Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine

 , ,
, , 
Abstract
:
1. Introduction
Computational Enzyme Redesign for Novel and Efficient aaRS Variants
2. Results
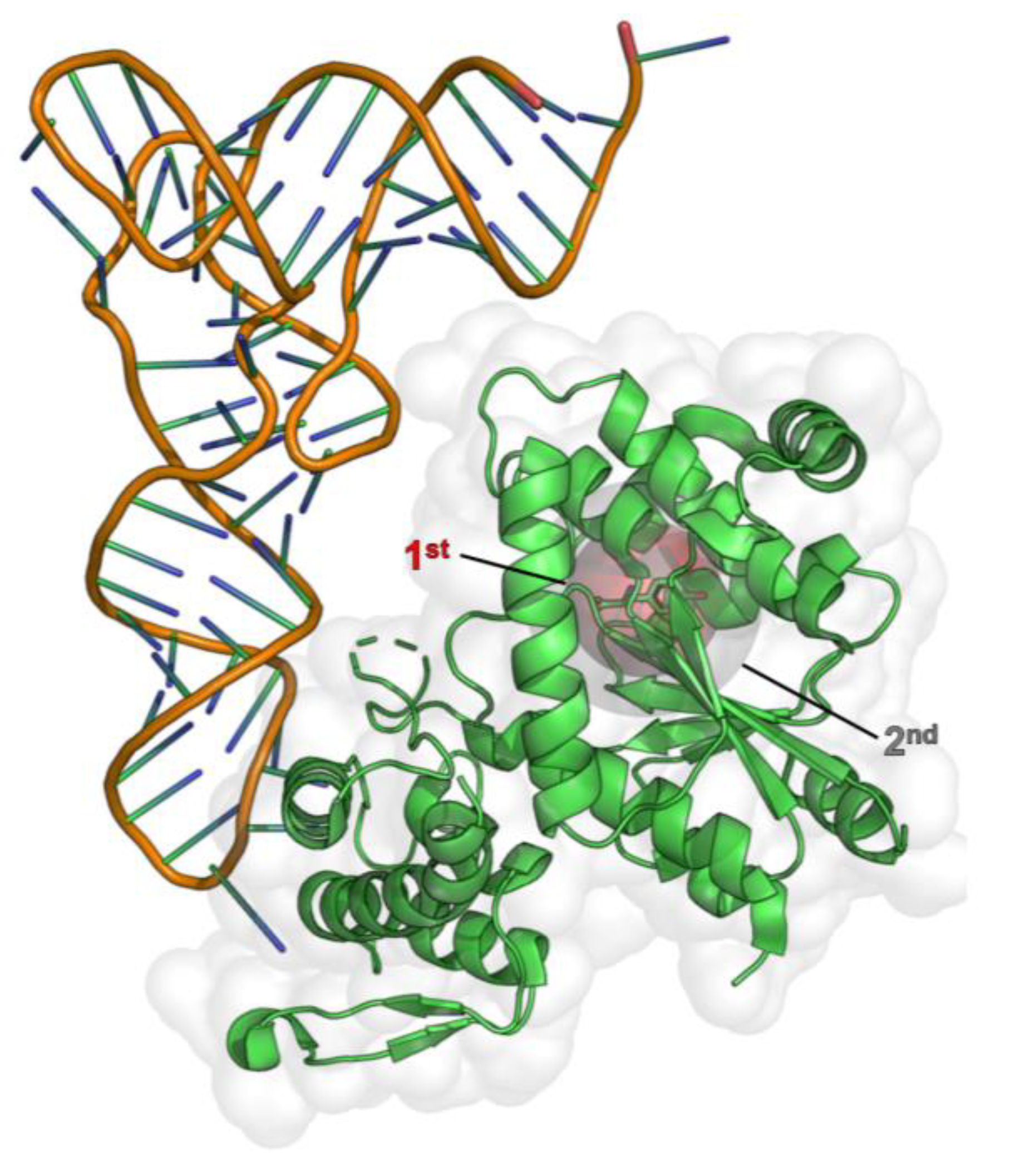
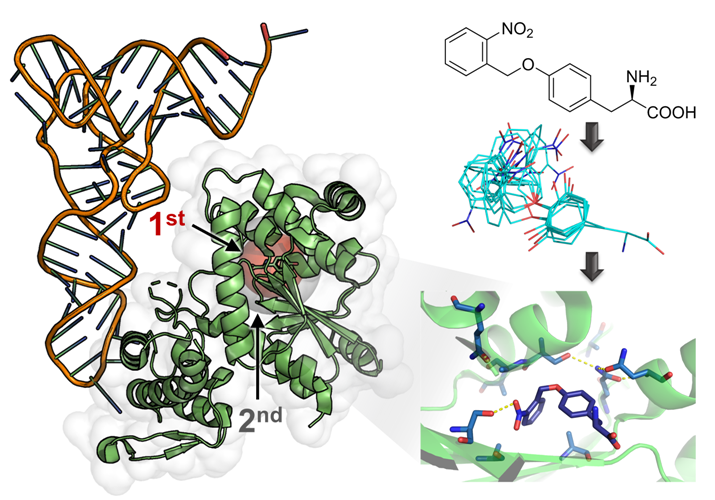
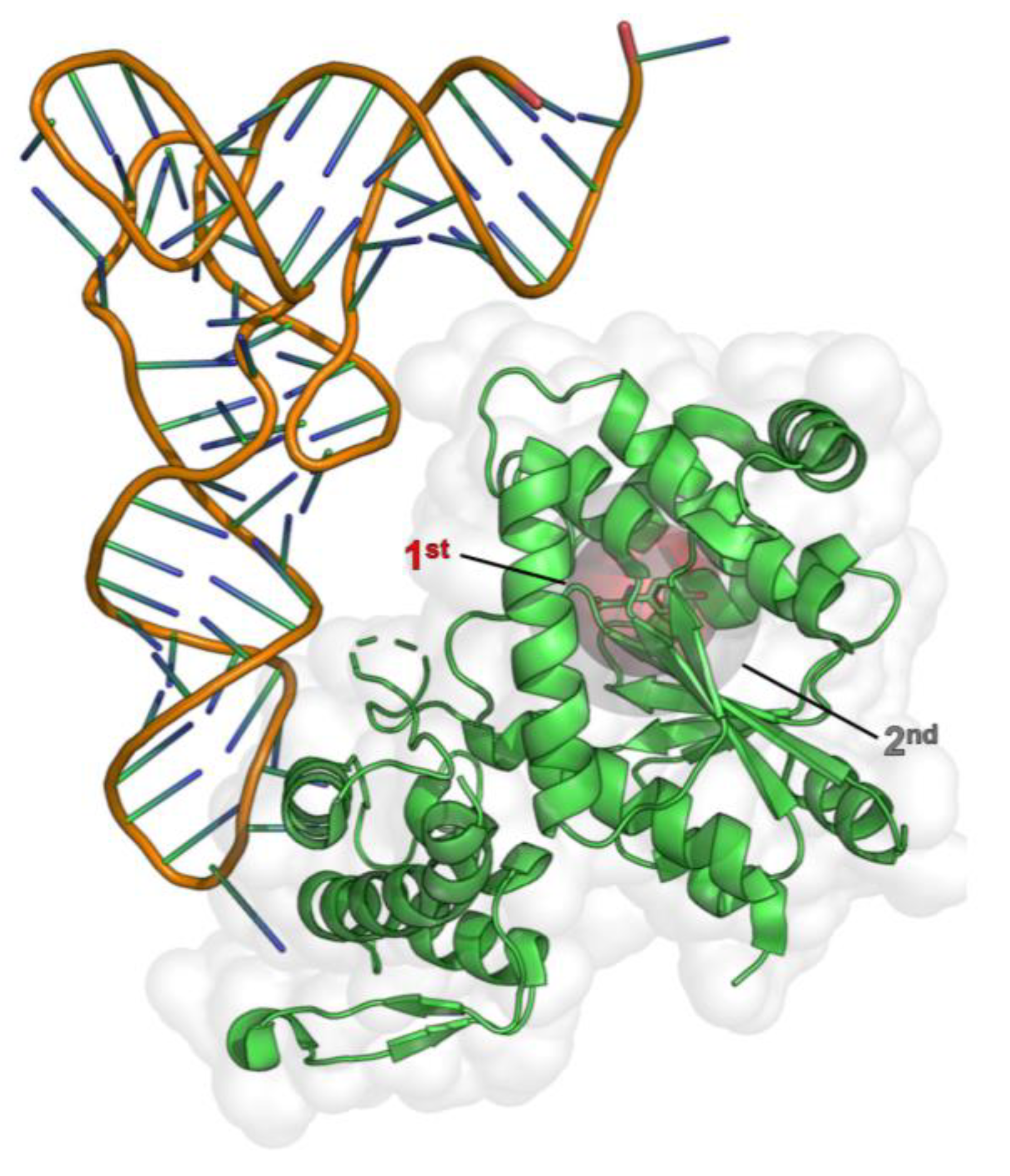
2.1. Computational Design of an aaRS Library
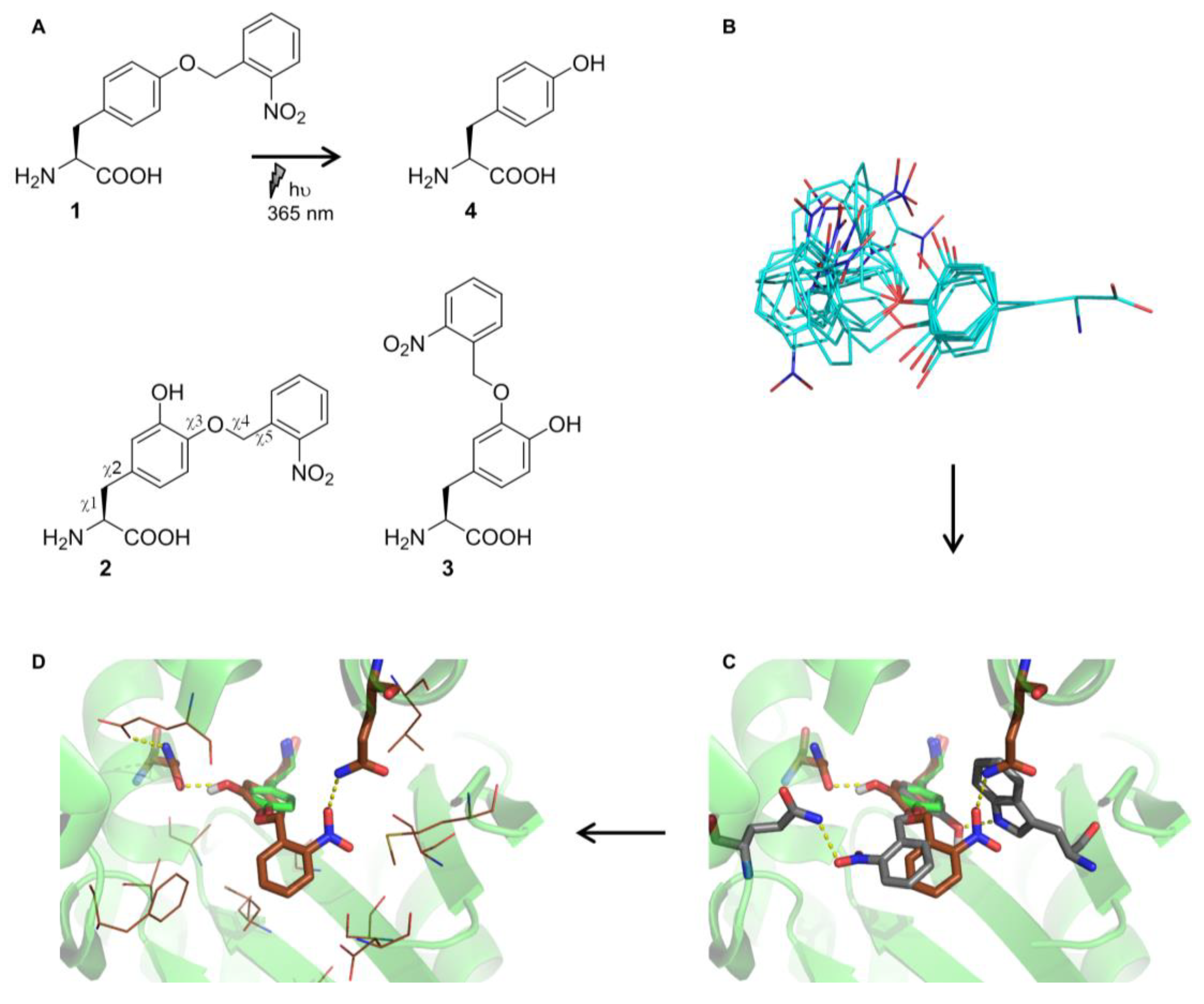
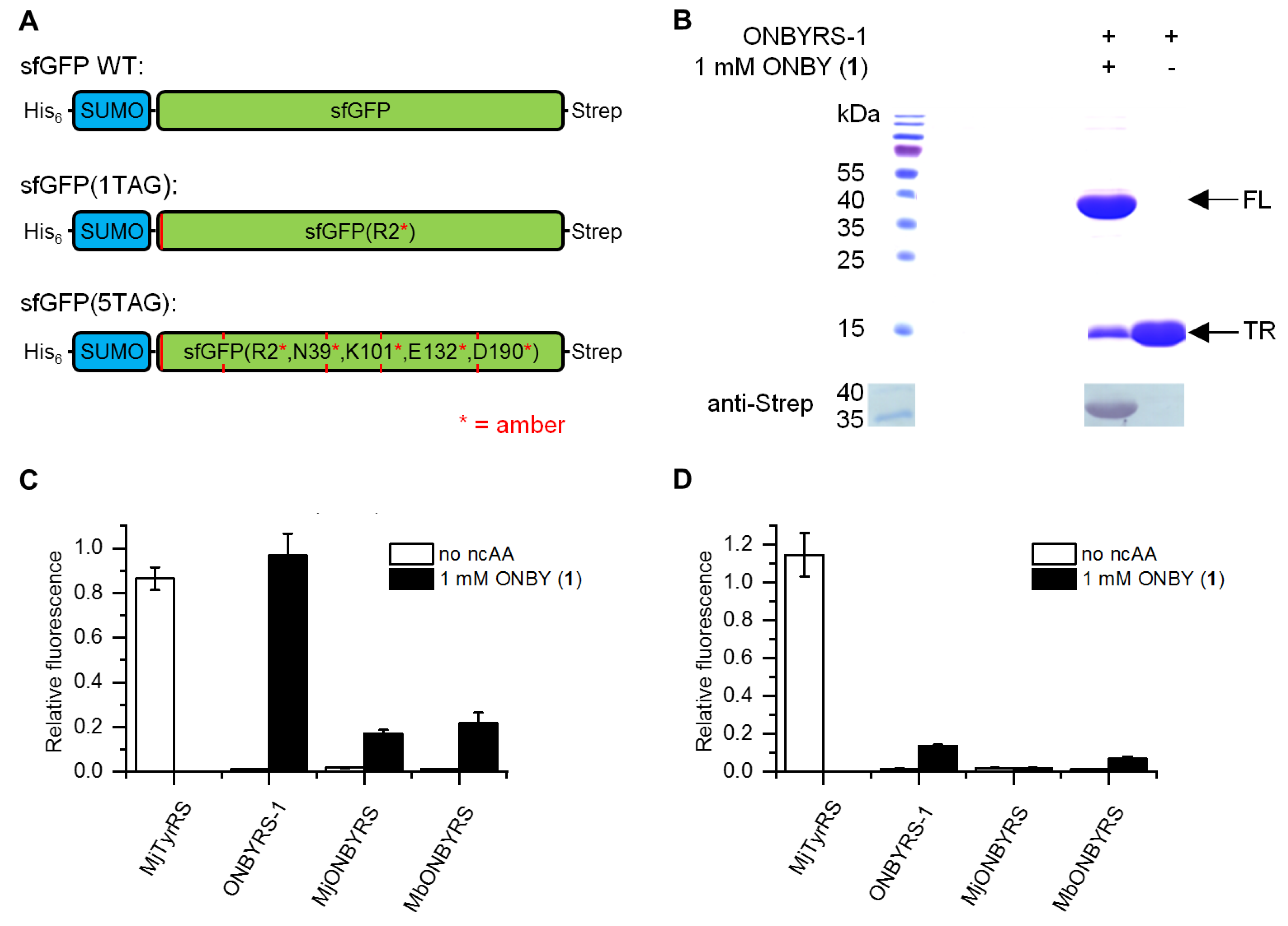
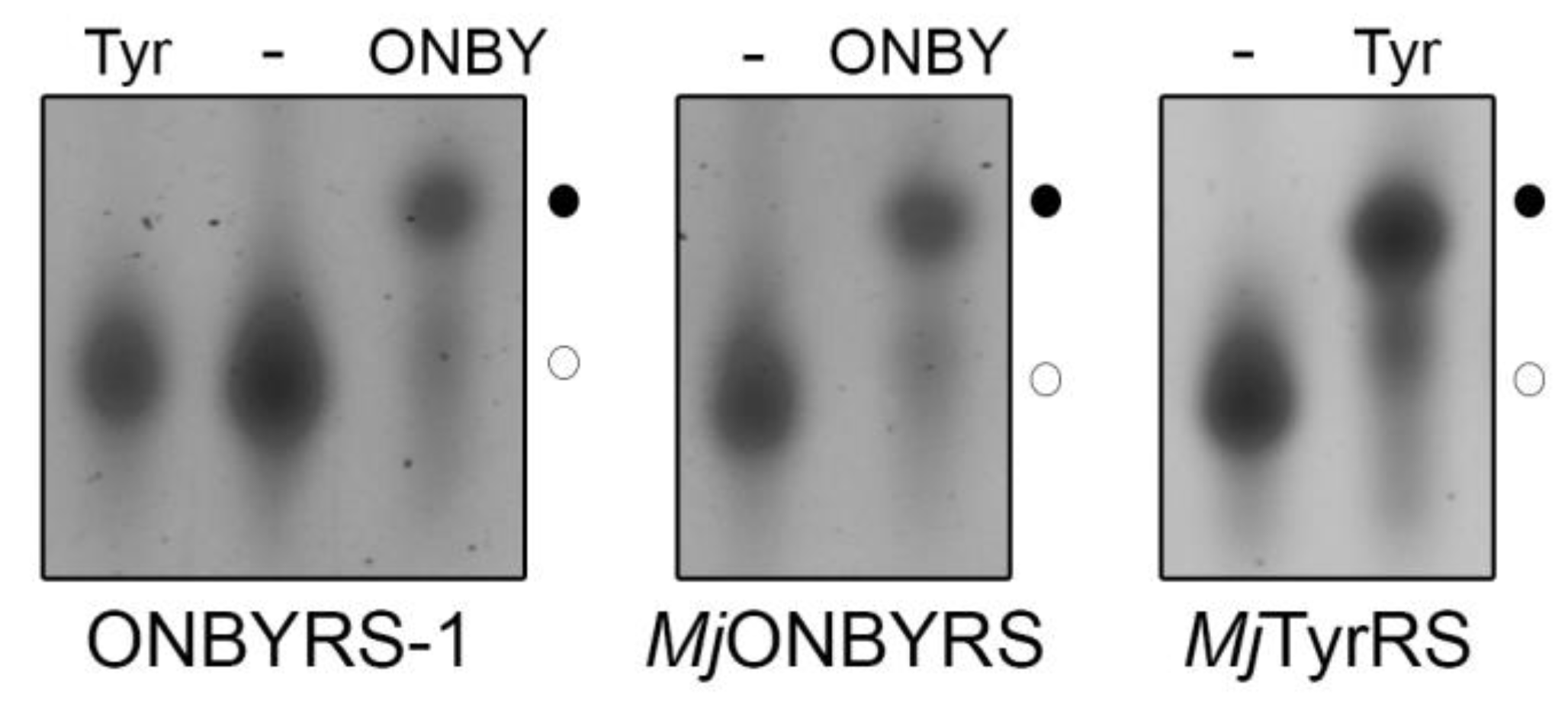
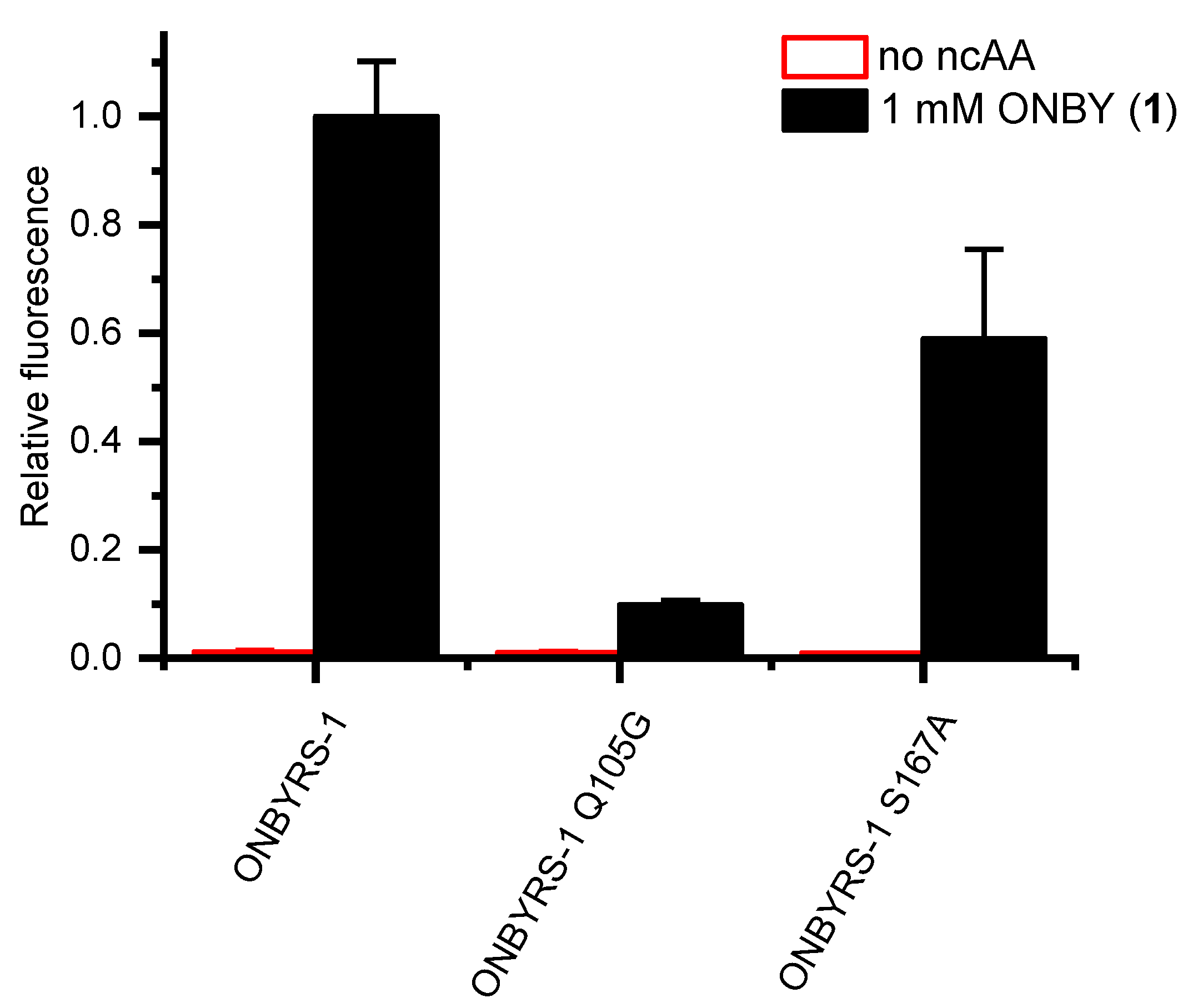
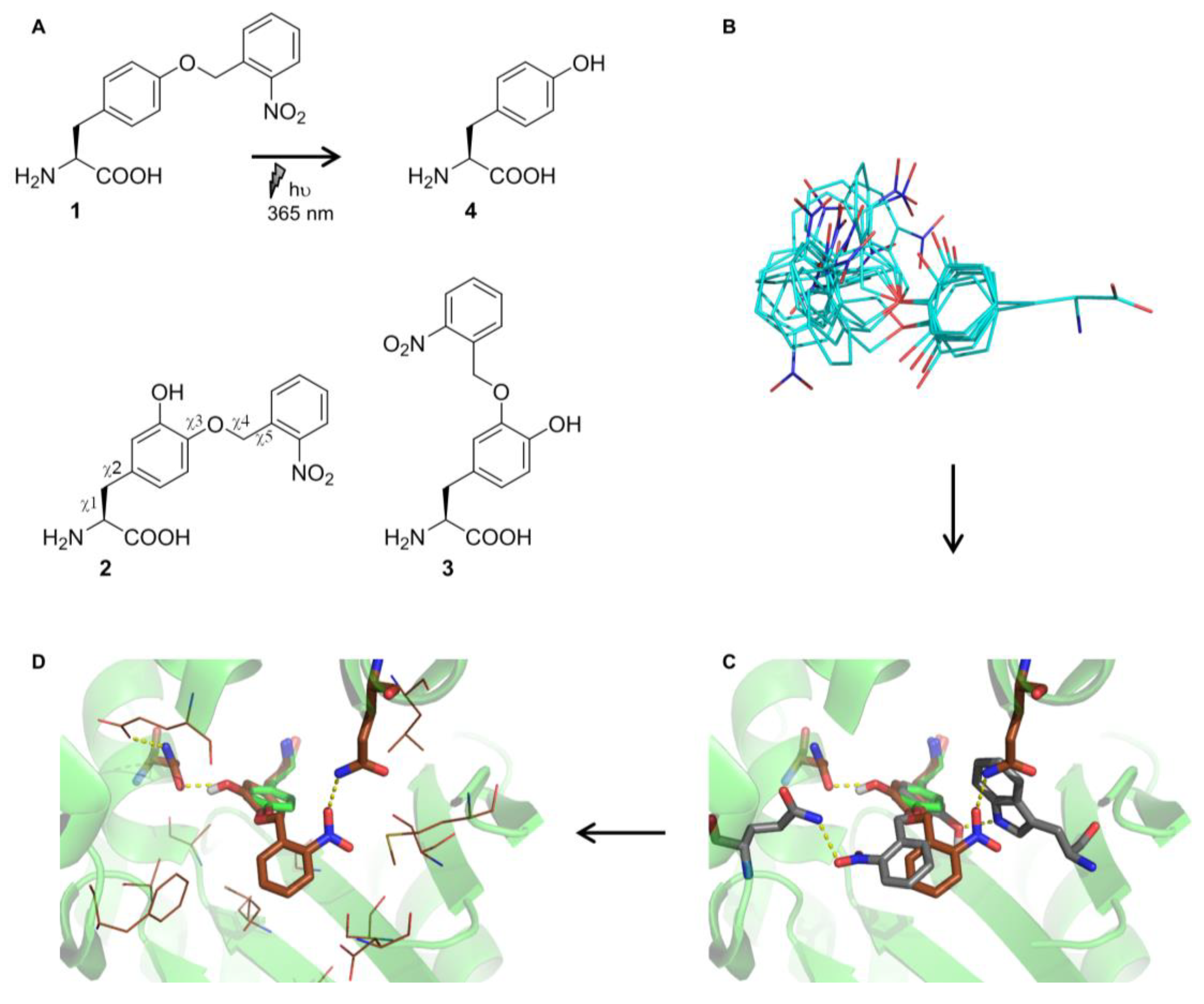
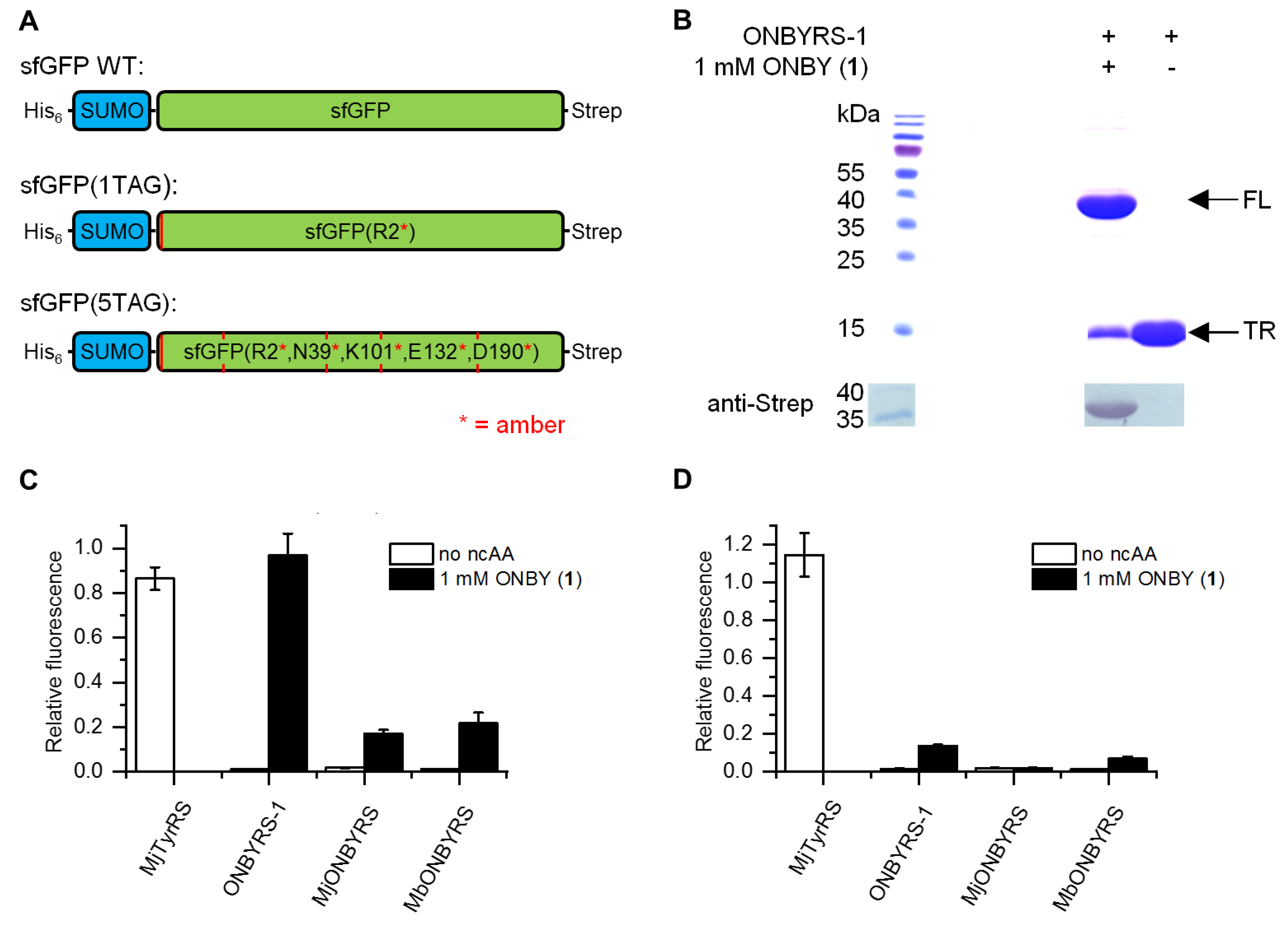
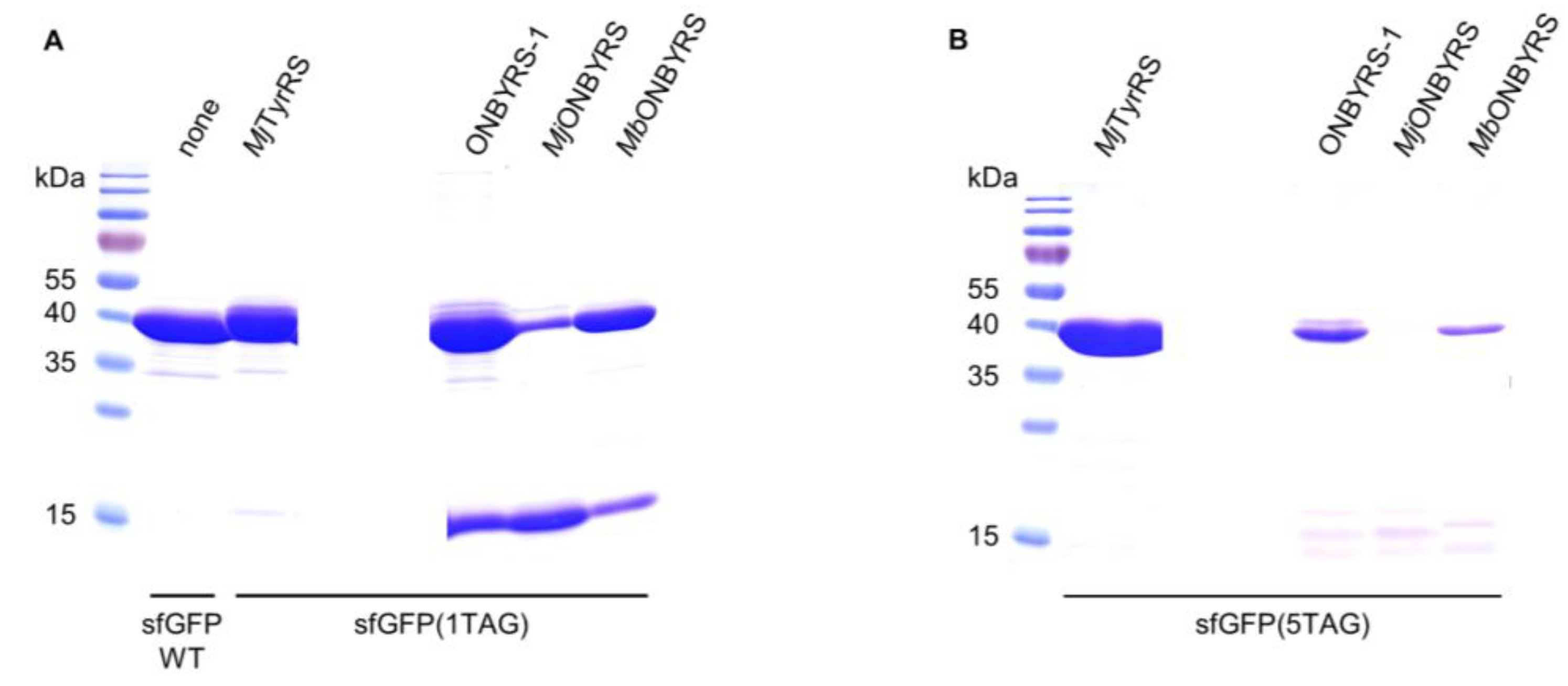
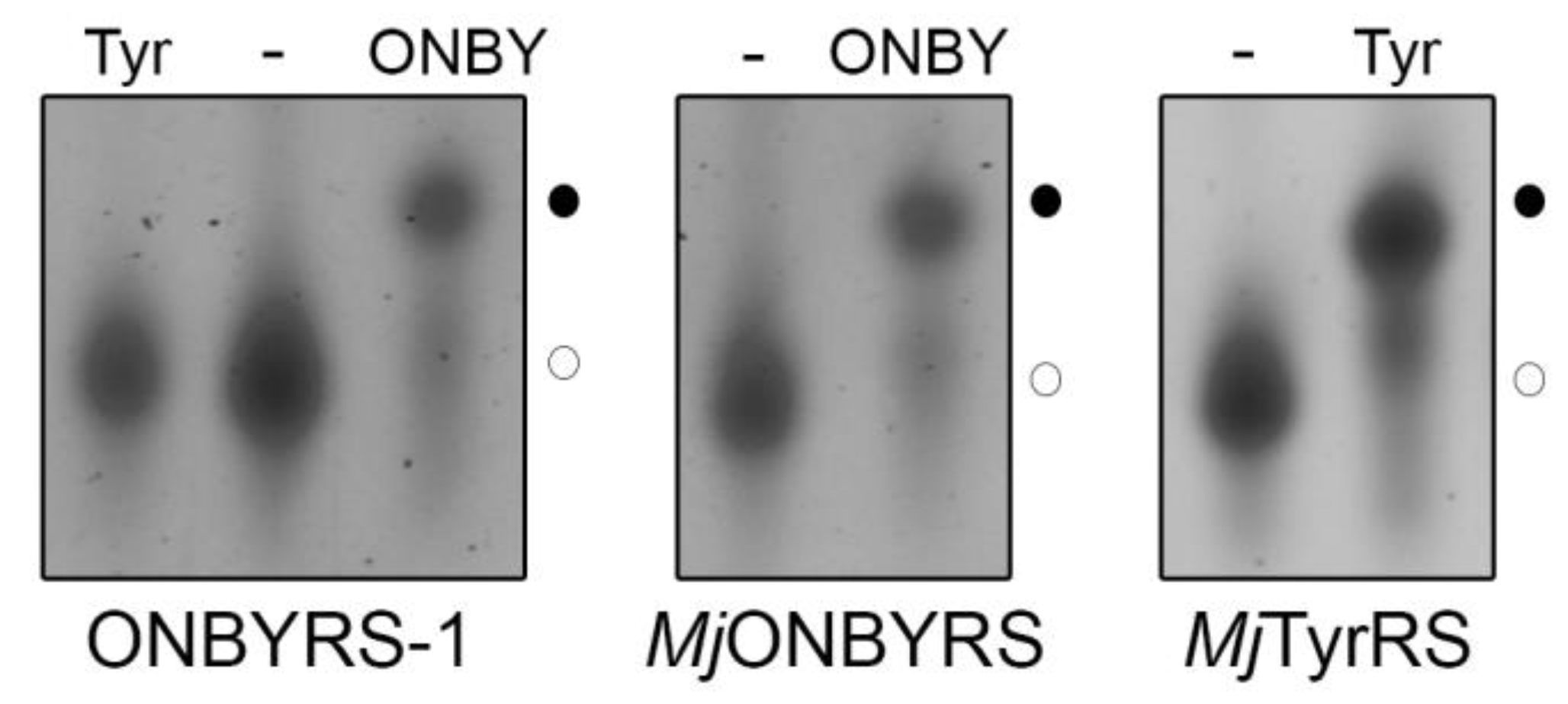
2.2. Selection and Characterization of an aaRS Specific for ONBY
2.3. Comparison to Previously Reported aaRSs
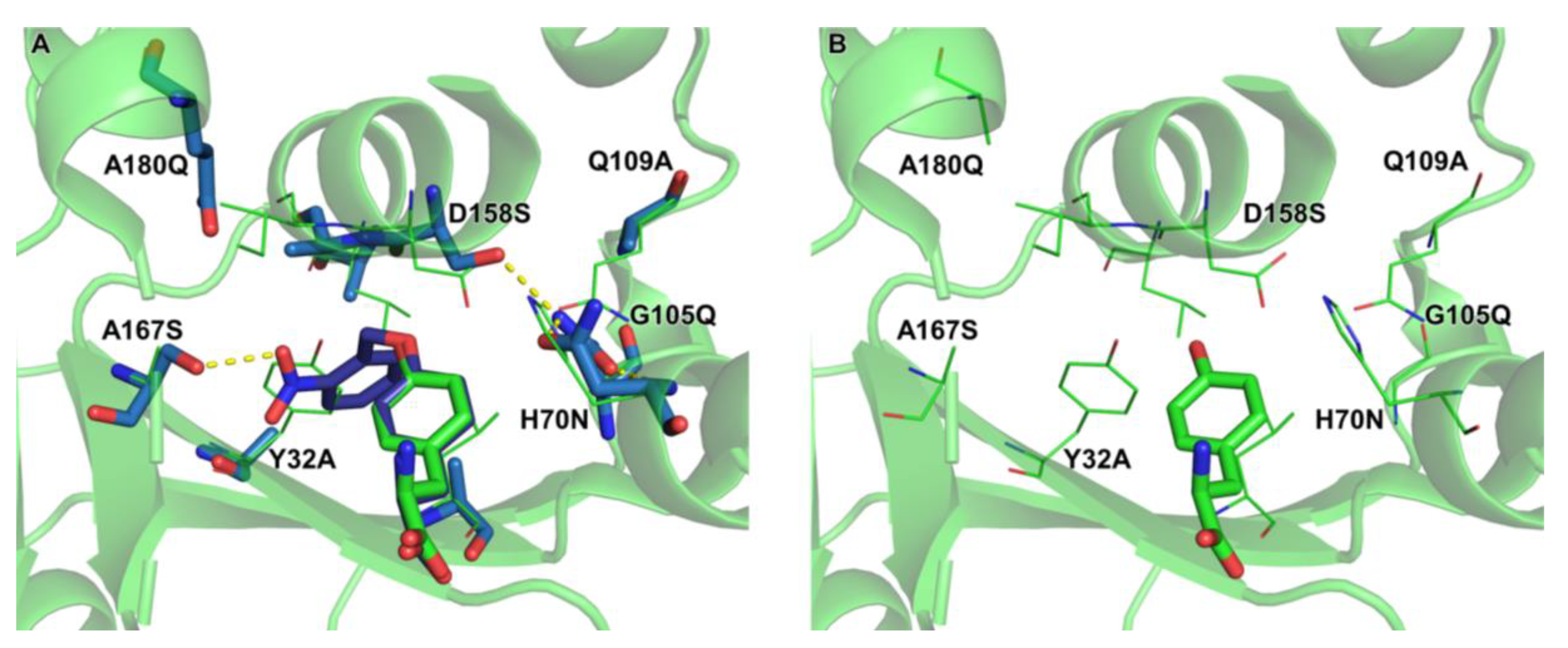
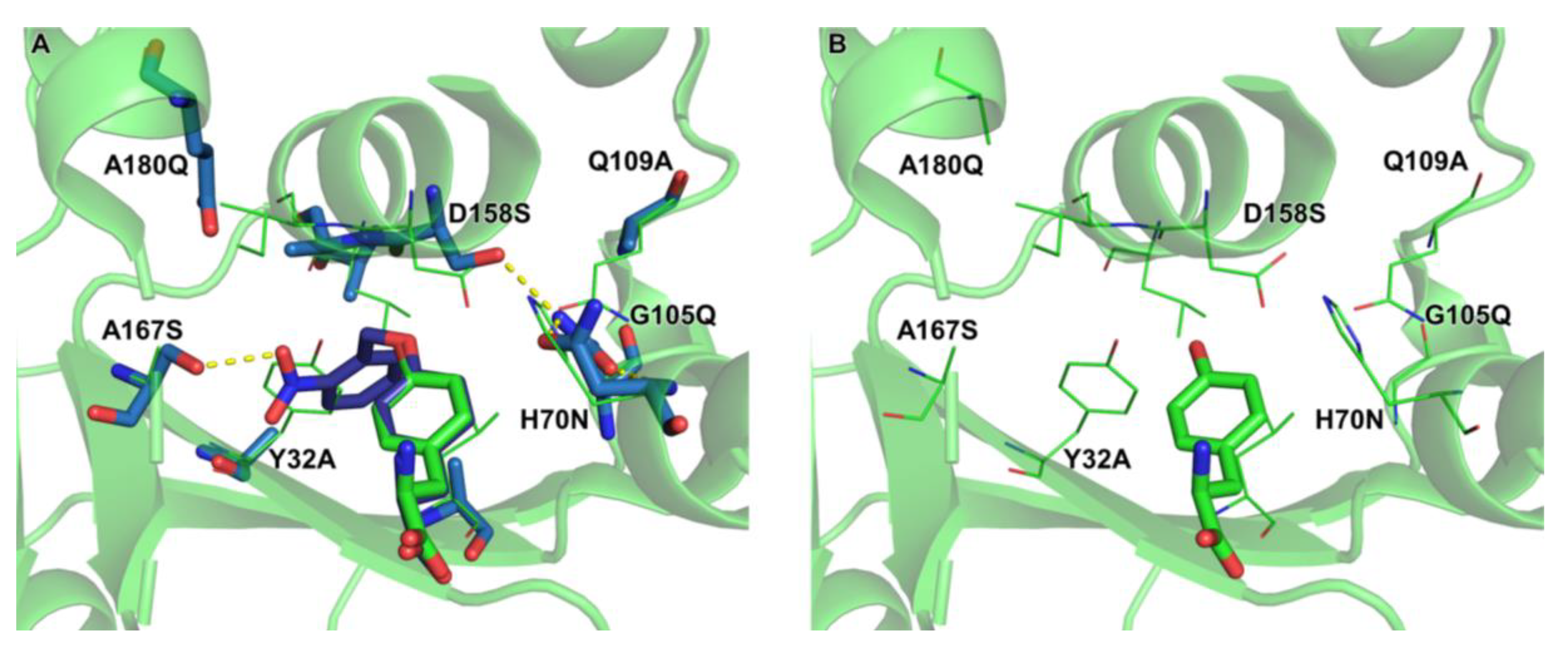
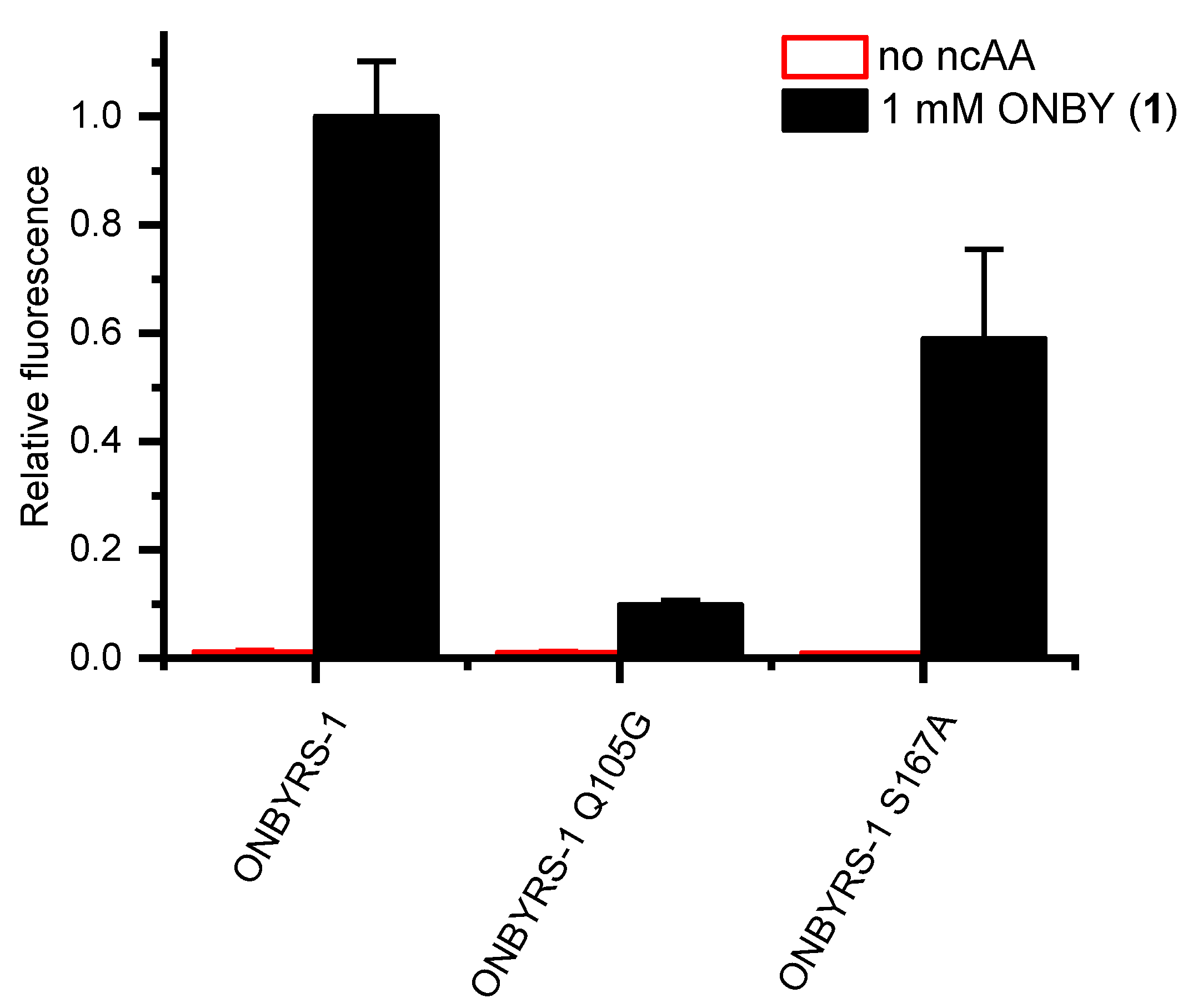
2.4. Active Site Modeling for ONBYRS-1 and Key Mutations
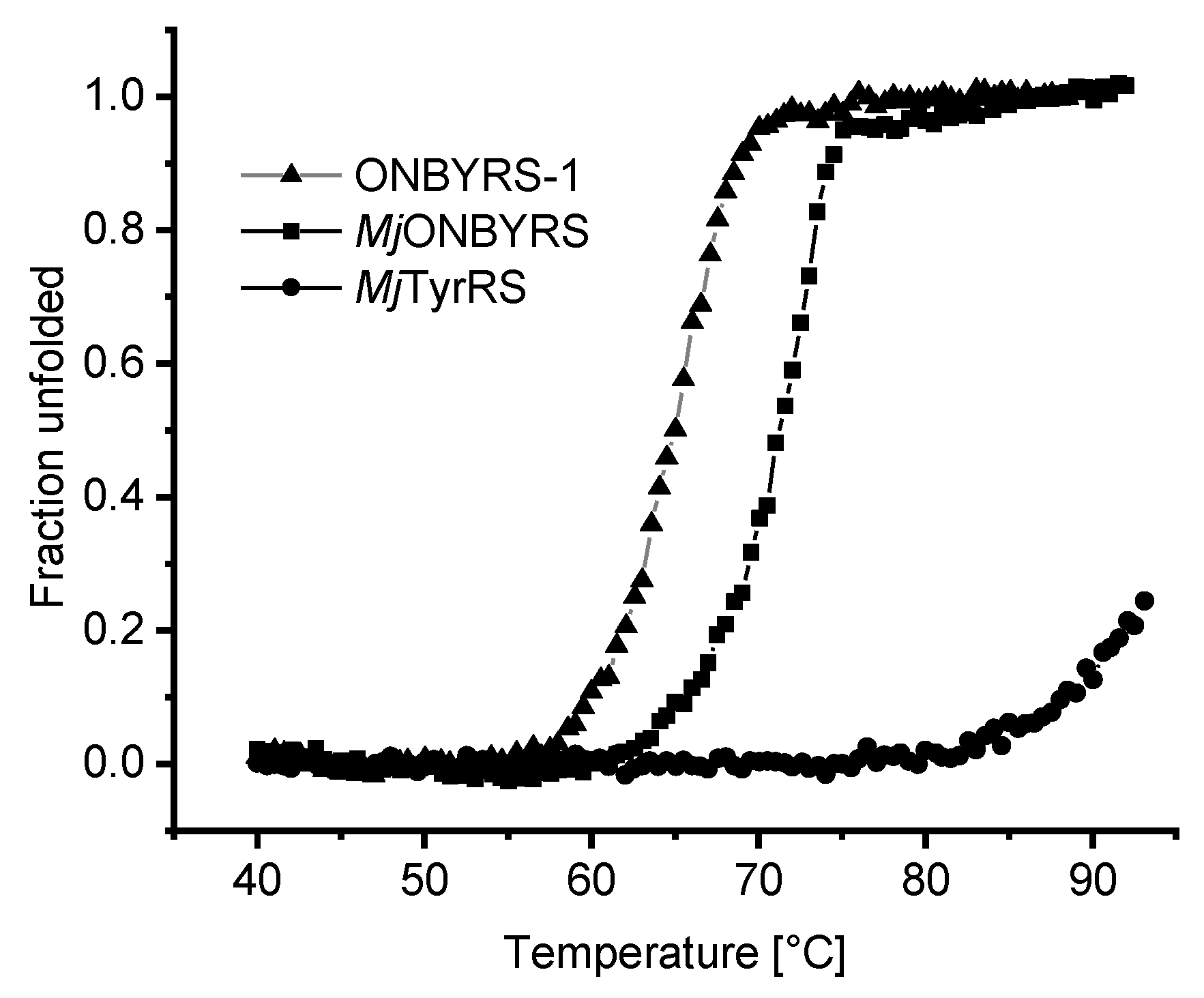
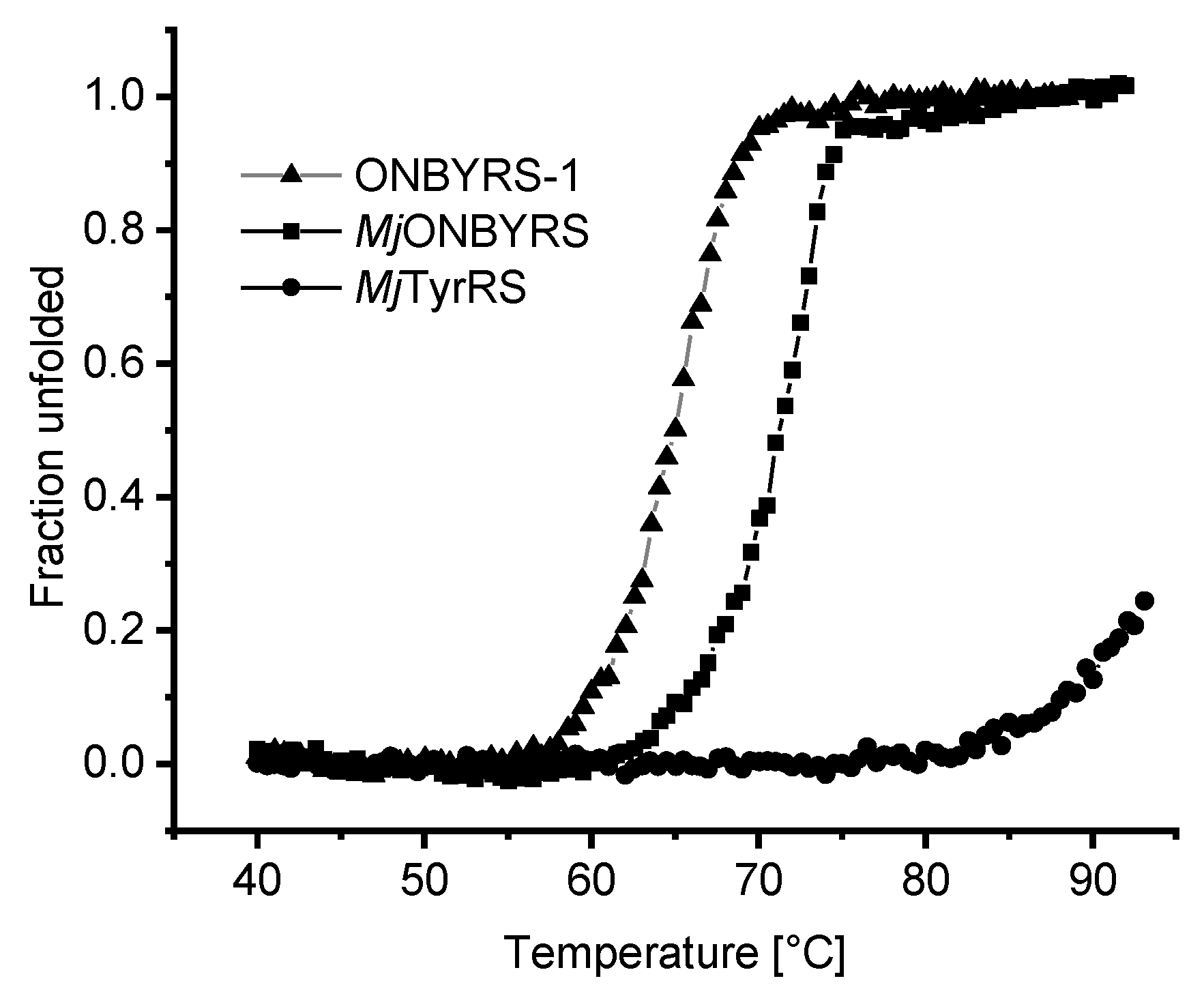
2.5. Impact of aaRS Mutations on Thermal Stability
3. Discussion
4. Materials and Methods
4.1. Computational Design Procedures
4.2. Noncanonical Amino Acid
4.3. Library Construction and Selection
4.4. Bacterial Strain Construction
4.5. Analysis of sfGFP Expression by Intact Cell Fluorescence
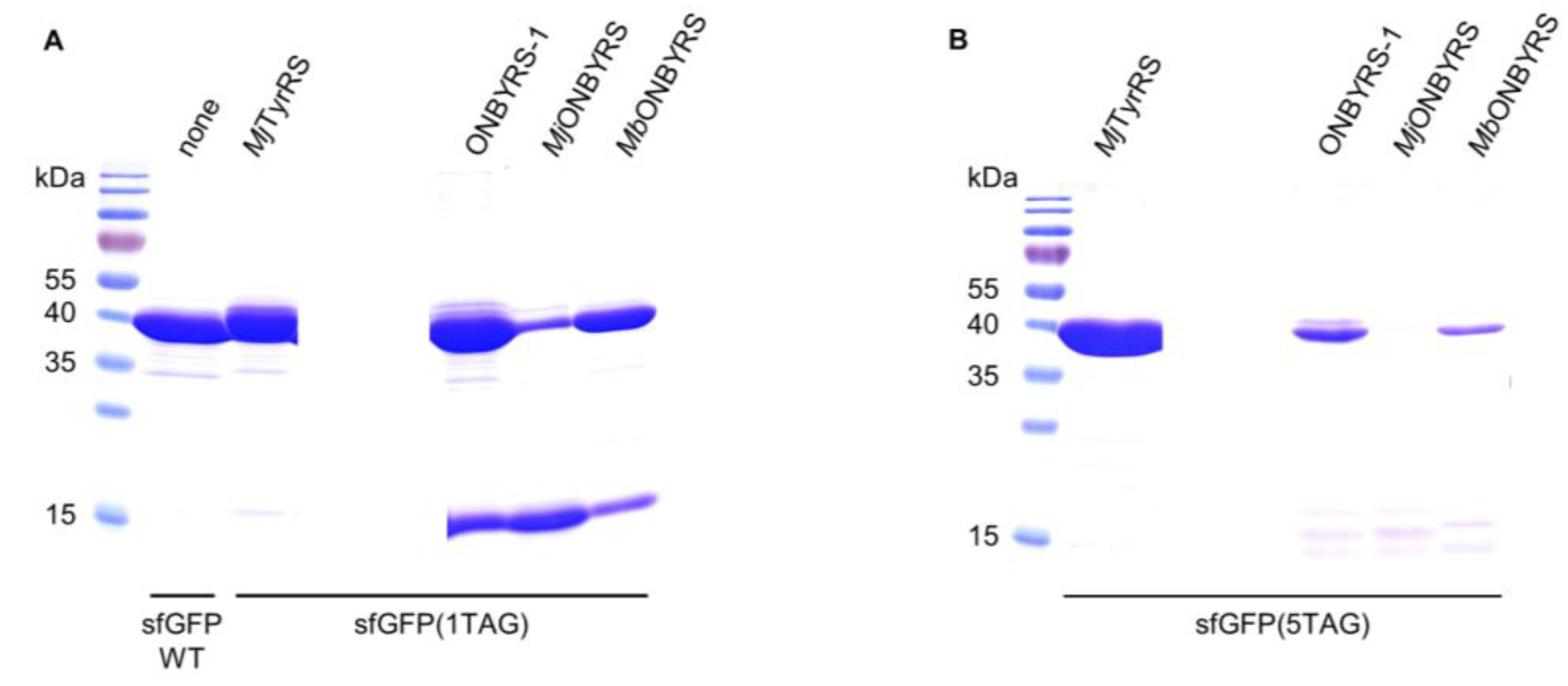
4.6. Protein Expression and Purification
4.7. Western Blot Analyses
4.8. Mass Spectrometry
4.9. Thermal Protein Unfolding
4.10. In Vitro Transcription and Aminoacylation of tRNA
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| aaRS | aminoacyl-tRNA synthetase |
| CAT | chloramphenicol acetyltransferase |
| CD | circular dichroism |
| CED | computational enzyme design |
| Cm | chloramphenicol |
| DOPA | L-3,4-dihydroxyphenylalanine |
| ELP | elastin-like polypeptide |
| Mb | Methanosarcina barkeri |
| Mj | Methanocaldococcus jannaschii |
| Mm | Methanosarcina mazei |
| ncAA | noncanonical amino acids |
| ONB-DOPA | ortho-nitrobenzyl L-3,4-dihydroxyphenylalanine |
| ONBY | ortho-nitrobenzyl L-tyrosine |
| o-pair | orthogonal pair |
| OTS | orthogonal translation system |
| PylRS | pyrrolysyl-tRNA synthetase |
| RF1 | release factor 1 |
| sfGFP | superfolder green fluorescent protein |
| SSM | site-saturation mutagenesis |
| SUMO | small ubiquitin-like modifier |
| Tyr | L-tyrosine |
| TyrRS | tyrosyl-tRNA synthetase |
| WT | wild type |
Appendix A
References
