The Cross-Talk Between the TNF-α and RASSF-Hippo Signalling Pathways

Abstract
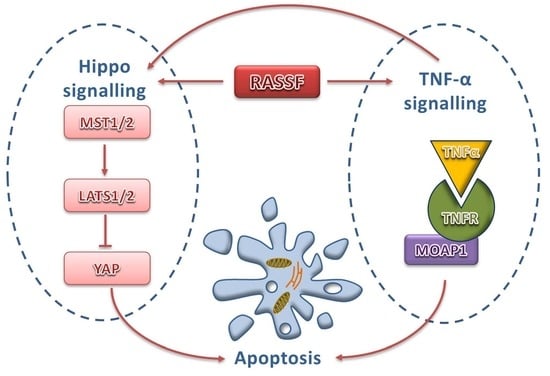
{kind=link}
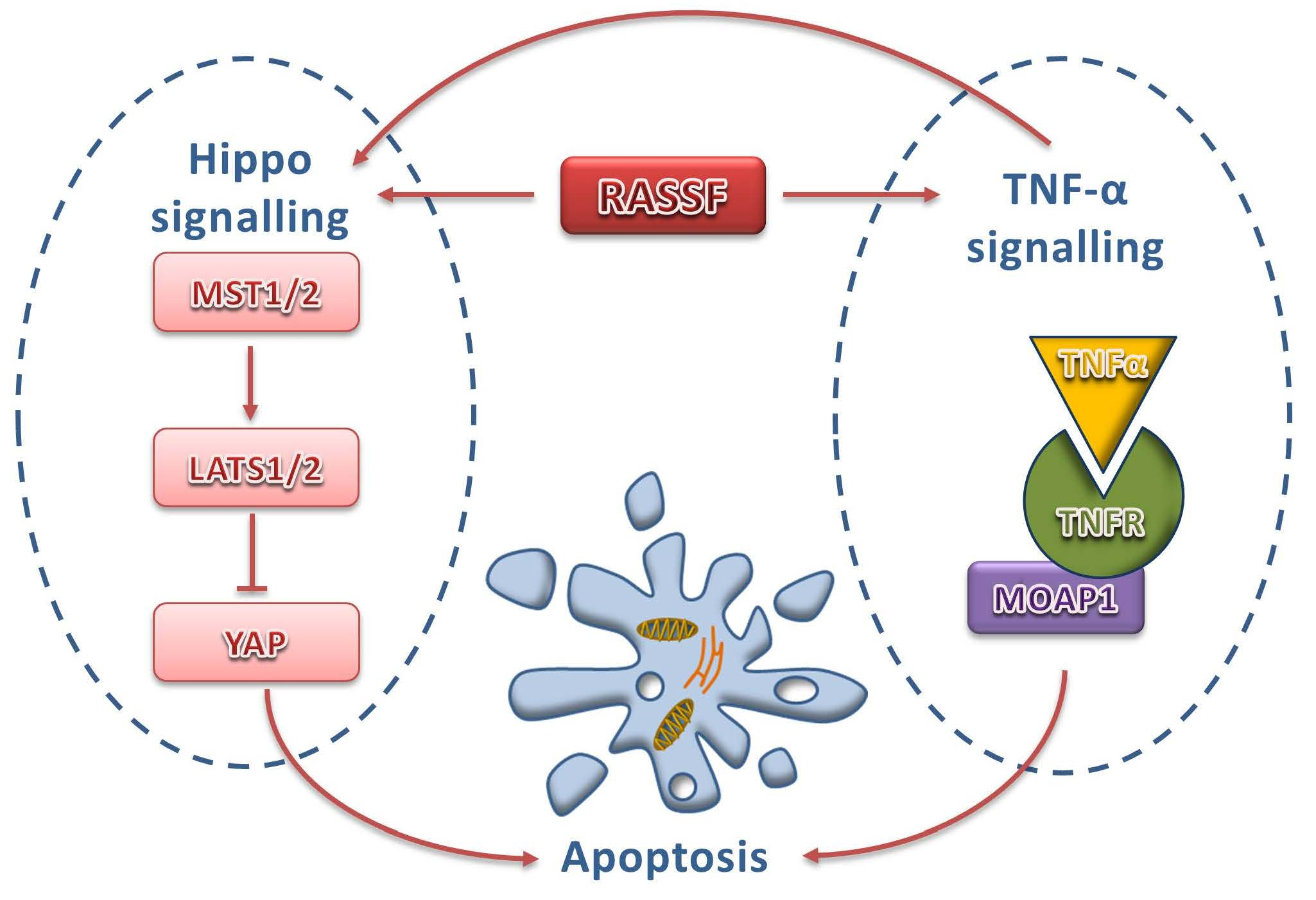
{kind=link}
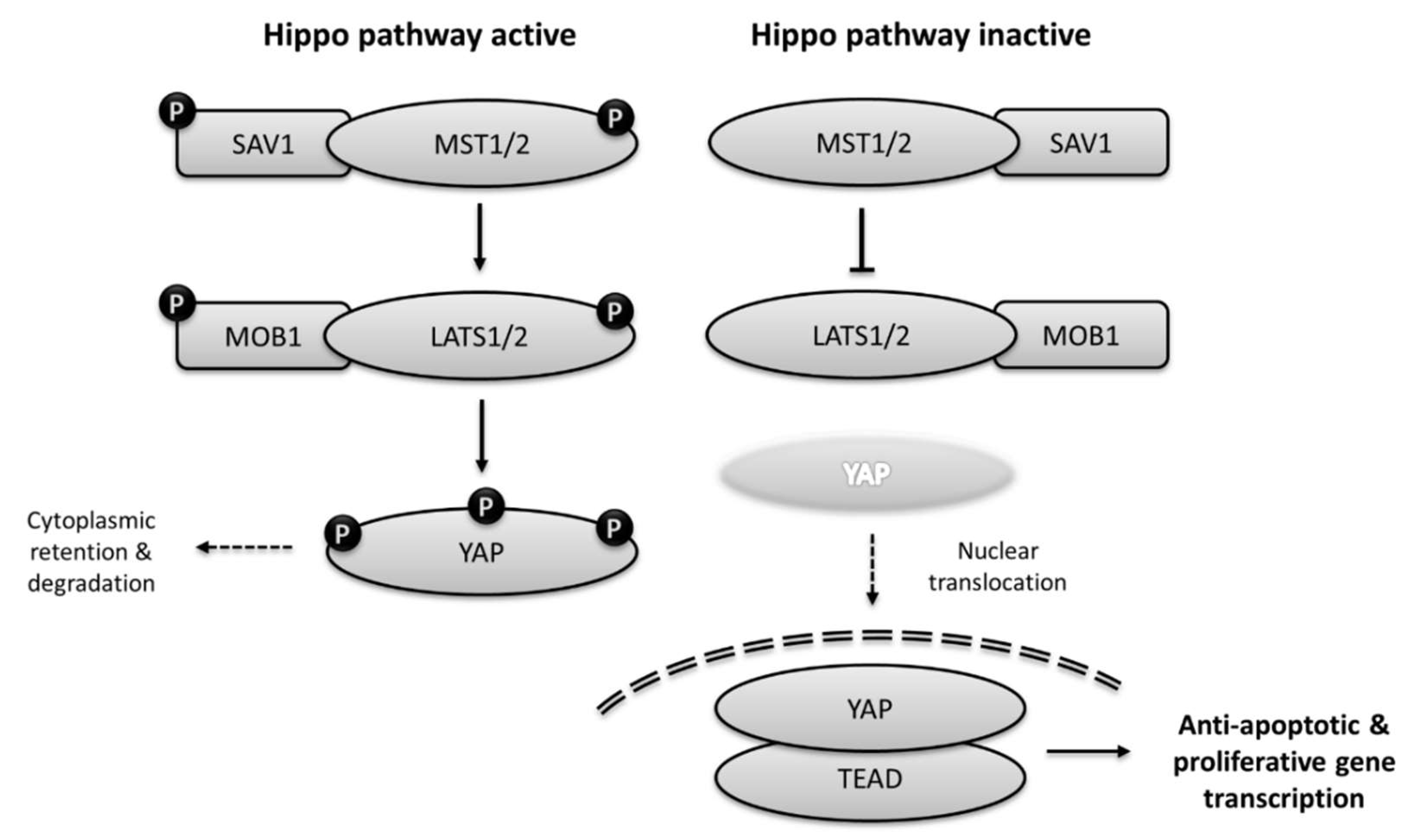
{kind=link}
{kind=link}
1. Introduction
2. The TNF-α Pathway
2.1. TNF-α and TNF Receptor Complex
2.2. TNF-α Dependent Apoptosis Pathway
2.3. Regulation of NF-κB Pathway
2.4. TNF-α Mediated Inflammatory Pathway
3. The Hippo signalling Pathway
3.1. Core Components of Hippo Pathway
3.2. Upstream Regulators of the Hippo Pathway
3.3. Roles of Hippo Pathway in Mediating Cellular Processes and Organ Functions
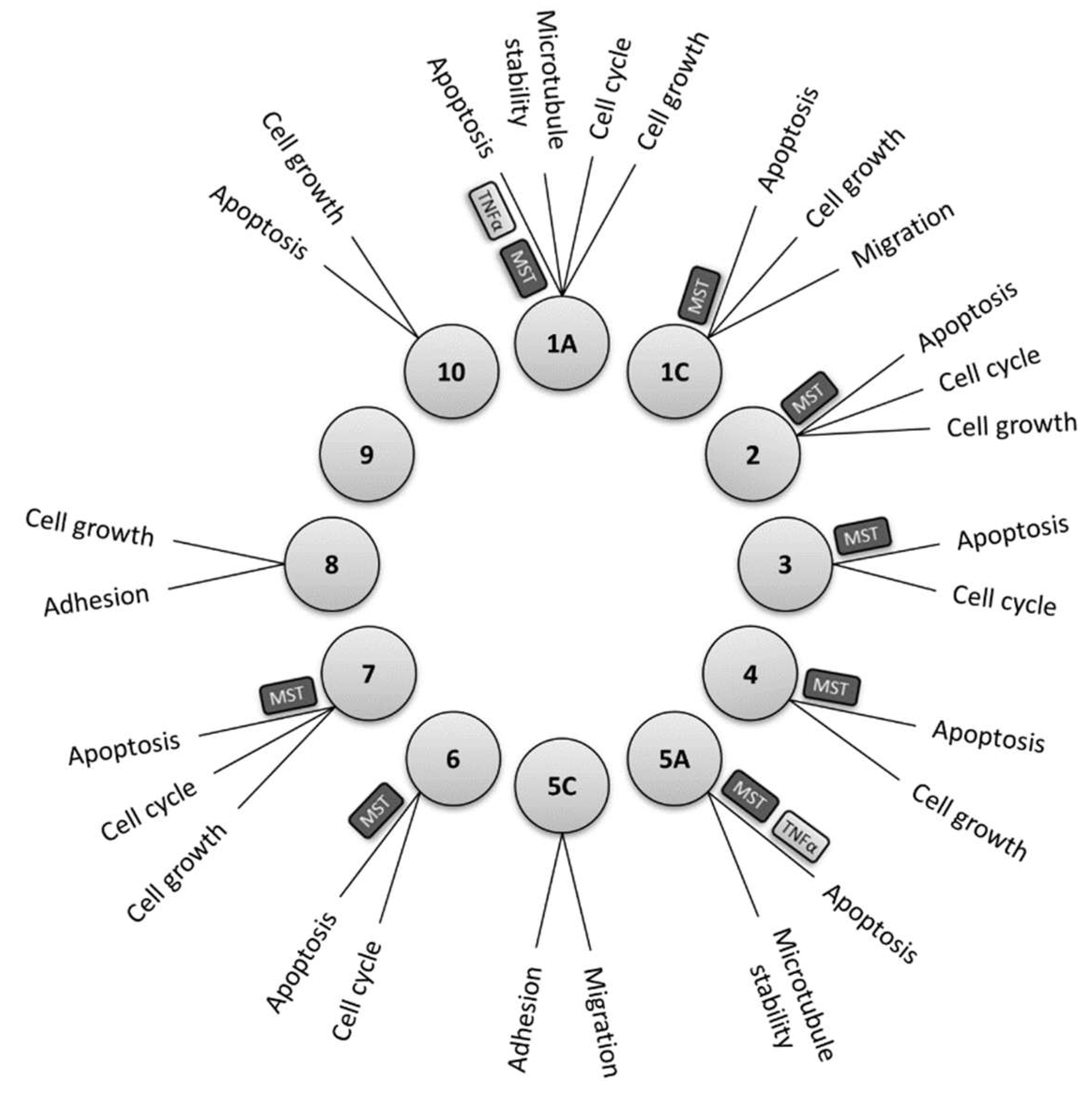
3.4. RASSF Family Proteins as Major Upstream Regulators of Hippo Pathway
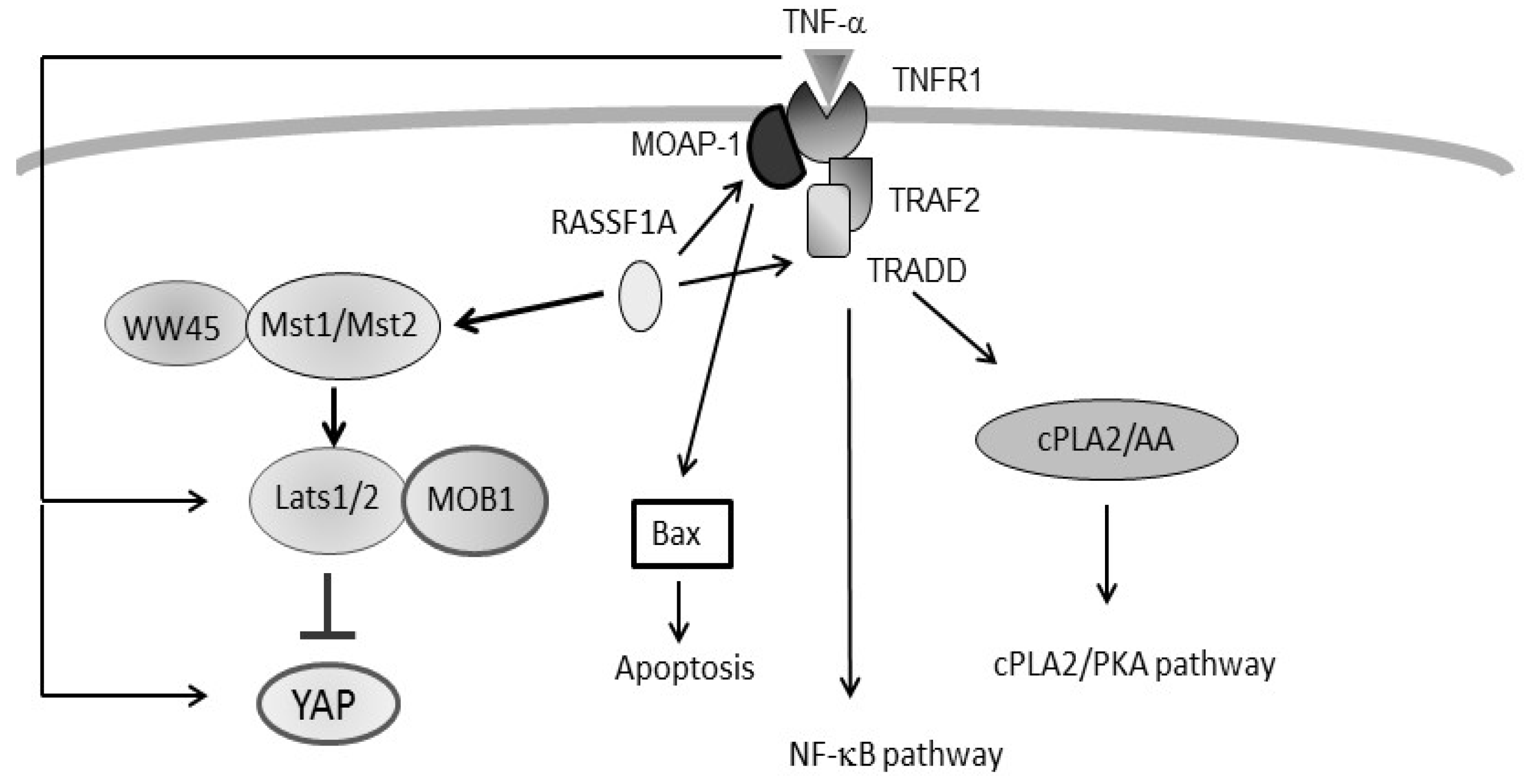
4. Regulation of TNF-α Signalling by RASSF1A
5. Intersection between TNF-α Signalling and the Hippo Pathway
5.1. Modulation of LATS2 by TNF-α
5.2. Modulation of YAP-TEAD Activity by TNF-α Cancer Cells and Chondrocytes
5.3. TNF-α Promotes YAP Activation in Endothelial Cells
6. Concluding Remarks
Funding
Conflicts of Interest
Abbreviations
| TNF-α | Tumour necrosis factor-alpha |
| RASSF | Ras-association domain family member |
| TRAIL | TNF-related apoptosis-inducing ligand |
| DISC | Death inducing signalling complex |
| Bcl-2 | B-cell lymphoma 2 |
| Apaf-1 | Apoptosis protease-activating factor 1 |
| ERK | extracellular signal–regulated kinase |
| YAP | Yes-associated protein |
| TAZ | Tafazzin |
| TEAD | TEA domain family member |
| MST | Mammalian Ste20-like kinase |
| LATS | Large tumour suppressor |
| TACE | TNF-α converting enzyme |
| TNFR | TNF receptor; DD, death domain |
| TRAD | TNFR-associated death domain protein |
| TRAF | TNFR-associated factor |
| RIPK1 | Receptor-interacting serine/threonine-protein kinase 1 |
| FADD | Fas associated protein with death domain |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| SODD | Silencer of death domain |
| MAPK | Mitogen-activated protein kinase |
| JNK | c-Jun-N-terminal kinase |
| IκB | inhibitor of kappa B |
| RIP | receptor interacting protein kinase |
| IKK | IκB kinase |
| 5-HETE | 5-hydroxyeicosatetraenoic acid |
| Yki | Yorkie |
| TAO1 | Serine/threonine-protein kinase TAO1 |
| SAV1 | Salvador homolog |
| MOB1 | MOB kinase activator 1 |
| AJ | adherens junction |
| TJ | tight junction |
