Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer



Abstract
1. Introduction
2. Endometrial Cancer: Molecular Classification of Subtypes
- POLE-ultramutated malignancies, accounting for 17.4% of high-grade endometrioid tumours, are characterized by a high mutational burden with somatic mutations in the exonuclease domain of POLE which encodes the catalytic subunit of polymerase epsilon. Despite the histological grade, this group is associated with good prognosis [29,30,31].
- Copy number-high serous-like tumours have serous or mixed histology. They have a low mutation rate and a small load of copy number aberrations. TP53 is commonly mutated (91.7%). They show HER2-amplification and deregulation of the cell cycle (in particular with CCNE1 overexpression [17]). Prognosis of these patients is poor [23,28].
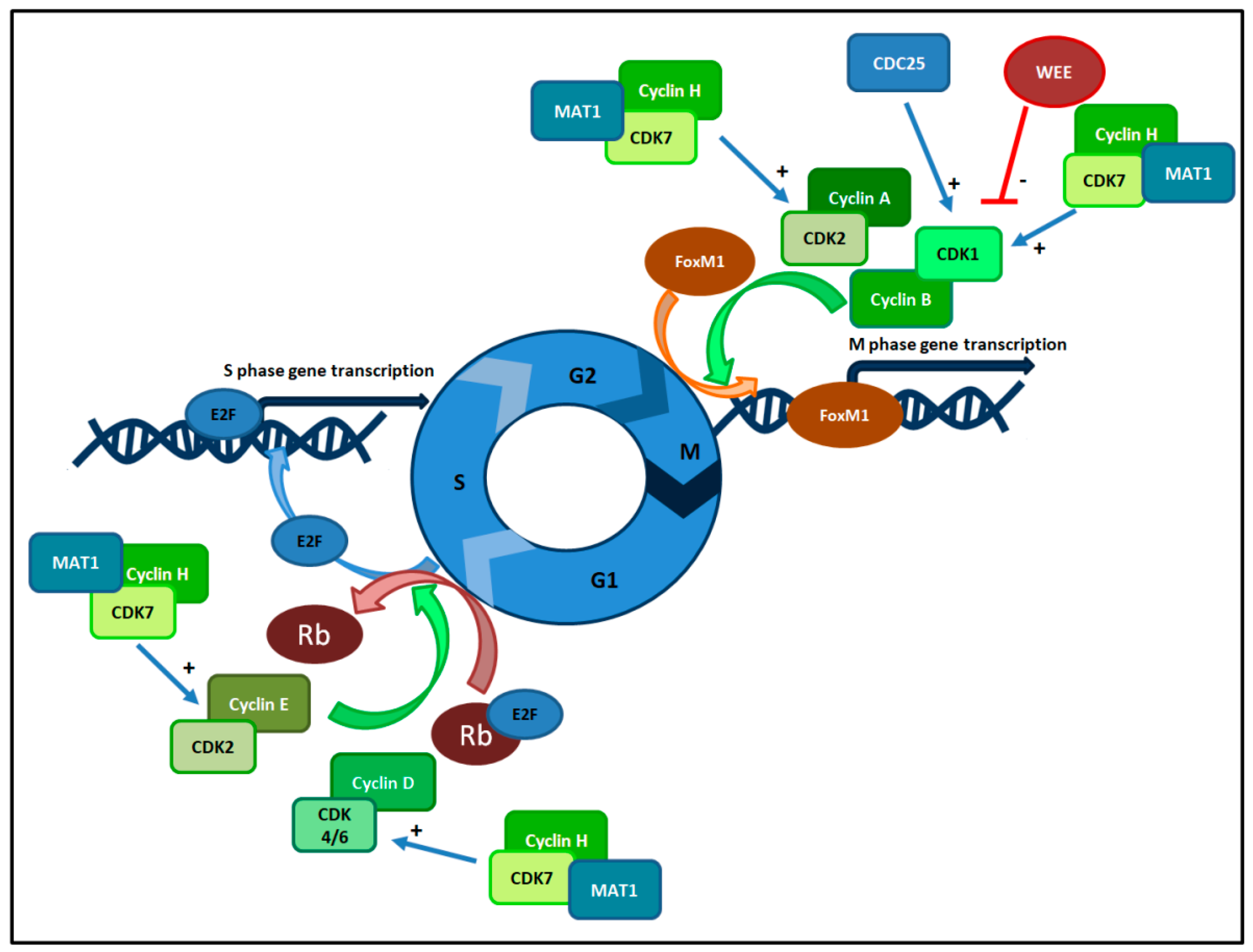
3. Cell Cycle Checkpoints and Cyclin Dependent Kinases Complexes
4. CDKis: Generations and Features
- First generation (e.g., alvocidib): characterized by low potency (pan-CDK), lack of specificity and off target toxic effects;
- Second generation (e.g., dinaciclib, CYC065): characterized by a non-selective inhibition of a wide range of CDKs [42] and equivalent activity in normal and tumour cells. Among these drugs, Dinaciclib inhibits Myc and shows interesting activity in triple negative BC cell lines, synergizing with PARP inhibitor niraparib and increasing DNA damage [43];
- Third generation (e.g., palbociclib, ribociclib and abemaciclib): characterized by selectivity for both a subset of CDKs and tumor cells compared to untransformed cells, as reported below;
- Fourth generation (e.g., ON-123300, TG02): characterized by higher potency than first generation and by a broad spectrum of activity against other pathways such as angiogenesis. These compounds are under investigation in preclinical and clinical settings.