- Yadavalli, S.S.; Ibba, M. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv. Protein Chem. Struct. Biol. 2012, 86, 1–43. [Google Scholar]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef]
- Dumas, A.; Lercher, L.; Spicer, C.D.; Davis, B.G. Designing logical codon reassignment – Expanding the chemistry in biology. Chem. Sci. 2015, 6, 50–69. [Google Scholar] [CrossRef]
- Vargas-Rodriguez, O.; Sevostyanova, A.; Söll, D.; Crnković, A. Upgrading aminoacyl-tRNA synthetases for genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 115–122. [Google Scholar] [CrossRef]
- Nehring, S.; Budisa, N.; Wiltschi, B. Performance analysis of orthogonal pairs designed for an expanded eukaryotic genetic code. PLoS ONE 2012, 7, e31992. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, P.; Ling, J.; Wang, Y.-S.; Söll, D. Upgrading protein synthesis for synthetic biology. Nat. Chem. Biol. 2013, 9, 594–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, T.; Kuratani, M.; Seki, E.; Hino, N.; Sakamoto, K.; Yokoyama, S. Structural Basis for Genetic-Code Expansion with Bulky Lysine Derivatives by an Engineered Pyrrolysyl-tRNA Synthetase. Cell Chem. Biol. 2019, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the genetic code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Perona, J.J.; Hadd, A. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry 2012, 51, 8705–8729. [Google Scholar] [CrossRef]
- Kille, S.; Acevedo-Rocha, C.G.; Parra, L.P.; Zhang, Z.-G.; Opperman, D.J.; Reetz, M.T.; Acevedo, J.P. Reducing codon redundancy and screening effort of combinatorial protein libraries created by saturation mutagenesis. ACS Synth. Biol. 2013, 2, 83–92. [Google Scholar] [CrossRef]
- Bullock, T.L.; Rodríguez-Hernández, A.; Corigliano, E.M.; Perona, J.J. A rationally engineered misacylating aminoacyl-tRNA synthetase. Proc. Natl. Acad. Sci. USA 2008, 105, 7428–7433. [Google Scholar] [CrossRef] [PubMed]
- Leaver-Fay, A.; Tyka, M.; Lewis, S.M.; Lange, O.F.; Thompson, J.; Jacak, R.; Kaufman, K.; Renfrew, P.D.; Smith, C.A.; Sheffler, W.; et al. ROSETTA3: An object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011, 487, 545–574. [Google Scholar] [PubMed]
- Richter, F.; Leaver-Fay, A.; Khare, S.D.; Bjelic, S.; Baker, D. De novo enzyme design using Rosetta3. PLoS ONE 2011, 6, e19230. [Google Scholar] [CrossRef]