| LKB1 | Liver kinase B1 |
| NPHP4 | Nephronophthisis 4 |
| AMOT | angiomotin |
| ECM | Extracellular matrix |
| GPCR | G-protein coupled receptor |
| ZO | Zona occludens |
| LPA | Lysophosphatidic acid |
| MOAP1 | Modulator of apoptosis 1 |
| Bax | Bcl-2-associated x protein |
| SARAH | Salvador RASSF Hippo |
| cPLA2 | phospholipase A2 |
| TLR | toll-like receptor |
| MyD88 | Myeloid differentiation primary response 88 |
| IRAK | Interleukin-1 receptor-associated kinase |
| Bcl-xL | B-cell lymphoma-extra large |
| STAT3 | Signal transducer and activator of transcription 3 |
| VEGF | Vascular endothelial growth factor |
| HUVEC | Human vascular endothelial cell |
| ROCK1 | Rho associated coiled-coil containing protein kinase 1 |
| VCAM1 | Vascular cell adhesion molecule 1 |
| ICAM1 | Intercellular adhesion molecule 1 |
| PKC | protein kinase C |
| ATM | ataxia telangiectasia mutated |
| TAK1 | Transforming growth factor beta-activated kinase 1 |
References
- Jacobson, M.D.; Weil, M.; Raff, M.C. Programmed cell death in animal development. Cell 1997, 88, 347–354. [Google Scholar] [CrossRef]
- Opferman, J.T. Apoptosis in the development of the immune system. Cell Death Differ. 2008, 15, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Miura, M. Programmed cell death in neurodevelopment. Dev. Cell 2015, 32, 478–490. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Van Empel, V.P.; Bertrand, A.T.; Hofstra, L.; Crijns, H.J.; Doevendans, P.A.; De Windt, L.J. Myocyte apoptosis in heart failure. Cardiovasc. Res. 2005, 67, 21–29. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and autoimmune diseases. Ann. N Y Acad. Sci. 2010, 1209, 10–16. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef] [PubMed]
- Misra, J.R.; Irvine, K.D. The Hippo Signaling Network and Its Biological Functions. Annu Rev. Genet. 2018, 52, 65–87. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Brenner, D.; Blaser, H.; Mak, T.W. Regulation of tumour necrosis factor signalling: Live or let die. Nat. Rev. Immunol. 2015, 15, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Takada, H.; Chen, N.J.; Mirtsos, C.; Suzuki, S.; Suzuki, N.; Wakeham, A.; Mak, T.W.; Yeh, W.C. Role of SODD in regulation of tumor necrosis factor responses. Mol. Cell Biol. 2003, 23, 4026–4033. [Google Scholar] [CrossRef] [PubMed]