5. CDKis Development and Approval
- In combination with fulvestrant, for the treatment of HR-positive, HER2-negative advanced or metastatic BC that progressed after ET on the basis of MONARCH 2 [66];
- As monotherapy for patients with HR-positive, HER2-negative advanced or metastatic BC that progressed after ET and previous chemotherapy;
- In combination with an AI as first-line ET for postmenopausal women with HR-positive HER2-negative advanced or metastatic BC on the basis of MONARCH 3 [67].
6. Predictors of Response and Resistance to CDKis
7. Preclinical Activity of CDKis in Endometrial Cancer
8. Current Development of CDKis in Endometrial Cancer
9. Discussion
- What role will CDKis play in ECs?
- What are the best partner drugs?
- How can we select patients that could benefit from CDKis?
- Are toxicities manageable in these patients?
- In EC, ET is an option in pretreated patients, above all in Type I EC that is characterized by strong HR-positivity, being a safe treatment in a setting of poor therapeutic options and in patients that usually have several comorbidities [16]. Nevertheless, clinical activity of ET is not fully elucidated and meta-analysis concluded that it does not improve OS, and we have insufficient data on its activity in reducing symptoms or improving Quality of Life [13]. Several efforts were done to optimize ET. In this setting, using deregulated pathways to enhance ET activity is an interesting option. Combinations with epigenetic modulators and drugs inhibiting the PI3K/AKT pathway or IGFR pathway are under development, but drugs that act on cell cycle checkpoints seem to be the most attractive strategy [21].
- Although different combinations are under development, doublet with an AI and a CDKi has the strongest preclinical rationale on the basis of cyclin D1 activity, which facilitates ER transcriptional activity, inducing ER-related gene expression. On the other hand, activation of cyclin D1 is downstream of the ER pathway [79,80]. Moreover, ECs express Rb in 70% of cases with frequent mutations or amplification of cyclin D [65,76]. In this context, combination of AIs with CDKis will use crosstalk and alteration of these two pathways to enhance activity of ET. This has been clinically demonstrated in BC and will probably play an important role in type I EC on the basis of its endocrine dependency; three phase II trial are developing this combination (NCT02730429, NCT02657928, NCT03134638). Other combinations are under development and BC is paving the way. Ongoing studies are combining CDKis with AI and IGFR inhibitors (NCT03099174) or different PI3K/AKT inhibitors, acting on pathways that are deregulated in ECs. Lastly, combinations of CDKis and immunotherapy are under development on the basis of CDKis preclinical activity in the tumour microenvironment (TME). Indeed, they induce a higher expression of PD-1, enhance antigen presentation and reduce proliferation of immunosuppressive T regulators. Ongoing clinical trials are evaluating these combinations in different settings including MSI-High solid tumours (NCT02791334), suggesting CDKis and immunotherapy could be a powerful combination in MSI-Hypermutated ECs [91,92].
- As described before, nowadays we have no clear predictor of response to CDKis with the exception of ER positivity in BC. Unfortunately, in ECs, ERs and PgRs, although having a role as prognostic factors, are not clear predictors of response to ET [21,93,94]. Moreover, response to ET in ECs is lower than in BCs, suggesting that drawing inference from BCs is not suitable for EC. Preclinical works suggest that different isoforms of ER and PgR could be related to different endocrine sensitivity [21], while mutations in ESR1, encoding for truncated forms of ER, recur in 20% of TGCA specimens [95]. Also, alteration of downstream pathways like the PI3K/AKT pathway [27], could play a role in endocrine resistance but further prospective studies are needed. In this context, some ongoing trials are selecting patients on the basis of ER positivity while others are conducting exploratory analysis to find predictors of response in EC. Lastly, BC development of CDKis is using window of opportunity trials in a neoadjuvant setting (i.e., NCT02441946). This could be applied to EC patients in the near future. Indeed a chemo-naïve population, receiving CDKis for a short period of time with the possibility of a tumour specimen after treatment (at time of surgery), could be the ideal setting to evaluate potential biomarkers of response in an homogeneous population.
- Dose limiting toxicities have been the major concern of first and second generation CDKis. A growing experience in management of CDK4/6 inhibitors in BC patients demonstrates that these are long-term well tolerated drugs in this setting with transient hematologic toxicities or diarrhea that recover with dose reduction [48]. Unfortunately, we have few data on long-term toxicities of CDK4/6 inhibitors and on adverse events that could occur with newest combinations and in EC populations. Indeed, one of the major concerns in the development of new drugs in ECs is toxicity of these compounds in a frail population. As described before, these patients have comorbidities like diabetes and hypertension and in this context hyperglycemia and hypercolesterolemia, which are PI3K inhibitors common AEs [24] or proteinuria and hypertension that are VEGF inhibitors common AEs could become unacceptable toxicities [96]. On the other hand, strategies intend to improve ET activity but not at the cost of a higher percentage of AEs, being one of the major pros of ET its manageable profile in these patents. Common toxicities of CDKis are haematological or gastrointestinal, suggesting they could be tolerated in EC patients, but ongoing studies will elucidate this aspect.