- Richter, F.; Baker, D. Computational Protein Design for Synthetic Biology. In Synthetic Biology; Zhao, H., Ed.; Academic Press: Boston, MA, USA, 2013; pp. 101–122. ISBN 978-0-12-394430-6. [Google Scholar]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Röthlisberger, D.; Khersonsky, O.; Wollacott, A.M.; Jiang, L.; DeChancie, J.; Betker, J.; Gallaher, J.L.; Althoff, E.A.; Zanghellini, A.; Dym, O.; et al. Kemp elimination catalysts by computational enzyme design. Nature 2008, 453, 190–195. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Althoff, E.A.; Clemente, F.R.; Doyle, L.; Röthlisberger, D.; Zanghellini, A.; Gallaher, J.L.; Betker, J.L.; Tanaka, F.; Barbas, C.F.; et al. De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.B.; Zanghellini, A.; Lovick, H.M.; Kiss, G.; Lambert, A.R.; St Clair, J.L.; Gallaher, J.L.; Hilvert, D.; Gelb, M.H.; Stoddard, B.L.; et al. Computational design of an enzyme catalyst for a stereoselective bimolecular Diels-Alder reaction. Science 2010, 329, 309–313. [Google Scholar] [CrossRef]
- Richter, F.; Blomberg, R.; Khare, S.D.; Kiss, G.; Kuzin, A.P.; Smith, A.J.T.; Gallaher, J.; Pianowski, Z.; Helgeson, R.C.; Grjasnow, A.; et al. Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. J. Am. Chem. Soc. 2012, 134, 16197–16206. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.S.; Nivón, L.G.; Richter, F.; Goldman, P.J.; Deerinck, T.J.; Yao, J.Z.; Richardson, D.; Phipps, W.S.; Ye, A.Z.; Ellisman, M.H.; et al. Computational design of a red fluorophore ligase for site-specific protein labeling in living cells. Proc. Natl. Acad. Sci. USA 2014, 111, E4551–E4559. [Google Scholar] [CrossRef] [Green Version]
- Heinisch, T.; Pellizzoni, M.; Dürrenberger, M.; Tinberg, C.E.; Köhler, V.; Klehr, J.; Häussinger, D.; Baker, D.; Ward, T.R. Improving the Catalytic Performance of an Artificial Metalloenzyme by Computational Design. J. Am. Chem. Soc. 2015, 137, 10414–10419. [Google Scholar] [CrossRef]
- Lippow, S.M.; Moon, T.S.; Basu, S.; Yoon, S.-H.; Li, X.; Chapman, B.A.; Robison, K.; Lipovšek, D.; Prather, K.L.J. Engineering enzyme specificity using computational design of a defined-sequence library. Chem. Biol. 2010, 17, 1306–1315. [Google Scholar] [CrossRef]
- Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J.W. Photocontrol of tyrosine phosphorylation in mammalian cells via genetic encoding of photocaged tyrosine. J. Am. Chem. Soc. 2012, 134, 11912–11915. [Google Scholar] [CrossRef]
- Wilkins, B.J.; Marionni, S.; Young, D.D.; Liu, J.; Wang, Y.; Di Salvo, M.L.; Deiters, A.; Cropp, T.A. Site-specific incorporation of fluorotyrosines into proteins in Escherichia coli by photochemical disguise. Biochemistry 2010, 49, 1557–1559. [Google Scholar] [CrossRef]