- Borghi, A.; Verstrepen, L.; Beyaert, R. TRAF2 multitasking in TNF receptor-induced signaling to NF-kappaB, MAP kinases and cell death. Biochem. Pharmacol. 2016, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.H.; Sun, S.C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor kappaB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Mohamed, T.M.; Zi, M.; Prehar, S.; Maqsood, A.; Abou-Leisa, R.; Nguyen, L.; Pfeifer, G.P.; Cartwright, E.J.; Neyses, L.; Oceandy, D. The tumour suppressor Ras-association domain family protein 1A (RASSF1A) regulates TNF-alpha signalling in cardiomyocytes. Cardiovasc. Res. 2014, 103, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Warzocha, K.; Bienvenu, J.; Coiffier, B.; Salles, G. Mechanisms of action of the tumor necrosis factor and lymphotoxin ligand-receptor system. Eur Cytokine Netw 1995, 6, 83–96. [Google Scholar] [PubMed]
- Yu, F.X.; Guan, K.L. The Hippo pathway: Regulators and regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol 2018, 15, 672–684. [Google Scholar] [CrossRef]
- Deng, Y.; Matsui, Y.; Zhang, Y.; Lai, Z.C. Hippo activation through homodimerization and membrane association for growth inhibition and organ size control. Dev. Biol. 2013, 375, 152–159. [Google Scholar] [CrossRef]
- Boggiano, J.C.; Vanderzalm, P.J.; Fehon, R.G. Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Dev. Cell 2011, 21, 888–895. [Google Scholar] [CrossRef]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef]
- Kim, N.G.; Koh, E.; Chen, X.; Gumbiner, B.M. E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 2011, 108, 11930–11935. [Google Scholar] [CrossRef] [PubMed]
- Das Thakur, M.; Feng, Y.; Jagannathan, R.; Seppa, M.J.; Skeath, J.B.; Longmore, G.D. Ajuba LIM proteins are negative regulators of the Hippo signaling pathway. Curr. Biol. 2010, 20, 657–662. [Google Scholar] [CrossRef]
- Nguyen, H.B.; Babcock, J.T.; Wells, C.D.; Quilliam, L.A. LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of Yap. Oncogene 2013, 32, 4100–4109. [Google Scholar] [CrossRef]
- Habbig, S.; Bartram, M.P.; Muller, R.U.; Schwarz, R.; Andriopoulos, N.; Chen, S.; Sagmuller, J.G.; Hoehne, M.; Burst, V.; Liebau, M.C.; et al. NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J. Cell Biol. 2011, 193, 633–642. [Google Scholar] [CrossRef]