10. Conclusions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Braun, M.M.; Overbeek-Wager, E.A.; Grumbo, R.J. Diagnosis and Management of Endometrial Cancer. Am. Fam Physician 2016, 93, 468–474. [Google Scholar]
- McAlpine, J.N.; Temkin, S.M.; Mackay, H.J. Endometrial cancer: Not your grandmother’s cancer. Cancer 2016, 122, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- Key, T.J.; Pike, M.C. The dose-effect relationship between ‘unopposed’ oestrogens and endometrial mitotic rate: Its central role in explaining and predicting endometrial cancer risk. Br. J. Cancer 1988, 57, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Schorge, J.O.; Rodabaugh, K.J.; Daniels, M.S.; Sun, C.C.; Soliman, P.T.; White, K.G.; Luthra, R.; Gershenson, D.M.; Broaddus, R.R. Prospective determination of prevalence of lynch syndrome in young women with endometrial cancer. J. Clin. Oncol. 2007, 25, 5158–5164. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, M.; Lissoni, A.A.; Cormio, G.; Katsaros, D.; Pellegrino, A.; Selvaggi, L.; Ghezzi, F.; Scambia, G.; Zola, P.; Grassi, R.; et al. Modified radical hysterectomy versus extrafascial hysterectomy in the treatment of stage I endometrial cancer: Results from the ILIADE randomized study. Ann. Surg. Oncol. 2009, 16, 3431–3441. [Google Scholar] [CrossRef]
- Nout, R.A.; Smit, V.T.; Putter, H.; Jürgenliemk-Schulz, I.M.; Jobsen, J.J.; Lutgens, L.C.; van der Steen-Banasik, E.M.; Mens, J.W.; Slot, A.; Kroese, M.C.; et al. Vaginal brachytherapy versus pelvic external beam radiotherapy for patients with endometrial cancer of high-intermediate risk (PORTEC-2): An open-label, non-inferiority, randomised trial. Lancet 2010, 375, 816–823. [Google Scholar] [CrossRef]
- de Boer, S.M.; Powell, M.E.; Mileshkin, L.; Katsaros, D.; Bessette, P.; Haie-Meder, C.; Ottevanger, P.B.; Ledermann, J.A.; Khaw, P.; Colombo, A.; et al. Adjuvant chemoradiotherapy versus radiotherapy alone for women with high-risk endometrial cancer (PORTEC-3): Final results of an international, open-label, multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 295–309. [Google Scholar] [CrossRef]
- Secord, A.A.; Geller, M.A.; Broadwater, G.; Holloway, R.; Shuler, K.; Dao, N.Y.; Gehrig, P.A.; O’Malley, D.M.; Finkler, N.; Havrilesky, L.J. A multicenter evaluation of adjuvant therapy in women with optimally resected stage IIIC endometrial cancer. Gynecol. Oncol. 2013, 128, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.J.; Viswanathan, A.N. Combined chemotherapy and radiation improves survival for node-positive endometrial cancer. Gynecol. Oncol. 2012, 127, 32–37. [Google Scholar] [CrossRef]
- Dowdy, S.C. Improving oncologic outcomes for women with endometrial cancer: Realigning our sights. Gynecol. Oncol. 2014, 133, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Colombo, N.; Creutzberg, C.; Amant, F.; Bosse, T.; González-Martín, A.; Ledermann, J.; Marth, C.; Nout, R.; Querleu, D.; Mirza, M.R.; et al. ESMO-ESGO-ESTRO Consensus Conference on Endometrial Cancer: Diagnosis, treatment and follow-up. Ann. Oncol. 2016, 27, 16–41. [Google Scholar] [CrossRef]
- Kokka, F.; Brockbank, E.; Oram, D.; Gallagher, C.; Bryant, A. Hormonal therapy in advanced or recurrent endometrial cancer. Cochrane Database Syst. Rev. 2010, CD007926. [Google Scholar] [CrossRef]
- Fleming, G.F. Second-Line Therapy for Endometrial Cancer: The Need for Better Options. J. Clin. Oncol 2015, 33, 3535–3540. [Google Scholar] [CrossRef] [PubMed]
- Huijgens, A.N.; Mertens, H.J. Factors predicting recurrent endometrial cancer. Facts Views Vis. Obgyn 2013, 5, 179–186. [Google Scholar] [PubMed]
- Ethier, J.L.; Desautels, D.N.; Amir, E.; MacKay, H. Is hormonal therapy effective in advanced endometrial cancer? A systematic review and meta-analysis. Gynecol Oncol 2017, 147, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl Acad Sci USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef] [PubMed]
- Thangavelu, A.; Hewitt, M.J.; Quinton, N.D.; Duffy, S.R. Neoadjuvant treatment of endometrial cancer using anastrozole: A randomised pilot study. Gynecol. Oncol. 2013, 131, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Berstein, L.; Maximov, S.; Gershfeld, E.; Meshkova, I.; Gamajunova, V.; Tsyrlina, E.; Larionov, A.; Kovalevskij, A.; Vasilyev, D. Neoadjuvant therapy of endometrial cancer with the aromatase inhibitor letrozole: Endocrine and clinical effects. Eur J. Obstet Gynecol. Reprod Biol. 2002, 105, 161–165. [Google Scholar] [CrossRef]