- Deiters, A.; Groff, D.; Ryu, Y.; Xie, J.; Schultz, P.G. A genetically encoded photocaged tyrosine. Angew. Chem. Int. Ed. Engl. 2006, 45, 2728–2731. [Google Scholar] [CrossRef]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Möglich, A.; et al. Photoactivatable Mussel-Based Underwater Adhesive Proteins by an Expanded Genetic Code. Chembiochem 2017, 18, 1819–1823. [Google Scholar] [CrossRef]
- Kobayashi, T.; Nureki, O.; Ishitani, R.; Yaremchuk, A.; Tukalo, M.; Cusack, S.; Sakamoto, K.; Yokoyama, S. Structural basis for orthogonal tRNA specificities of tyrosyl-tRNA synthetases for genetic code expansion. Nat. Struct. Biol. 2003, 10, 425–432. [Google Scholar] [CrossRef]
- Zeymer, C.; Hilvert, D. Directed Evolution of Protein Catalysts. Annu. Rev. Biochem. 2018, 87, 131–157. [Google Scholar] [CrossRef]
- Lajoie, M.J.; Rovner, A.J.; Goodman, D.B.; Aerni, H.-R.; Haimovich, A.D.; Kuznetsov, G.; Mercer, J.A.; Wang, H.H.; Carr, P.A.; Mosberg, J.A.; et al. Genomically recoded organisms expand biological functions. Science 2013, 342, 357–360. [Google Scholar] [CrossRef]
- Johnson, D.B.F.; Xu, J.; Shen, Z.; Takimoto, J.K.; Schultz, M.D.; Schmitz, R.J.; Xiang, Z.; Ecker, J.R.; Briggs, S.P.; Wang, L. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat. Chem. Biol. 2011, 7, 779–786. [Google Scholar] [CrossRef]
- Kirchner, S.; Ignatova, Z. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet. 2015, 16, 98–112. [Google Scholar] [CrossRef]
- Hall, K.B.; Sampson, J.R.; Uhlenbeck, O.C.; Redfield, A.G. Structure of an unmodified tRNA molecule. Biochemistry 1989, 28, 5794–5801. [Google Scholar] [CrossRef]
- Byrne, R.T.; Konevega, A.L.; Rodnina, M.V.; Antson, A.A. The crystal structure of unmodified tRNAPhe from Escherichia coli. Nucleic Acids Res. 2010, 38, 4154–4162. [Google Scholar] [CrossRef]
- Sampson, J.R.; Uhlenbeck, O.C. Biochemical and physical characterization of an unmodified yeast phenylalanine transfer RNA transcribed in vitro. Proc. Natl. Acad. Sci. USA 1988, 85, 1033–1037. [Google Scholar] [CrossRef]
- Liu, W.; Alfonta, L.; Mack, A.V.; Schultz, P.G. Structural basis for the recognition of para-benzoyl-L-phenylalanine by evolved aminoacyl-tRNA synthetases. Angew. Chem. Int. Ed. Engl. 2007, 46, 6073–6075. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Russell, W.K.; Wang, Z.; Wan, W.; Dodd, L.E.; Pai, P.-J.; Russell, D.H.; Liu, W.R. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol. Biosyst. 2011, 7, 714–717. [Google Scholar] [CrossRef]
- Wang, P.; Vaidehi, N.; Tirrell, D.A.; Goddard, W.A. Virtual screening for binding of phenylalanine analogues to phenylalanyl-tRNA synthetase. J. Am. Chem. Soc. 2002, 124, 14442–14449. [Google Scholar] [CrossRef]