- Oka, T.; Remue, E.; Meerschaert, K.; Vanloo, B.; Boucherie, C.; Gfeller, D.; Bader, G.D.; Sidhu, S.S.; Vandekerckhove, J.; Gettemans, J.; et al. Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem. J. 2010, 432, 461–472. [Google Scholar] [CrossRef]
- Remue, E.; Meerschaert, K.; Oka, T.; Boucherie, C.; Vandekerckhove, J.; Sudol, M.; Gettemans, J. TAZ interacts with zonula occludens-1 and -2 proteins in a PDZ-1 dependent manner. FEBS Lett. 2010, 584, 4175–4180. [Google Scholar] [CrossRef]
- Oka, T.; Schmitt, A.P.; Sudol, M. Opposing roles of angiomotin-like-1 and zona occludens-2 on pro-apoptotic function of YAP. Oncogene 2012, 31, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lu, Q.; Wang, L.H.; Liu, C.Y.; Lei, Q.; Guan, K.L. Angiomotin is a novel Hippo pathway component that inhibits YAP oncoprotein. Genes Dev. 2011, 25, 51–63. [Google Scholar] [CrossRef]
- Chakraborty, S.; Hong, W. Linking Extracellular Matrix Agrin to the Hippo Pathway in Liver Cancer and Beyond. Cancers 2018, 10, 45. [Google Scholar] [CrossRef]
- Xiong, W.C.; Mei, L. Agrin to YAP in Cancer and Neuromuscular Junctions. Trends Cancer 2017, 3, 247–248. [Google Scholar] [CrossRef] [PubMed]
- Bassat, E.; Mutlak, Y.E.; Genzelinakh, A.; Shadrin, I.Y.; Baruch Umansky, K.; Yifa, O.; Kain, D.; Rajchman, D.; Leach, J.; Riabov Bassat, D.; et al. The extracellular matrix protein agrin promotes heart regeneration in mice. Nature 2017, 547, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Furth, N.; Aylon, Y. The LATS1 and LATS2 tumor suppressors: Beyond the Hippo pathway. Cell Death Differ. 2017, 24, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Thompson, B.J.; Sahai, E. MST kinases in development and disease. J. Cell Biol. 2015, 210, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.H.; Camargo, F.D.; Yimlamai, D. Hippo Signaling in the Liver Regulates Organ Size, Cell Fate, and Carcinogenesis. Gastroenterology 2017, 152, 533–545. [Google Scholar] [CrossRef]
- Wong, J.S.; Meliambro, K.; Ray, J.; Campbell, K.N. Hippo signaling in the kidney: The good and the bad. Am. J. Physiol. Renal. Physiol. 2016, 311, F241–F248. [Google Scholar] [CrossRef]
- Hara, H.; Takeda, N.; Kondo, M.; Kubota, M.; Saito, T.; Maruyama, J.; Fujiwara, T.; Maemura, S.; Ito, M.; Naito, A.T.; et al. Discovery of a Small Molecule to Increase Cardiomyocytes and Protect the Heart After Ischemic Injury. JACC Basic Transl. Sci. 2018, 3, 639–653. [Google Scholar] [CrossRef]