- Arnold, M.; Pandeya, N.; Byrnes, G.; Renehan, P.A.G.; Stevens, G.A.; Ezzati, P.M.; Ferlay, J.; Miranda, J.J.; Romieu, I.; Dikshit, R.; et al. Global burden of cancer attributable to high body-mass index in 2012: A population-based study. Lancet Oncol. 2015, 16, 36–46. [Google Scholar] [CrossRef]
- Jerzak, K.J.; Duska, L.; MacKay, H.J. Endocrine therapy in endometrial cancer: An old dog with new tricks. Gynecol. Oncol. 2019. [Google Scholar] [CrossRef]
- Aghajanian, C.; Filiaci, V.; Dizon, D.S.; Carlson, J.W.; Powell, M.A.; Secord, A.A.; Tewari, K.S.; Bender, D.P.; O’Malley, D.M.; Stuckey, A.; et al. A phase II study of frontline paclitaxel/carboplatin/bevacizumab, paclitaxel/carboplatin/temsirolimus, or ixabepilone/carboplatin/bevacizumab in advanced/recurrent endometrial cancer. Gynecol. Oncol. 2018, 150, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Aglietta, M.; Genta, S.; Valabrega, G. Checkpoint inhibitors in endometrial cancer: Preclinical rationale and clinical activity. Oncotarget 2017, 8, 90532–90544. [Google Scholar] [CrossRef]
- Slomovitz, B.M.; Jiang, Y.; Yates, M.S.; Soliman, P.T.; Johnston, T.; Nowakowski, M.; Levenback, C.; Zhang, Q.; Ring, K.; Munsell, M.F.; et al. Phase II study of everolimus and letrozole in patients with recurrent endometrial carcinoma. J. Clin. Oncol. 2015, 33, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Bokhman, J.V. Two pathogenetic types of endometrial carcinoma. Gynecol. Oncol. 1983, 15, 10–17. [Google Scholar] [CrossRef]
- Murali, R.; Soslow, R.A.; Weigelt, B. Classification of endometrial carcinoma: More than two types. Lancet Oncol 2014, 15, e268–e278. [Google Scholar] [CrossRef]
- Le Gallo, M.; Bell, D.W. The emerging genomic landscape of endometrial cancer. Clin. Chem. 2014, 60, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; Benz, C.C.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, E.; Bosse, T.; Nout, R.A.; MacKay, H.J.; Church, D.N.; Nijman, H.W.; Leary, A.; Edmondson, R.J.; Powell, M.E.; Crosbie, E.J.; et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod. Pathol. 2015, 28, 836–844. [Google Scholar] [CrossRef]
- Church, D.N.; Stelloo, E.; Nout, R.A.; Valtcheva, N.; Depreeuw, J.; ter Haar, N.; Noske, A.; Amant, F.; Tomlinson, I.P.; Wild, P.J.; et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J. Natl. Cancer Inst. 2015, 107, 402. [Google Scholar] [CrossRef]
- McConechy, M.K.; Talhouk, A.; Leung, S.; Chiu, D.; Yang, W.; Senz, J.; Reha-Krantz, L.J.; Lee, C.H.; Huntsman, D.G.; Gilks, C.B.; et al. Endometrial Carcinomas with POLE Exonuclease Domain Mutations Have a Favorable Prognosis. Clin. Cancer Res. 2016, 22, 2865–2873. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, P.; Della Pepa, C.; Berardi, S.; Califano, D.; Scala, S.; Buonaguro, L.; Ciliberto, G.; Brauchli, P.; Pignata, S. Tumor genotype and immune microenvironment in POLE-ultramutated and MSI-hypermutated Endometrial Cancers: New candidates for checkpoint blockade immunotherapy? Cancer Treat. Rev. 2016, 48, 61–68. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]
- Lin, Z.P.; Zhu, Y.L.; Ratner, E.S. Targeting Cyclin-Dependent Kinases for Treatment of Gynecologic Cancers. Front. Oncol. 2018, 8, 303. [Google Scholar] [CrossRef]
- Chohan, T.A.; Qayyum, A.; Rehman, K.; Tariq, M.; Akash, M.S.H. An insight into the emerging role of cyclin-dependent kinase inhibitors as potential therapeutic agents for the treatment of advanced cancers. Biomed. Pharmacother 2018, 107, 1326–1341. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Harlow, E.; Hunt, T.; Hunter, T.; Lahti, J.M.; Manning, G.; Morgan, D.O.; Tsai, L.H.; Wolgemuth, D.J. Cyclin-dependent kinases: A family portrait. Nat. Cell Biol. 2009, 11, 1275–1276. [Google Scholar] [CrossRef]
- Harbour, J.W.; Luo, R.X.; Dei Santi, A.; Postigo, A.A.; Dean, D.C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [PubMed]
- Schneider, E.; Kartarius, S.; Schuster, N.; Montenarh, M. The cyclin H/cdk7/Mat1 kinase activity is regulated by CK2 phosphorylation of cyclin H. Oncogene 2002, 21, 5031–5037. [Google Scholar] [CrossRef]
- Romano, D. Comparison of preclinical data across CDK 4-6 inhibitors. In Proceedings of the ESMO Symposium on Signalling Pathways in Cancer 2018, Barcelona, Spain, 23 March 2018. [Google Scholar]
