In-Cell Synthesis of Bioorthogonal Alkene Tag S-Allyl-Homocysteine and Its Coupling with Reprogrammed Translation



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
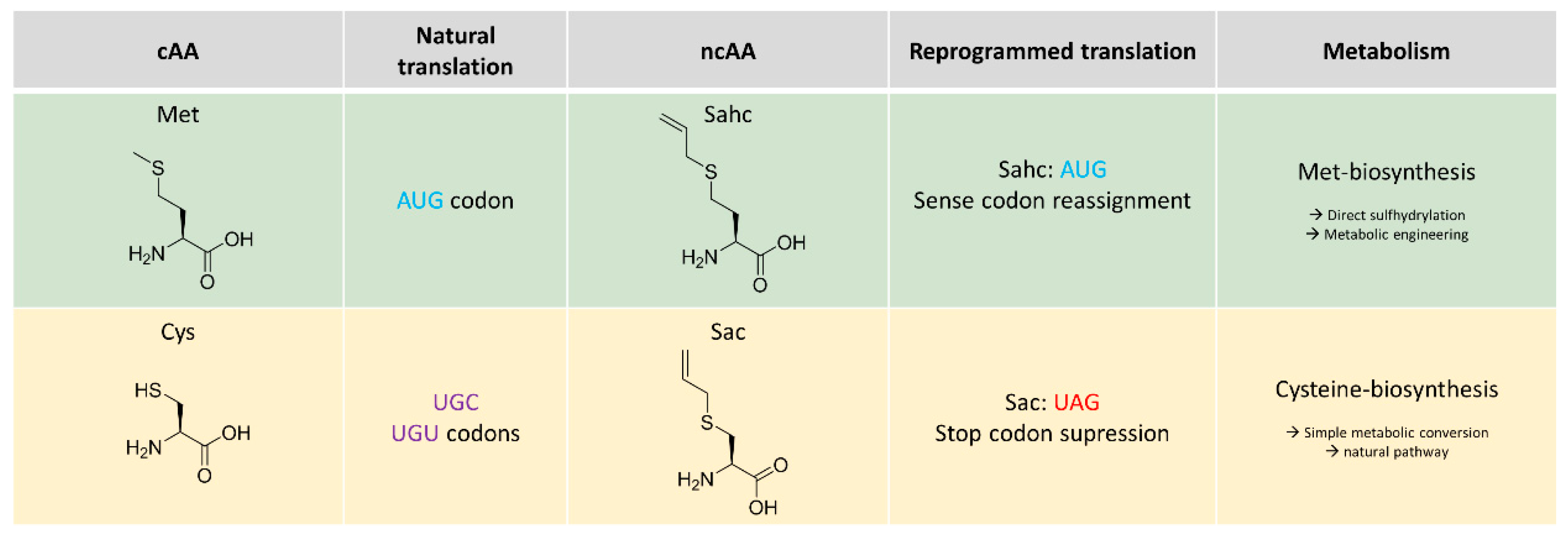
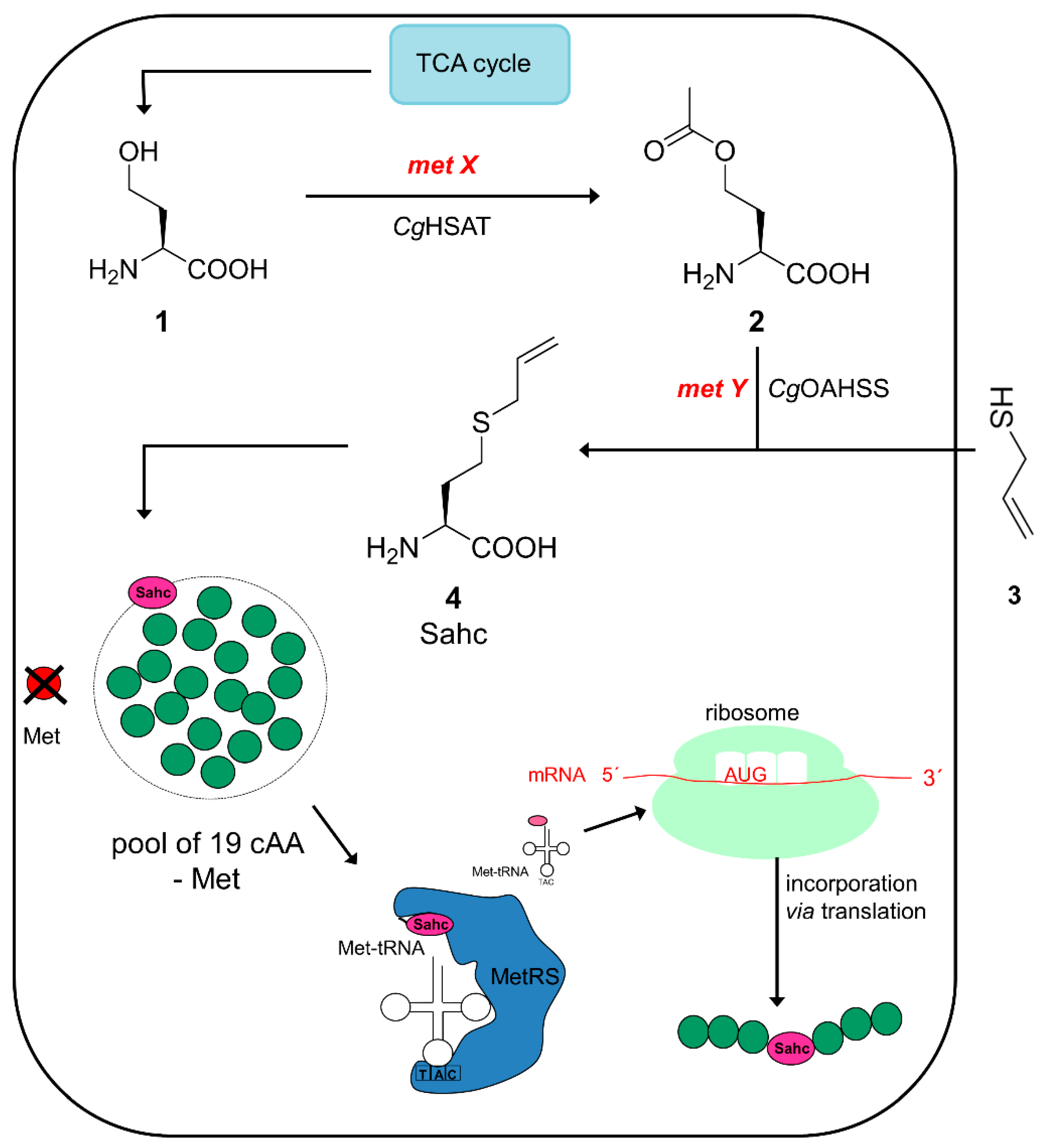
2.1. Metabolic Configuration of E. coli for In Situ Production of Sahc
2.2. Expression Experiments and Analytics of Sahc-Incorporation
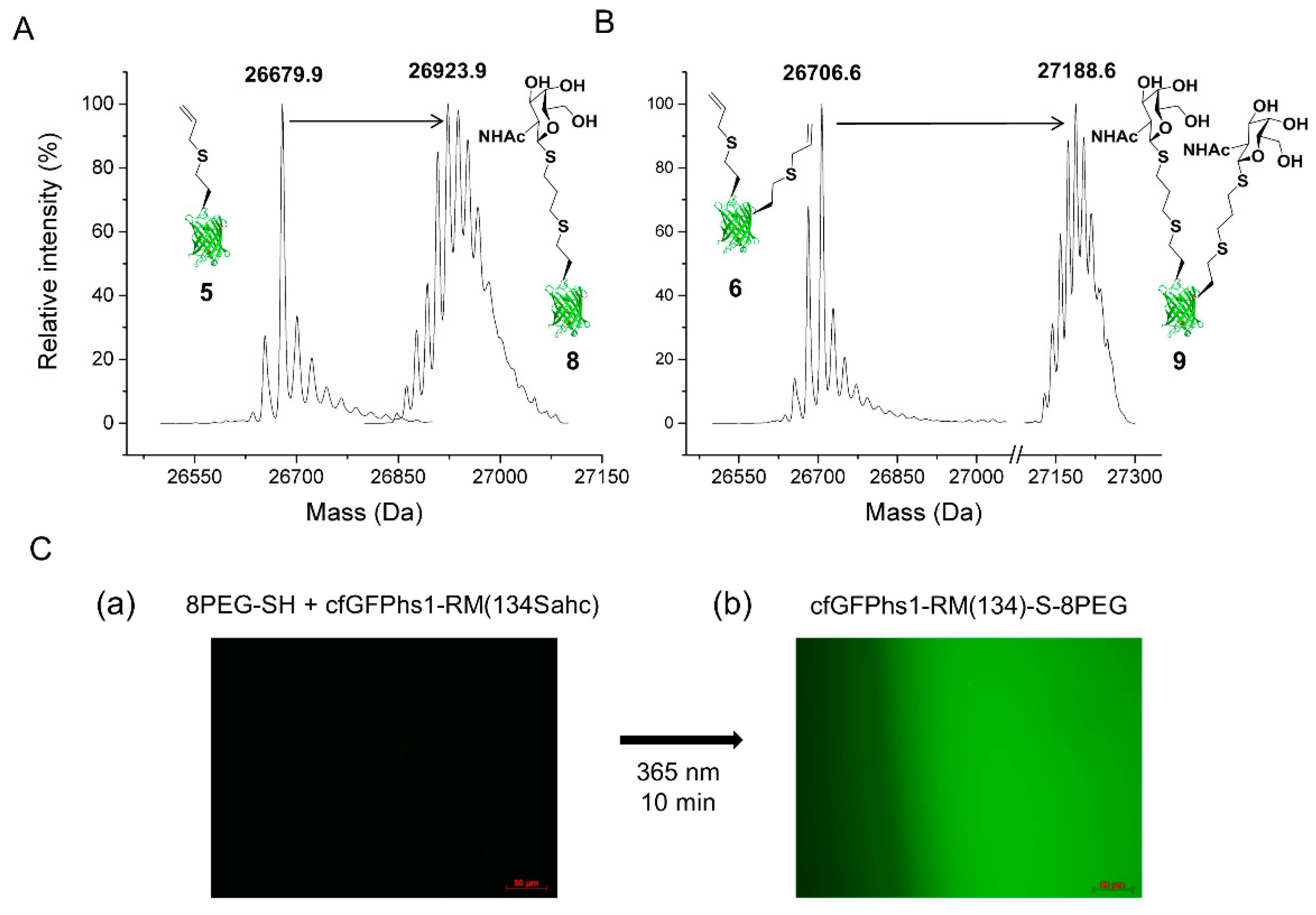
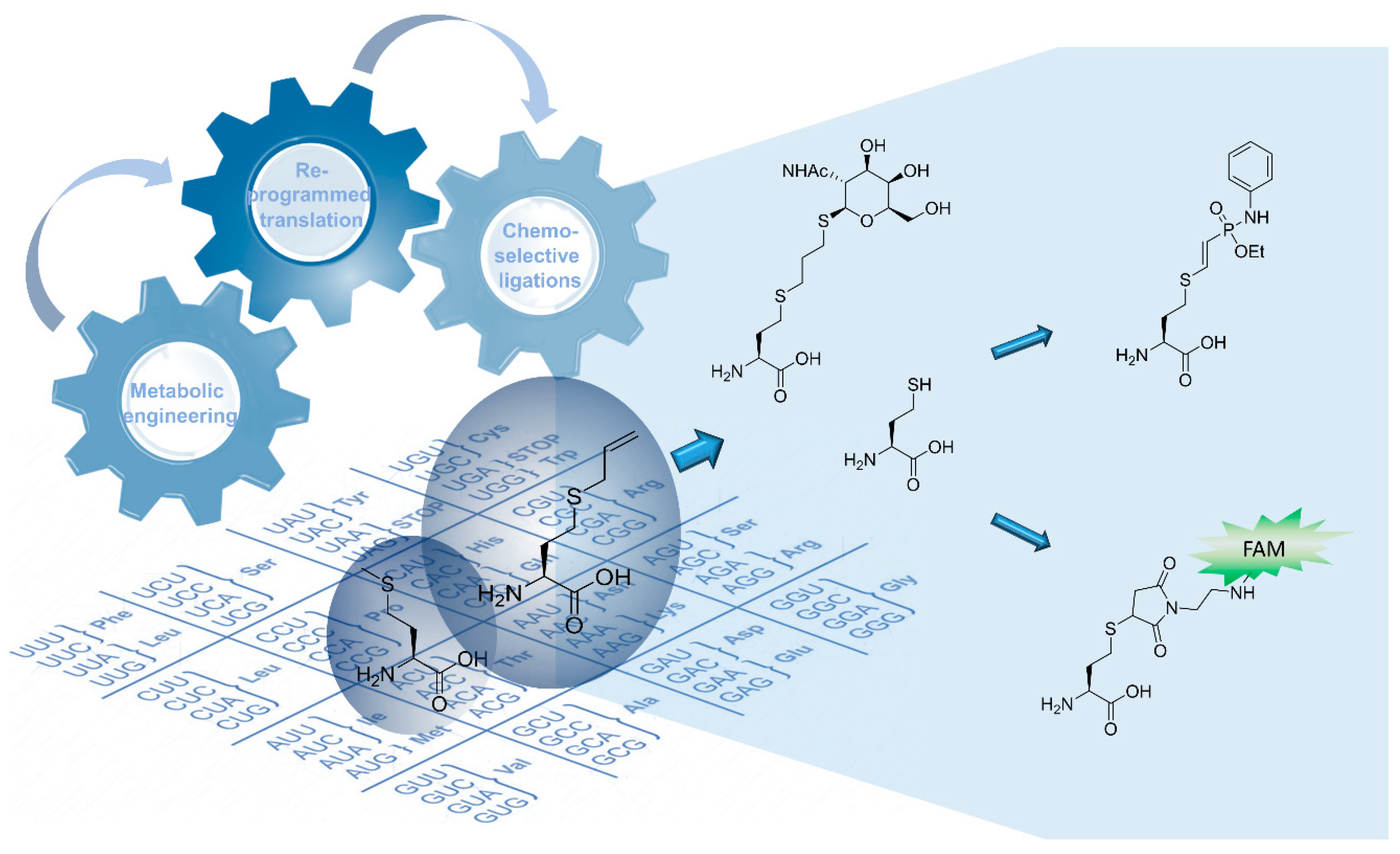
2.2.1. Thiol-Ene Conjugation Experiments
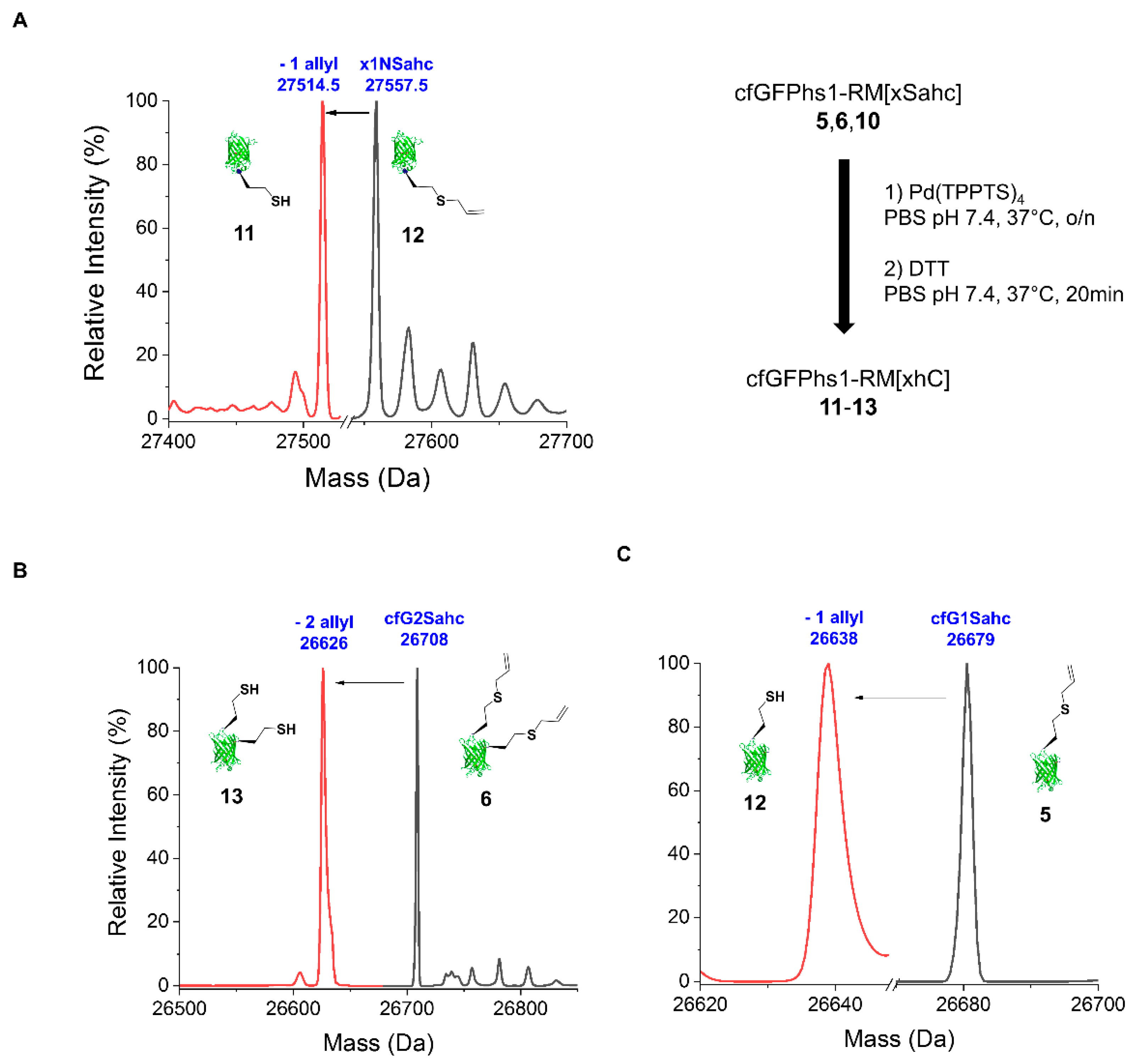
2.2.2. Deprotection of the Sahc Allyl Group
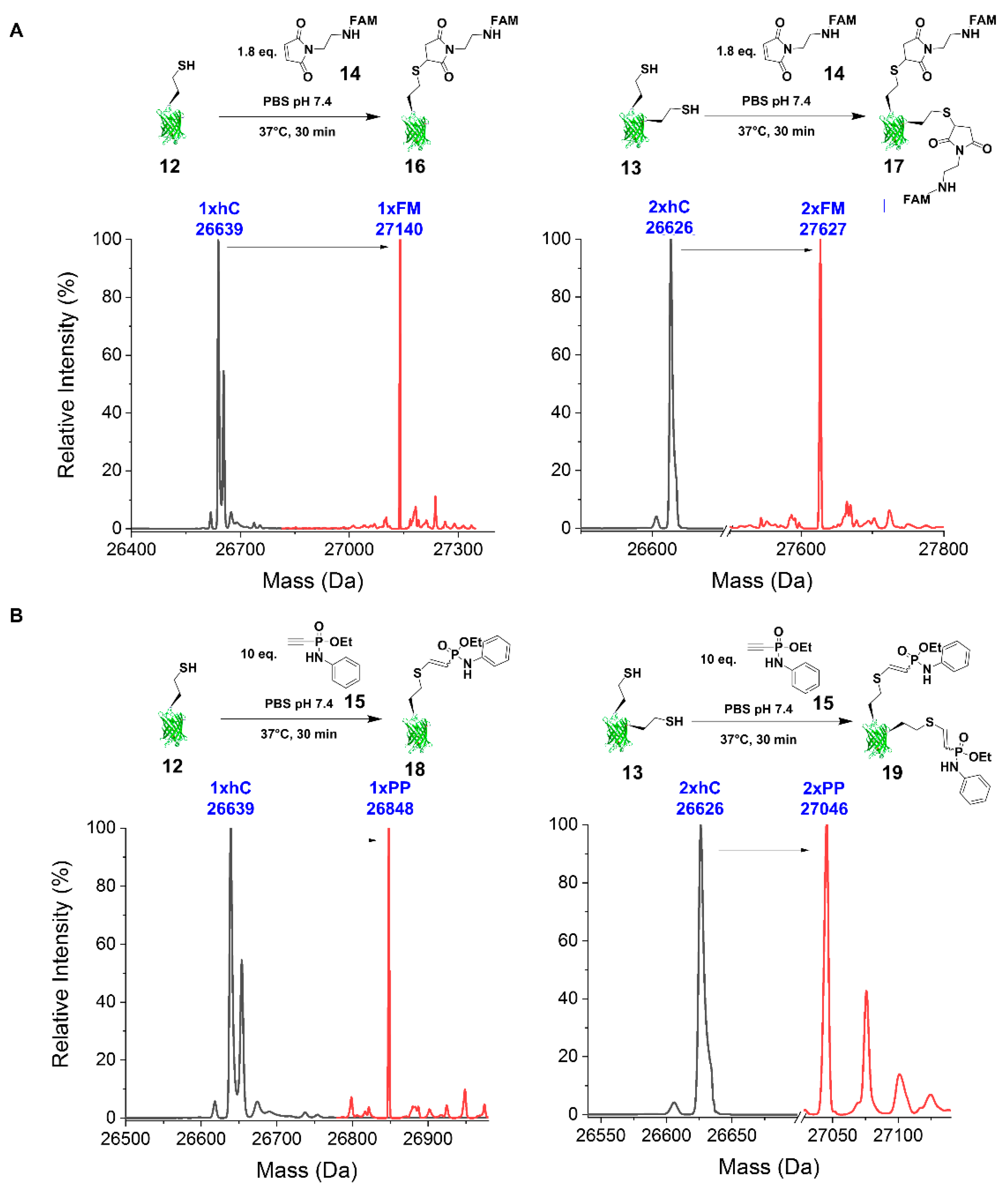
2.2.3. Reaction with FAM-Maleimide and Phosphonamidate Coupling
3. Discussion
3.1. Attributes and Perspectives of Sahc as Handle for Selective Protein Conjugation
3.2. Challenges and Possible Solutions to Intracellular Sahc Production
4. Materials and Methods
4.1. Media and Fermentation Procedure
4.2. Strains and Plasmids Used