- Lin, Z.; von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.A.; van Gorp, P.R.; et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef]
- Leach, J.P.; Heallen, T.; Zhang, M.; Rahmani, M.; Morikawa, Y.; Hill, M.C.; Segura, A.; Willerson, J.T.; Martin, J.F. Hippo pathway deficiency reverses systolic heart failure after infarction. Nature 2017, 550, 260. [Google Scholar] [CrossRef]
- Zhou, D.; Zhang, Y.; Wu, H.; Barry, E.; Yin, Y.; Lawrence, E.; Dawson, D.; Willis, J.E.; Markowitz, S.D.; Camargo, F.D.; et al. Mst1 and Mst2 protein kinases restrain intestinal stem cell proliferation and colonic tumorigenesis by inhibition of Yes-associated protein (Yap) overabundance. Proc. Natl. Acad. Sci. USA 2011, 108, E1312–E1320. [Google Scholar] [CrossRef]
- Lu, L.; Li, Y.; Kim, S.M.; Bossuyt, W.; Liu, P.; Qiu, Q.; Wang, Y.; Halder, G.; Finegold, M.J.; Lee, J.S.; et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc. Natl. Acad. Sci. USA 2010, 107, 1437–1442. [Google Scholar] [CrossRef]
- Kim, T.S.; Lee, D.H.; Kim, S.K.; Shin, S.Y.; Seo, E.J.; Lim, D.S. Mammalian sterile 20-like kinase 1 suppresses lymphoma development by promoting faithful chromosome segregation. Cancer Res. 2012, 72, 5386–5395. [Google Scholar] [CrossRef]
- Pan, W.W.; Moroishi, T.; Koo, J.H.; Guan, K.L. Cell type-dependent function of LATS1/2 in cancer cell growth. Oncogene 2018. [Google Scholar] [CrossRef]
- Moroishi, T.; Hayashi, T.; Pan, W.W.; Fujita, Y.; Holt, M.V.; Qin, J.; Carson, D.A.; Guan, K.L. The Hippo Pathway Kinases LATS1/2 Suppress Cancer Immunity. Cell 2016, 167, 1525–1539. [Google Scholar] [CrossRef]
- Xu, B.; Sun, D.; Wang, Z.; Weng, H.; Wu, D.; Zhang, X.; Zhou, Y.; Hu, W. Expression of LATS family proteins in ovarian tumors and its significance. Hum. Pathol. 2015, 46, 858–867. [Google Scholar] [CrossRef]
- Van Haele, M.; Moya, I.M.; Karaman, R.; Rens, G.; Snoeck, J.; Govaere, O.; Nevens, F.; Verslype, C.; Topal, B.; Monbaliu, D.; et al. YAP and TAZ Heterogeneity in Primary Liver Cancer: An Analysis of Its Prognostic and Diagnostic Role. Int. J. Mol. Sci. 2019, 20, 638. [Google Scholar] [CrossRef]
- Rozengurt, E.; Sinnett-Smith, J.; Eibl, G. Yes-associated protein (YAP) in pancreatic cancer: At the epicenter of a targetable signaling network associated with patient survival. Signal. Transduct Target. Ther. 2018, 3, 11. [Google Scholar] [CrossRef]