- Zhang, D.; Vaidehi, N.; Goddard, W.A.; Danzer, J.F.; Debe, D. Structure-based design of mutant Methanococcus jannaschii tyrosyl-tRNA synthetase for incorporation of O-methyl-L-tyrosine. Proc. Natl. Acad. Sci. USA 2002, 99, 6579–6584. [Google Scholar] [CrossRef] [Green Version]
- Xuan, W.; Collins, D.; Koh, M.; Shao, S.; Yao, A.; Xiao, H.; Garner, P.; Schultz, P.G. Site-Specific Incorporation of a Thioester Containing Amino Acid into Proteins. ACS Chem. Biol. 2018, 13, 578–581. [Google Scholar] [CrossRef]
- Saravanan Prabhu, N.; Ayyadurai, N.; Deepankumar, K.; Chung, T.; Lim, D.J.; Yun, H. Reassignment of sense codons: Designing and docking of proline analogs for Escherichia coli prolyl-tRNA synthetase to expand the genetic code. J. Mol. Catal. B Enzym. 2012, 78, 57–64. [Google Scholar] [CrossRef]
- Antonczak, A.K.; Simova, Z.; Yonemoto, I.T.; Bochtler, M.; Piasecka, A.; Czapinska, H.; Brancale, A.; Tippmann, E.M. Importance of single molecular determinants in the fidelity of expanded genetic codes. Proc. Natl. Acad. Sci. USA 2011, 108, 1320–1325. [Google Scholar] [CrossRef]
- Nadarajan, S.P.; Mathew, S.; Deepankumar, K.; Yun, H. An in silico approach to evaluate the polyspecificity of methionyl-tRNA synthetases. J. Mol. Graph. Model. 2013, 39, 79–86. [Google Scholar] [CrossRef]
- Chou, C.; Deiters, A. Light-activated gene editing with a photocaged zinc-finger nuclease. Angew. Chem. Int. Ed. Engl. 2011, 50, 6839–6842. [Google Scholar] [CrossRef]
- Groff, D.; Thielges, M.C.; Cellitti, S.; Schultz, P.G.; Romesberg, F.E. Efforts toward the direct experimental characterization of enzyme microenvironments: tyrosine100 in dihydrofolate reductase. Angew. Chem. Int. Ed. Engl. 2009, 48, 3478–3481. [Google Scholar] [CrossRef]
- Chou, C.; Young, D.D.; Deiters, A. A light-activated DNA polymerase. Angew. Chem. Int. Ed. Engl. 2009, 48, 5950–5953. [Google Scholar] [CrossRef]
- Chou, C.; Young, D.D.; Deiters, A. Photocaged t7 RNA polymerase for the light activation of transcription and gene function in pro- and eukaryotic cells. Chembiochem 2010, 11, 972–977. [Google Scholar] [CrossRef]
- Larson, A.S.; Hergenrother, P.J. Light activation of Staphylococcus aureus toxin YoeBSa1 reveals guanosine-specific endoribonuclease activity. Biochemistry 2014, 53, 188–201. [Google Scholar] [CrossRef]
- Böcker, J.K.; Friedel, K.; Matern, J.C.J.; Bachmann, A.-L.; Mootz, H.D. Generation of a genetically encoded, photoactivatable intein for the controlled production of cyclic peptides. Angew. Chem. Int. Ed. Engl. 2015, 54, 2116–2120. [Google Scholar] [CrossRef]