4.3. Model Protein Cysteine-Free GFPhs1-RM (cfGFPhs1-RM): Generation and Mutagenesis
4.4. Expression and Purification of cfGFPhs1-RM(1Sahc), cfGFPhs1-RM(134Sahc) and cfGFPhs1-RM(134Sahc:143Sahc)
4.5. Mass Spectrometry
4.6. Deprotection Reactions
4.7. Conjugation Reactions
4.7.1. Thiol-Ene Conjugations
4.7.2. 8PEG-SH Gel Thiol-Ene Reaction—Immobilisation of cfGFPhs1-RM(134Sahc) on Hydrogels
4.7.3. FAM-Maleimide
4.7.4. Phenyl Phosphonamidate
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aha | azidohomoalanine |
| cAA | canonical amino acid |
| Cg | Corynobacterium glutamicum |
| Cys | cysteine |
| cfGFP | cysteine-free green fluorescent protein |
| E. coli | Escherichia coli |
| GalNAc | galactosamineacetate |
| HCys | l-homocysteine |
| Met | l-methionine |
| MetRS | methionyl-tRNA synthetase |
| ncAA | non-canonical amino acid |
| Oahs | O-acetyl-l-homoserine |
| Sac | S-allyl-l-cysteine |
| Sahc | S-allyl-l-homocysteine |