- Parry, D.; Guzi, T.; Shanahan, F.; Davis, N.; Prabhavalkar, D.; Wiswell, D.; Seghezzi, W.; Paruch, K.; Dwyer, M.P.; Doll, R.; et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol. Cancer Ther. 2010, 9, 2344–2353. [Google Scholar] [CrossRef]
- Carey, J.P.W.; Karakas, C.; Bui, T.; Chen, X.; Vijayaraghavan, S.; Zhao, Y.; Wang, J.; Mikule, K.; Litton, J.K.; Hunt, K.K.; et al. Synthetic Lethality of PARP Inhibitors in Combination with MYC Blockade Is Independent of BRCA Status in Triple-Negative Breast Cancer. Cancer Res. 2018, 78, 742–757. [Google Scholar] [CrossRef] [PubMed]
- Christian, B.A.; Grever, M.R.; Byrd, J.C.; Lin, T.S. Flavopiridol in the treatment of chronic lymphocytic leukemia. Curr. Opin. Oncol. 2007, 19, 573–578. [Google Scholar] [CrossRef]
- Phelps, M.A.; Lin, T.S.; Johnson, A.J.; Hurh, E.; Rozewski, D.M.; Farley, K.L.; Wu, D.; Blum, K.A.; Fischer, B.; Mitchell, S.M.; et al. Clinical response and pharmacokinetics from a phase 1 study of an active dosing schedule of flavopiridol in relapsed chronic lymphocytic leukemia. Blood 2009, 113, 2637–2645. [Google Scholar] [CrossRef] [PubMed]
- Blum, W.; Phelps, M.A.; Klisovic, R.B.; Rozewski, D.M.; Ni, W.; Albanese, K.A.; Rovin, B.; Kefauver, C.; Devine, S.M.; Lucas, D.M.; et al. Phase I clinical and pharmacokinetic study of a novel schedule of flavopiridol in relapsed or refractory acute leukemias. Haematologica 2010, 95, 1098–1105. [Google Scholar] [CrossRef]
- Ghia, P.; Scarfò, L.; Perez, S.; Pathiraja, K.; Derosier, M.; Small, K.; McCrary Sisk, C.; Patton, N. Efficacy and safety of dinaciclib vs. ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017, 129, 1876–1878. [Google Scholar] [CrossRef]
- Spring, L.M.; Zangardi, M.L.; Moy, B.; Bardia, A. Clinical Management of Potential Toxicities and Drug Interactions Related to Cyclin-Dependent Kinase 4/6 Inhibitors in Breast Cancer: Practical Considerations and Recommendations. Oncologist 2017, 22, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Tolaney, S.M.; Rugo, H.S.; Cortés, J.; Diéras, V.; Patt, D.; Wildiers, H.; Hudis, C.A.; O’Shaughnessy, J.; Zamora, E.; et al. MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR. Clin. Cancer Res. 2017, 23, 5218–5224. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat.Rev.Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- DeCaprio, J.A.; Ludlow, J.W.; Lynch, D.; Furukawa, Y.; Griffin, J.; Piwnica-Worms, H.; Huang, C.M.; Livingston, D.M. The product of the retinoblastoma susceptibility gene has properties of a cell cycle regulatory element. Cell 1989, 58, 1085–1095. [Google Scholar] [CrossRef]
- Barnes, D.M.; Gillett, C.E. Cyclin D1 in breast cancer. Breast Cancer Res.Treat. 1998, 52, 1–15. [Google Scholar]
- Ertel, A.; Dean, J.L.; Rui, H.; Liu, C.; Witkiewicz, A.K.; Knudsen, K.E.; Knudsen, E.S. RB-pathway disruption in breast cancer: Differential association with disease subtypes, disease-specific prognosis and therapeutic response. Cell Cycle 2010, 9, 4153–4163. [Google Scholar] [CrossRef]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl Acad Sci USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [PubMed]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A.; et al. Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentiallyinhibits proliferation of luminal estrogen receptor-positive humanbreast cancer cell lines in vitro. Breast Cancer Res. 2009, 11, R77. [Google Scholar] [CrossRef]
- O’Brien, N.A.; Tomaso, E.D.; Ayala, R.; Tong, L. Abstract 4756: In vivo efficacy of combined targeting of CDK 4/6, ER and PI3K signaling in ER+ breast cancer. Cancer Res. 2014, 74. [Google Scholar] [CrossRef]
- Thangavel, C.; Dean, J.L.; Ertel, A.; Knudsen, K.E.; Aldaz, C.M.; Witkiewicz, A.K.; Clarke, R.; Knudsen, E.S. Therapeutically activating RB: Reestablishing cell cycle control in endocrine therapy-resistant breast cancer. Endocr-Relat. Cancer 2011, 18, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)†. Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- De Luca, A.; Maiello, M.R.; D’Alessio, A.; Frezzetti, D.; Gallo, M.; Carotenuto, M.; Normanno, N. Pharmacokinetic drug evaluation of palbociclib for the treatment of breast cancer. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 891–900. [Google Scholar] [CrossRef] [PubMed]