- Debaugnies, M.; Sanchez-Danes, A.; Rorive, S.; Raphael, M.; Liagre, M.; Parent, M.A.; Brisebarre, A.; Salmon, I.; Blanpain, C. YAP and TAZ are essential for basal and squamous cell carcinoma initiation. EMBO Rep. 2018, 19, e45809. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Donninger, H.; Schmidt, M.L.; Mezzanotte, J.; Barnoud, T.; Clark, G.J. Ras signaling through RASSF proteins. Semin Cell Dev. Biol. 2016, 58, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Richter, A.M.; Pfeifer, G.P.; Dammann, R.H. The RASSF proteins in cancer; from epigenetic silencing to functional characterization. Biochim. Biophys. Acta 2009, 1796, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Volodko, N.; Gordon, M.; Salla, M.; Ghazaleh, H.A.; Baksh, S. RASSF tumor suppressor gene family: Biological functions and regulation. FEBS Lett. 2014, 588, 2671–2684. [Google Scholar] [CrossRef]
- Lock, F.E.; Underhill-Day, N.; Dunwell, T.; Matallanas, D.; Cooper, W.; Hesson, L.; Recino, A.; Ward, A.; Pavlova, T.; Zabarovsky, E.; et al. The RASSF8 candidate tumor suppressor inhibits cell growth and regulates the Wnt and NF-kappaB signaling pathways. Oncogene 2010, 29, 4307–4316. [Google Scholar] [CrossRef]
- Oceandy, D.; Pickard, A.; Prehar, S.; Zi, M.; Mohamed, T.M.; Stanley, P.J.; Baudoin-Stanley, F.; Nadif, R.; Tommasi, S.; Pfeifer, G.P.; et al. Tumor suppressor Ras-association domain family 1 isoform A is a novel regulator of cardiac hypertrophy. Circulation 2009, 120, 607–616. [Google Scholar] [CrossRef]
- Praskova, M.; Khoklatchev, A.; Ortiz-Vega, S.; Avruch, J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004, 381, 453–462. [Google Scholar] [CrossRef]
- Ikeda, M.; Kawata, A.; Nishikawa, M.; Tateishi, Y.; Yamaguchi, M.; Nakagawa, K.; Hirabayashi, S.; Bao, Y.; Hidaka, S.; Hirata, Y.; et al. Hippo pathway-dependent and -independent roles of RASSF6. Sci. Signal. 2009, 2, ra59. [Google Scholar] [CrossRef]
- Foley, C.J.; Freedman, H.; Choo, S.L.; Onyskiw, C.; Fu, N.Y.; Yu, V.C.; Tuszynski, J.; Pratt, J.C.; Baksh, S. Dynamics of RASSF1A/MOAP-1 association with death receptors. Mol. Cell Biol. 2008, 28, 4520–4535. [Google Scholar] [CrossRef] [PubMed]