| SPI | Selective Pressure Incorporation |
Appendix A

References
- Schindeldecker, M.; Moosmann, B. Protein-borne methionine residues as structural antioxidants in mitochondria. Amino Acids 2015, 47, 1421–1432. [Google Scholar] [CrossRef]
- Wolschner, C.; Giese, A.; Kretzschmar, H.A.; Huber, R.; Moroder, L.; Budisa, N. Design of anti- and pro-aggregation variants to assess the effects of methionine oxidation in human prion protein. Proc. Natl. Acad. Sci. USA 2009, 106, 7756–7761. [Google Scholar] [CrossRef]
- Gilles, A.M.; Marlière, P.; Rose, T.; Sarfati, R.; Longin, R.; Meier, A.; Fermandjian, S.; Monnot, M.; Cohen, G.N.; Bârzu, O. Conservative replacement of methionine by norleucine in Escherichia coli adenylate kinase. J. Biol. Chem. 1988, 263, 8204–8209. [Google Scholar]
- Cowie, D.B.; Cohen, G.N. Biosynthesis by Escherichia coli of active altered proteins containing selenium instead of sulfur. Biochim. Biophys. Acta 1957, 26, 252–261. [Google Scholar] [CrossRef]
- Besse, D.; Budisa, N.; Karnbrock, W.; Minks, C.; Musiol, H.J.; Pegoraro, S.; Siedler, F.; Weyher, E.; Moroder, L. Chalcogen-analogs of amino acids. Their use in X-ray crystallographic and folding studies of peptides and proteins. Biol. Chem. 1997, 378, 211–218. [Google Scholar]
- De Simone, A.; Acevedo-Rocha, C.G.; Hoesl, M.G.; Budisa, N. Towards Reassignment of the Methionine Codon AUG to Two Different Noncanonical Amino Acids in Bacterial Translation. Croat. Chem. Acta 2016, 89, 243–253. [Google Scholar] [CrossRef]
- Yoshida, A.; Yamasaki, M. Studies on the mechanism of protein synthesis; incorporation of ethionine into alpha-amylase of Bacillus subtilis. Biochim. Biophys. Acta 1959, 34, 158–165. [Google Scholar] [CrossRef]
- Tang, Y.; Tirrell, D.A. Attenuation of the editing activity of the Escherichia coli leucyl-tRNA synthetase allows incorporation of novel amino acids into proteins in vivo. Biochemistry 2002, 41, 10635–10645. [Google Scholar] [CrossRef] [PubMed]