- Toogood, P.L.; Harvey, P.J.; Repine, J.T.; Sheehan, D.J.; VanderWel, S.N.; Zhou, H.; Keller, P.R.; McNamara, D.J.; Sherry, D.; Zhu, T.; et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J. Med. Chem. 2005, 48, 2388–2406. [Google Scholar] [CrossRef]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.; Im, S.A.; Gelmon, K.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.; et al. Palbociclib and Letrozole in Advanced Breast Cancer. N. Engl. J. Med. 2016, 375, 1925–1936. [Google Scholar] [CrossRef]
- O’Leary, B.; Cutts, R.J.; Liu, Y.; Hrebien, S.; Huang, X.; Fenwick, K.; André, F.; Loibl, S.; Loi, S.; Garcia-Murillas, I.; et al. The Genetic Landscape and Clonal Evolution of Breast Cancer Resistance to Palbociclib plus Fulvestrant in the PALOMA-3 Trial. Cancer Discov. 2018, 8, 1390–1403. [Google Scholar] [CrossRef]
- Corona, S.P.; Generali, D. Abemaciclib: A CDK4/6 inhibitor for the treatment of HR+/HER2- advanced breast cancer. Drug Des. Devel. Ther. 2018, 12, 321–330. [Google Scholar] [CrossRef]
- Gong, X.; Litchfield, L.M.; Webster, Y.; Chio, L.C.; Wong, S.S.; Stewart, T.R.; Dowless, M.; Dempsey, J.; Zeng, Y.; Torres, R.; et al. Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 2017, 32, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W.; Toi, M.; Neven, P.; Sohn, J.; Inoue, K.; Pivot, X.; Burdaeva, O.; Okera, M.; Masuda, N.; Kaufman, P.A.; et al. MONARCH 2: Abemaciclib in Combination With Fulvestrant in Women With HR+/HER2- Advanced Breast Cancer Who Had Progressed While Receiving Endocrine Therapy. J. Clin. Oncol. 2017, 35, 2875–2884. [Google Scholar] [CrossRef]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N. Ribociclib for the first-line treatment of advanced hormone receptor-positive breast cancer: A review of subgroup analyses from the MONALEESA-2 trial. Breast Cancer Res. 2018, 20, 123. [Google Scholar] [CrossRef]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.L.; Winer, E.P.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2018, 29, 1541–1547. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Huang, D.; Yang, F.; Guan, X. Potential biomarkers of CDK4/6 inhibitors in hormone receptor-positive advanced breast cancer. Breast Cancer Res. Treat. 2018, 168, 287–297. [Google Scholar] [CrossRef] [PubMed]
- DeMichele, A.; Clark, A.S.; Tan, K.S.; Heitjan, D.F.; Gramlich, K.; Gallagher, M.; Lal, P.; Feldman, M.; Zhang, P.; Colameco, C.; et al. CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: Phase II activity, safety, and predictive biomarker assessment. Clin. Cancer Res. 2015, 21, 995–1001. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef] [PubMed]
- Wander, S.A.; Cohen, O.; Johnson, G.N.; Kim, D.; Luo, F.; Mao, P.; Nayar, U.; Helvie, K.; Marini, L.; Freeman, S.; et al. Whole exome sequencing (WES) in hormone-receptor positive (HR+) metastatic breast cancer (MBC) to identify mediators of resistance to cyclin-dependent kinase 4/6 inhibitors (CDK4/6i). J. Clin. Oncol. 2018, 36. [Google Scholar] [CrossRef]
- Tsuda, H.; Yamamoto, K.; Inoue, T.; Uchiyama, I.; Umesaki, N. The role of p16-cyclin d/CDK-pRb pathway in the tumorigenesis of endometrioid-type endometrial carcinoma. Br. J. Cancer 2000, 82, 675–682. [Google Scholar] [CrossRef]
- Santala, S.; Talvensaari-Mattila, A.; Soini, Y.; Honkavuori-Toivola, M.; Santala, M. High expression of cyclin A is associated with poor prognosis in endometrial endometrioid adenocarcinoma. Tumour Biol. 2014, 35, 5395–5399. [Google Scholar] [CrossRef]
- Tanaka, T.; Terai, Y.; Ashihara, K.; Fujiwara, S.; Tanaka, Y.; Sasaki, H.; Tsunetoh, S.; Ohmichi, M. The efficacy of the cyclin-dependent kinase 4/6 inhibitor in endometrial cancer. PLoS ONE 2017, 12, e0177019. [Google Scholar] [CrossRef]
- Dosil, M.A.; Mirantes, C.; Eritja, N.; Felip, I.; Navaridas, R.; Gatius, S.; Santacana, M.; Colàs, E.; Moiola, C.; Schoenenberger, J.A.; et al. Palbociclib has antitumour effects on Pten-deficient endometrial neoplasias. J. Pathol. 2017, 242, 152–164. [Google Scholar] [CrossRef]
- Huang, K.T.; Pavlides, S.C.; Lecanda, J.; Blank, S.V.; Mittal, K.R.; Gold, L.I. Estrogen and progesterone regulate p27kip1 levels via the ubiquitin-proteasome system: Pathogenic and therapeutic implications for endometrial cancer. PLoS ONE 2012, 7, e46072. [Google Scholar] [CrossRef]
- Butt, A.J.; McNeil, C.M.; Musgrove, E.A.; Sutherland, R.L. Downstream targets of growth factor and oestrogen signalling and endocrine resistance: The potential roles of c-Myc, cyclin D1 and cyclin E. Endocr. Relat. Cancer 2005, 12, S47–S59. [Google Scholar] [CrossRef]