- Böcker, J.K.; Dörner, W.; Mootz, H.D. Light-control of the ultra-fast Gp41-1 split intein with preserved stability of a genetically encoded photo-caged amino acid in bacterial cells. Chem. Commun. 2019, 55, 1287–1290. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Mahesh, M.; Elsässer, S.J.; Hancock, S.M.; Uttamapinant, C.; Chin, J.W. Genetic encoding of photocaged cysteine allows photoactivation of TEV protease in live mammalian cells. J. Am. Chem. Soc. 2014, 136, 2240–2243. [Google Scholar] [CrossRef]
- Ankenbruck, N.; Courtney, T.; Naro, Y.; Deiters, A. Optochemical Control of Biological Processes in Cells and Animals. Angew. Chem. Int. Ed. Engl. 2018, 57, 2768–2798. [Google Scholar] [CrossRef]
- Courtney, T.; Deiters, A. Recent advances in the optical control of protein function through genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 99–107. [Google Scholar] [CrossRef]
- Zhang, Z.; Smith, B.A.C.; Wang, L.; Brock, A.; Cho, C.; Schultz, P.G. A new strategy for the site-specific modification of proteins in vivo. Biochemistry 2003, 42, 6735–6746. [Google Scholar] [CrossRef]
- Hamano-Takaku, F.; Iwama, T.; Saito-Yano, S.; Takaku, K.; Monden, Y.; Kitabatake, M.; Soll, D.; Nishimura, S. A mutant Escherichia coli tyrosyl-tRNA synthetase utilizes the unnatural amino acid azatyrosine more efficiently than tyrosine. J. Biol. Chem. 2000, 275, 40324–40328. [Google Scholar] [CrossRef]
- Bloom, J.D.; Labthavikul, S.T.; Otey, C.R.; Arnold, F.H. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. USA 2006, 103, 5869–5874. [Google Scholar] [CrossRef] [Green Version]
- Kavran, J.M.; Gundllapalli, S.; O’Donoghue, P.; Englert, M.; Söll, D.; Steitz, T.A. Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. USA 2007, 104, 11268–11273. [Google Scholar] [CrossRef]
- Mukai, T.; Hoshi, H.; Ohtake, K.; Takahashi, M.; Yamaguchi, A.; Hayashi, A.; Yokoyama, S.; Sakamoto, K. Highly reproductive Escherichia coli cells with no specific assignment to the UAG codon. Sci. Rep. 2015, 5, 9699. [Google Scholar] [CrossRef] [Green Version]
- Amiram, M.; Haimovich, A.D.; Fan, C.; Wang, Y.; Aerni, H.-R.; Ntai, I.; Moonan, D.W.; Ma, N.J.; Rovner, A.J.; Hong, S.H.; et al. Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol. 2015, 33, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Renfrew, P.D.; Choi, E.J.; Bonneau, R.; Kuhlman, B. Incorporation of noncanonical amino acids into Rosetta and use in computational protein-peptide interface design. PLoS ONE 2012, 7, e32637. [Google Scholar] [CrossRef]
