin-156 Lowers Tumor Burden in a Murine Model of Diethylnitrosamine-Induced Hepatocellular Carcinoma. Int. J. Mol. Sci. 2020, 21, 252. https://doi.org/10.3390/ijms21010252
Haberl EM, Pohl R, Rein-Fischboeck L, Feder S, Sinal CJ, Bruckmann A, Hoering M, Krautbauer S, Liebisch G, Buechler C. Overexpression of Hepatocyte Chemerin-156 Lowers Tumor Burden in a Murine Model of Diethylnitrosamine-Induced Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2020; 21(1):252. https://doi.org/10.3390/ijms21010252
Chicago/Turabian StyleHaberl, Elisabeth M., Rebekka Pohl, Lisa Rein-Fischboeck, Susanne Feder, Christopher J. Sinal, Astrid Bruckmann, Marcus Hoering, Sabrina Krautbauer, Gerhard Liebisch, and Christa Buechler. 2020. "Overexpression of Hepatocyte Chemerin-156 Lowers Tumor Burden in a Murine Model of Diethylnitrosamine-Induced Hepatocellular Carcinoma" International Journal of Molecular Sciences 21, no. 1: 252. https://doi.org/10.3390/ijms21010252
APA StyleHaberl, E. M., Pohl, R., Rein-Fischboeck, L., Feder, S., Sinal, C. J., Bruckmann, A., Hoering, M., Krautbauer, S., Liebisch, G., & Buechler, C. (2020). Overexpression of Hepatocyte Chemerin-156 Lowers Tumor Burden in a Murine Model of Diethylnitrosamine-Induced Hepatocellular Carcinoma. International Journal of Molecular Sciences, 21(1), 252. https://doi.org/10.3390/ijms21010252