- Altucci, L.; Addeo, R.; Cicatiello, L.; Germano, D.; Pacilio, C.; Battista, T.; Cancemi, M.; Petrizzi, V.B.; Bresciani, F.; Weisz, A. Estrogen induces early and timed activation of cyclin-dependent kinases 4, 5, and 6 and increases cyclin messenger ribonucleic acid expression in rat uterus. Endocrinology 1997, 138, 978–984. [Google Scholar] [CrossRef]
- Hamilton, C.A.; Cheung, M.K.; Osann, K.; Chen, L.; Teng, N.N.; Longacre, T.A.; Powell, M.A.; Hendrickson, M.R.; Kapp, D.S.; Chan, J.K. Uterine papillary serous and clear cell carcinomas predict for poorer survival compared to grade 3 endometrioid corpus cancers. Br. J. Cancer 2006, 94, 642–646. [Google Scholar] [CrossRef]
- Milde-Langosch, K.; Bamberger, A.M.; Goemann, C.; Rössing, E.; Rieck, G.; Kelp, B.; Löning, T. Expression of cell-cycle regulatory proteins in endometrial carcinomas: Correlations with hormone receptor status and clinicopathologic parameters. J. Cancer Res. Clin. Oncol. 2001, 127, 537–544. [Google Scholar] [CrossRef]
- Cocco, E.; Lopez, S.; Black, J.; Bellone, S.; Bonazzoli, E.; Predolini, F.; Ferrari, F.; Schwab, C.L.; Menderes, G.; Zammataro, L.; et al. Dual CCNE1/PIK3CA targeting is synergistic in CCNE1-amplified/PIK3CA-mutated uterine serous carcinomas in vitro and in vivo. Br. J. Cancer 2016, 115, 303–311. [Google Scholar] [CrossRef]
- Flaherty, K.T.; Lorusso, P.M.; Demichele, A.; Abramson, V.G.; Courtney, R.; Randolph, S.S.; Shaik, M.N.; Wilner, K.D.; O’Dwyer, P.J.; Schwartz, G.K. Phase I, dose-escalation trial of the oral cyclin-dependent kinase 4/6 inhibitor PD 0332991, administered using a 21-day schedule in patients with advanced cancer. Clin. Cancer Res. 2012, 18, 568–576. [Google Scholar] [CrossRef]
- Infante, J.R.; Cassier, P.A.; Gerecitano, J.F.; Witteveen, P.O.; Chugh, R.; Ribrag, V.; Chakraborty, A.; Matano, A.; Dobson, J.R.; Crystal, A.S.; et al. A Phase I Study of the Cyclin-Dependent Kinase 4/6 Inhibitor Ribociclib (LEE011) in Patients with Advanced Solid Tumors and Lymphomas. Clin. Cancer Res. 2016, 22, 5696–5705. [Google Scholar] [CrossRef]
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753. [Google Scholar] [CrossRef]
- Benson, C.; White, J.; De Bono, J.; O’Donnell, A.; Raynaud, F.; Cruickshank, C.; McGrath, H.; Walton, M.; Workman, P.; Kaye, S.; et al. A phase I trial of the selective oral cyclin-dependent kinase inhibitor seliciclib (CYC202; R-Roscovitine), administered twice daily for 7 days every 21 days. Br. J. Cancer 2007, 96, 29–37. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Frederick, H.J.; M, C.J. Phase 1 study of sapacitabine and seliciclib in patients with advanced solid tumors. J. Clin. Oncol. 2017, 34, 2503. [Google Scholar] [CrossRef]
- Miller, T.W.; Balko, J.M.; Fox, E.M.; Ghazoui, Z.; Dunbier, A.; Anderson, H.; Dowsett, M.; Jiang, A.; Smith, R.A.; Maira, S.M.; et al. ERα-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov. 2011, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.R.; Juric, D.; Kim, N.; Mino-Kenudson, M.; Huynh, T.; Costa, C.; Lockerman, E.L.; Pollack, S.F.; Liu, M.; Li, X.; et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell 2014, 26, 136–149. [Google Scholar] [CrossRef]
- Teh, J.L.F.; Aplin, A.E. Arrested Developments: CDK4/6 Inhibitor Resistance and Alterations in the Tumor Immune Microenvironment. Clin. Cancer Res. 2019, 25, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Ameratunga, M.; Kipps, E.; Okines, A.F.C.; Lopez, J.S. To Cycle or Fight-CDK4/6 Inhibitors at the Crossroads of Anticancer Immunity. Clin. Cancer Res. 2019, 25, 21–28. [Google Scholar] [CrossRef]
- Karnezis, A.N.; Leung, S.; Magrill, J.; McConechy, M.K.; Yang, W.; Chow, C.; Kobel, M.; Lee, C.H.; Huntsman, D.G.; Talhouk, A.; et al. Evaluation of endometrial carcinoma prognostic immunohistochemistry markers in the context of molecular classification. J. Pathol. Clin. Res. 2017, 3, 279–293. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, D.; Gong, C.; Zhang, F.; He, J.; Zhang, W.; Zhao, Y.; Sun, J. Prognostic role of hormone receptors in endometrial cancer: A systematic review and meta-analysis. World J. Surg Oncol. 2015, 13, 208. [Google Scholar] [CrossRef] [PubMed]
- Holst, F.; Hoivik, E.A.; Gibson, W.J.; Taylor-Weiner, A.; Schumacher, S.E.; Asmann, Y.W.; Grossmann, P.; Trovik, J.; Necela, B.M.; Thompson, E.A.; et al. Recurrent hormone-binding domain truncated ESR1 amplifications in primary endometrial cancers suggest their implication in hormone independent growth. Sci. Rep. 2016, 6, 25521. [Google Scholar] [CrossRef] [PubMed]