- Wang, L. Engineering the Genetic Code in Cells and Animals: Biological Considerations and Impacts. Acc. Chem. Res. 2017, 50, 2767–2775. [Google Scholar] [CrossRef]
- Guo, L.-T.; Wang, Y.-S.; Nakamura, A.; Eiler, D.; Kavran, J.M.; Wong, M.; Kiessling, L.L.; Steitz, T.A.; O’Donoghue, P.; Söll, D. Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 16724–16729. [Google Scholar] [CrossRef]
- Bryson, D.I.; Fan, C.; Guo, L.-T.; Miller, C.; Söll, D.; Liu, D.R. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol. 2017, 13, 1253–1260. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Zeng, Y.; Wang, W.W.; Qiao, Y.; Kurra, Y.; Liu, W.R. Evolving the N-Terminal Domain of Pyrrolysyl-tRNA Synthetase for Improved Incorporation of Noncanonical Amino Acids. Chembiochem 2018, 19, 26–30. [Google Scholar] [CrossRef]
- Gan, R.; Perez, J.G.; Carlson, E.D.; Ntai, I.; Isaacs, F.J.; Kelleher, N.L.; Jewett, M.C. Translation system engineering in Escherichia coli enhances non-canonical amino acid incorporation into proteins. Biotechnol. Bioeng. 2017, 114, 1074–1086. [Google Scholar] [CrossRef]
- Wong, J.T. Membership mutation of the genetic code: Loss of fitness by tryptophan. Proc. Natl. Acad. Sci. USA 1983, 80, 6303–6306. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Skillman, A.G.; Warren, G.L.; Ellingson, B.A.; Stahl, M.T. Conformer generation with OMEGA: Algorithm and validation using high quality structures from the Protein Databank and Cambridge Structural Database. J. Chem. Inf. Model. 2010, 50, 572–584. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Leaver-Fay, A.; Corn, J.E.; Strauch, E.-M.; Khare, S.D.; Koga, N.; Ashworth, J.; Murphy, P.; Richter, F.; Lemmon, G.; et al. RosettaScripts: A scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE 2011, 6, e20161. [Google Scholar] [CrossRef]
- Richter, F. Custom python script. Available online: https://github.com/flosopher/floscripts/blob/master/genutils/SequenceProfile.py (accessed on 2 April 2019).
- Young, T.S.; Ahmad, I.; Yin, J.A.; Schultz, P.G. An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 2010, 395, 361–374. [Google Scholar] [CrossRef]
- Budisa, N.; Steipe, B.; Demange, P.; Eckerskorn, C.; Kellermann, J.; Huber, R. High-level biosynthetic substitution of methionine in proteins by its analogs 2-aminohexanoic acid, selenomethionine, telluromethionine and ethionine in Escherichia coli. Eur. J. Biochem. 1995, 230, 788–796. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef]