- Baksh, S.; Tommasi, S.; Fenton, S.; Yu, V.C.; Martins, L.M.; Pfeifer, G.P.; Latif, F.; Downward, J.; Neel, B.G. The tumor suppressor RASSF1A and MAP-1 link death receptor signaling to Bax conformational change and cell death. Mol. Cell 2005, 18, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Kudo, T.; Ikeda, M.; Nishikawa, M.; Yang, Z.; Ohno, K.; Nakagawa, K.; Hata, Y. The RASSF3 candidate tumor suppressor induces apoptosis and G1-S cell-cycle arrest via p53. Cancer Res. 2012, 72, 2901–2911. [Google Scholar] [CrossRef]
- Allen, N.P.; Donninger, H.; Vos, M.D.; Eckfeld, K.; Hesson, L.; Gordon, L.; Birrer, M.J.; Latif, F.; Clark, G.J. RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene 2007, 26, 6203–6211. [Google Scholar] [CrossRef]
- Gordon, M.; El-Kalla, M.; Zhao, Y.; Fiteih, Y.; Law, J.; Volodko, N.; Anwar-Mohamed, A.; El-Kadi, A.O.; Liu, L.; Odenbach, J.; et al. The tumor suppressor gene, RASSF1A, is essential for protection against inflammation -induced injury. PLoS One 2013, 8, e75483. [Google Scholar] [CrossRef]
- Del Re, D.P.; Matsuda, T.; Zhai, P.; Gao, S.; Clark, G.J.; Van Der Weyden, L.; Sadoshima, J. Proapoptotic Rassf1A/Mst1 signaling in cardiac fibroblasts is protective against pressure overload in mice. J. Clin. Invest. 2010, 120, 3555–3567. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Pei, J.; Xia, H.; Ke, H.; Wang, H.; Tao, W. Lats2, a putative tumor suppressor, inhibits G1/S transition. Oncogene 2003, 22, 4398–4405. [Google Scholar] [CrossRef]
- Ke, H.; Pei, J.; Ni, Z.; Xia, H.; Qi, H.; Woods, T.; Kelekar, A.; Tao, W. Putative tumor suppressor Lats2 induces apoptosis through downregulation of Bcl-2 and Bcl-x(L). Exp. Cell Res. 2004, 298, 329–338. [Google Scholar] [CrossRef]
- Aylon, Y.; Michael, D.; Shmueli, A.; Yabuta, N.; Nojima, H.; Oren, M. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev. 2006, 20, 2687–2700. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wei, K.J.; Zhang, W.B.; Sun, H.; Pan, H.Y.; Zhang, L. LATS2 induced by TNF-alpha and inhibited cell proliferation and invasion by phosphorylating YAP in oral squamous cell carcinoma. J. Oral Pathol. Med. 2015, 44, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yang, Y.; Yuan, F.; Huang, J.; Xu, W.; Mao, B.; Yuan, Z.; Bi, W. TNFalpha-YAP/p65-HK2 axis mediates breast cancer cell migration. Oncogenesis 2017, 6, e383. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chen, X.; Wang, Q.; Xu, X.; Xu, B. TNFalpha promotes glioblastoma A172 cell mitochondrial apoptosis via augmenting mitochondrial fission and repression of MAPK-ERK-YAP signaling pathways. Onco. Targets Ther. 2018, 11, 7213–7227. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-kappaB regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef]