- Bogliolo, S.; Cassani, C.; Gardella, B.; Musacchi, V.; Babilonti, L.; Venturini, P.L.; Ferrero, S.; Spinillo, A. Current opinion on bevacizumab on endometrial cancer treatment. Expert. Opin. Biol. Ther. 2015, 15, 299–307. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| First Generation (e.g., Alvocidib) | Second Generation (e.g., Dinaciclib, CYC065) | Third Generation (e.g., Abemaciclib, Ribociclib, Palbociclib) | Fourth Generation (e.g., TG02) | |
|---|---|---|---|---|
| Mode of action | Multi-serine/threonine CDKis | Inhibitor of a wide range of CDKs | Inhibitor of CDK4 and CDK6 | Pyrimidine-based multi-kinase inhibitor (together with JAK2 and FLT3) |
| Side effects | -Tumour lyses syndrome (in CCL, reversible) -Myelosuppression -Diarrhea | -Neutropenia -Thrombocytopenia -Pneumonia -Sepsis | -Neutropenia -Fatigue -GE toxicity (diarrhea) | -early clinical development, side effects not yet available |
| Advantages | -First to demonstrate clinical activity in vitro | -activity shown in Multiple Myeloma -Enhance PARPis activity in vitro (Dinaciclib) | -Higher potency -Selective activity intumour cells -Cross BBB (abemaciclib) -Manageable safety profile -oral administration -administered once daily (palbociclib and ribociclib) | -Higher potency (non pan-CDK) -Large spectrum of activity (e.g., also angiogenesis) -Potent anti-proliferative effects in tumour cell lines |
| Limitations | -Low potency (pan-CDK) -Lack of specificity -Off target toxic effects -intravenous administration | -Non selective activity -Equivalent activity on normal and tumour cells -intravenous administration | -Prolonged QT interval (mandatory ECG before and during treatment with ribociclib) | -Under investigation in preclinical and clinical setting -dose-dependent |
| Description | Condition | Line of Therapy | Primary Endpoint | Phase | Status | Trial Identifier |
|---|---|---|---|---|---|---|
| Palbociclib | ||||||
| Palbociclib | Ovarian teratomas, GCTs or tumours with alteration at the G1/S checkpoint. | na | ORR, Safety | II | Recruiting | NCT01037790 |
| Palbociclib+ cisplatin or carboplatin | Solid tumours | na | %AEs, DLT, RP2D | I | Recruiting | NCT02897375 |
| Palbociclib | Solid tumours with alteration at the G1/S checkpoint. | >2 line | ORR | II | Recruiting | NCT02465060 |
| Letrozole+ Palbociclib/Letrozole +placebo | Metastatic EC | No more than 1 prior line of ET | PFS | II | Recruiting | NCT02730429 |
| Palbociclib + Gedatolisib | Solid tumours | na | %AEs, DLT, RP2D | I | Recruiting | NCT03065062 |
| RIBOCICLIB | ||||||
| Ribociclib +Letrozole | OC and EC | na | %PFS at 12 weeks | II | Active, not recruiting | NCT02657928 |
| Ribociclib+ everolimus + Letrozole | EC | ≤ 3 line | DLT/CBR | I/II | Recruiting | NCT03008408 |
| ABEMACICLIB | ||||||
| LY3023414 (PI3Ki) and Abemaciclib +/− Letrozole | EC | na | PFS at 6 months, ORR | II | Not yet recruiting | NCT03675893 |
| Fulvestrant+ Abemaciclib | EC | 1 or 2 line | ORR | II | Recruiting | NCT03643510 |
| OTHER CDKIS | ||||||
| Dinaciclib + Veliparib | Solid tumours with BRCA mutation | na | RP2D | I | Recruiting | NCT01434316 |
| Seliciclib+ Sapacitabine | Solid tumours | na | MTD | I | Unknown | NCT00999401 |
| CYC065 | Solid tumours or lymphomas | na | DLTs | I | Recruiting | NCT02552953 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannone, G.; Tuninetti, V.; Ghisoni, E.; Genta, S.; Scotto, G.; Mittica, G.; Valabrega, G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. Int. J. Mol. Sci. 2019, 20, 2353. https://doi.org/10.3390/ijms20092353
Giannone G, Tuninetti V, Ghisoni E, Genta S, Scotto G, Mittica G, Valabrega G. Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. International Journal of Molecular Sciences. 2019; 20(9):2353. https://doi.org/10.3390/ijms20092353
Chicago/Turabian StyleGiannone, Gaia, Valentina Tuninetti, Eleonora Ghisoni, Sofia Genta, Giulia Scotto, Gloria Mittica, and Giorgio Valabrega. 2019. "Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer" International Journal of Molecular Sciences 20, no. 9: 2353. https://doi.org/10.3390/ijms20092353
APA StyleGiannone, G., Tuninetti, V., Ghisoni, E., Genta, S., Scotto, G., Mittica, G., & Valabrega, G. (2019). Role of Cyclin-Dependent Kinase Inhibitors in Endometrial Cancer. International Journal of Molecular Sciences, 20(9), 2353. https://doi.org/10.3390/ijms20092353