- Pédelacq, J.-D.; Cabantous, S.; Tran, T.; Terwilliger, T.C.; Waldo, G.S. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 2006, 24, 79–88. [Google Scholar] [CrossRef]
- Pace, C.N.; Scholtz, J.M. Measuring the conformational stability of a protein. In Protein Structure, A Practical Approach; Creighton, T.E., Ed.; Oxford University Press: New York, NY, USA, 1997; pp. 299–321. [Google Scholar]
- Zaborske, J.; Pan, T. Genome-wide analysis of aminoacylation (charging) levels of tRNA using microarrays. J. Vis. Exp. 2010. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MjTyrRS Position | Library | ONBYRS-1 |
|---|---|---|
| Y32 | A, S | A |
| L65 | A, I, L, F, S | A |
| A67 | A, Q | A |
| L69 | L, K, G, W | L |
| H70 | A, N, S | N |
| G105 | G, A, Q | Q |
| F108 | F, L | F |
| Q109 | A, Q, Y | A |
| M154 | E, M, T, G | M |
| D158 | A, G, S | S |
| I159 | A, G, S, I | A |
| L162 | A, M | A |
| V164 | A, T, V | V |
| A167 | N, Q, G, S | S |
| H177 | A, H, Q, Y | H |
| A180 | A, Q | Q |
| V188 | N, Q, T, V | V |
| Protein Construct | E. coli Strain | O-Pair | ONBY | Yield (mg·L−1) |
|---|---|---|---|---|
| sfGFP WT | BL21(DE3) | - | - | 89 ± 16 |
| sfGFP(1TAG) | BL21(DE3) | MjTyrRS | - | 81 ± 23 |
| ONBYRS-1 | + | 93 ± 9 | ||
| MjONBYRS | + | 7 ± 2 | ||
| MbONBYRS | + | 21 ± 2 | ||
| sfGFP(5TAG) | C321.ΔA.exp(DE3) | MjTyrRS | - | 89 ± 12 |
| ONBYRS-1 | + | 13 ± 2 | ||
| MjONBYRS | + | n.d. | ||
| MbONBYRS | + | 5 ± 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baumann, T.; Hauf, M.; Richter, F.; Albers, S.; Möglich, A.; Ignatova, Z.; Budisa, N. Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine. Int. J. Mol. Sci. 2019, 20, 2343. https://doi.org/10.3390/ijms20092343
Baumann T, Hauf M, Richter F, Albers S, Möglich A, Ignatova Z, Budisa N. Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine. International Journal of Molecular Sciences. 2019; 20(9):2343. https://doi.org/10.3390/ijms20092343
Chicago/Turabian StyleBaumann, Tobias, Matthias Hauf, Florian Richter, Suki Albers, Andreas Möglich, Zoya Ignatova, and Nediljko Budisa. 2019. "Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine" International Journal of Molecular Sciences 20, no. 9: 2343. https://doi.org/10.3390/ijms20092343
APA StyleBaumann, T., Hauf, M., Richter, F., Albers, S., Möglich, A., Ignatova, Z., & Budisa, N. (2019). Computational Aminoacyl-tRNA Synthetase Library Design for Photocaged Tyrosine. International Journal of Molecular Sciences, 20(9), 2343. https://doi.org/10.3390/ijms20092343