- Kim, J.; Kim, Y.H.; Kim, J.; Park, D.Y.; Bae, H.; Lee, D.H.; Kim, K.H.; Hong, S.P.; Jang, S.P.; Kubota, Y.; et al. YAP/TAZ regulates sprouting angiogenesis and vascular barrier maturation. J. Clin. Invest. 2017, 127, 3441–3461. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Bao, Q.; Zhang, Y.; Liu, M.; Lv, H.; Liu, Y.; Yao, L.; Li, B.; Zhang, C.; He, S.; et al. Yes-Associated Protein Promotes Angiogenesis via Signal Transducer and Activator of Transcription 3 in Endothelial Cells. Circ. Res. 2018, 122, 591–605. [Google Scholar] [CrossRef]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev. Cell 2017, 42, 462–478. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Kim, N.E.; Kim, B.M.; Seo, M.; Heo, J.H. TNF-alpha-Induced YAP/TAZ Activity Mediates Leukocyte-Endothelial Adhesion by Regulating VCAM1 Expression in Endothelial Cells. Int. J. Mol. Sci. 2018, 19, 3428. [Google Scholar] [CrossRef]
- Kono, K.; Tamashiro, D.A.; Alarcon, V.B. Inhibition of RHO-ROCK signaling enhances ICM and suppresses TE characteristics through activation of Hippo signaling in the mouse blastocyst. Dev. Biol. 2014, 394, 142–155. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oceandy, D.; Amanda, B.; Ashari, F.Y.; Faizah, Z.; Aziz, M.A.; Stafford, N. The Cross-Talk Between the TNF-α and RASSF-Hippo Signalling Pathways. Int. J. Mol. Sci. 2019, 20, 2346. https://doi.org/10.3390/ijms20092346
Oceandy D, Amanda B, Ashari FY, Faizah Z, Aziz MA, Stafford N. The Cross-Talk Between the TNF-α and RASSF-Hippo Signalling Pathways. International Journal of Molecular Sciences. 2019; 20(9):2346. https://doi.org/10.3390/ijms20092346
Chicago/Turabian StyleOceandy, Delvac, Bella Amanda, Faisal Yusuf Ashari, Zakiyatul Faizah, M Aminudin Aziz, and Nicholas Stafford. 2019. "The Cross-Talk Between the TNF-α and RASSF-Hippo Signalling Pathways" International Journal of Molecular Sciences 20, no. 9: 2346. https://doi.org/10.3390/ijms20092346
APA StyleOceandy, D., Amanda, B., Ashari, F. Y., Faizah, Z., Aziz, M. A., & Stafford, N. (2019). The Cross-Talk Between the TNF-α and RASSF-Hippo Signalling Pathways. International Journal of Molecular Sciences, 20(9), 2346. https://doi.org/10.3390/ijms20092346