- Kiick, K.L.; Saxon, E.; Tirrell, D.A.; Bertozzi, C.R. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. USA 2002, 99, 19–24. [Google Scholar] [CrossRef]
- Bhushan, B.; Lin, Y.A.; Bak, M.; Phanumartwiwath, A.; Yang, N.; Bilyard, M.K.; Tanaka, T.; Hudson, K.L.; Lercher, L.; Stegmann, M.; et al. Genetic Incorporation of Olefin Cross-Metathesis Reaction Tags for Protein Modification. J. Am. Chem. Soc. 2018, 140, 14599–14603. [Google Scholar] [CrossRef]
- Truong, F. Expanding Protein Sequence Space through Incorporation of Non-Canonical Amino Acids. Ph.D. Thesis, California Institute of Technology, Pasedana, CA, USA, 2013. [Google Scholar]
- Law, B.J.C.; Struck, A.-W.; Bennett, M.R.; Wilkinson, B.; Micklefield, J. Site-specific bioalkylation of rapamycin by the RapM 16-O-methyltransferase. Chem. Sci. 2015, 6, 2885–2892. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zheng, W.; Luo, M. A sensitive mass spectrum assay to characterize engineered methionine adenosyltransferases with S-alkyl methionine analogues as substrates. Anal. Biochem. 2014, 450, 11–19. [Google Scholar] [CrossRef]
- Wang, F.; Singh, S.; Zhang, J.; Huber, T.D.; Helmich, K.E.; Sunkara, M.; Hurley, K.A.; Goff, R.D.; Bingman, C.A.; Morris, A.J.; et al. Understanding molecular recognition of promiscuity of thermophilic methionine adenosyltransferase sMAT from Sulfolobus solfataricus. FEBS J. 2014, 281, 4224–4239. [Google Scholar] [CrossRef]
- Singh, S.; Zhang, J.; Huber, T.D.; Sunkara, M.; Hurley, K.; Goff, R.D.; Wang, G.; Zhang, W.; Liu, C.; Rohr, J.; et al. Facile chemoenzymatic strategies for the synthesis and utilization of S-adenosyl-L-methionine analogues. Angew. Chem. Int. Ed. Engl. 2014, 53, 3965–3969. [Google Scholar] [CrossRef] [PubMed]
- Meinnel, T.; Mechulam, Y.; Blanquet, S. Methionine as translation start signal: A review of the enzymes of the pathway in Escherichia coli. Biochimie 1993, 75, 1061–1075. [Google Scholar] [CrossRef]
- Hatono, S.; Jimenez, A.; Wargovich, M.J. Chemopreventive effect of S-allylcysteine and its relationship to the detoxification enzyme glutathione S-transferase. Carcinogenesis 1996, 17, 1041–1044. [Google Scholar] [CrossRef] [PubMed]
- Borlinghaus, J.; Albrecht, F.; Gruhlke, M.; Nwachukwu, I.; Slusarenko, A. Allicin: Chemistry and Biological Properties. Molecules 2014, 19, 12591–12618. [Google Scholar] [CrossRef]
- Weiss, N.; Ide, N.; Abahji, T.; Nill, L.; Keller, C.; Hoffmann, U. Aged Garlic Extract Improves Homocysteine-Induced Endothelial Dysfunction in Macro- and Microcirculation. J. Nutr. 2006, 136, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.M.; Johnson, C.A.; Watanabe, R. Preparation of S-allyl-DL-homocysteine and related compounds and tests of their growth effects in rats. J. Biol. Chem. 1955, 212, 49–57. [Google Scholar]
- Völler, J.-S.; Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr. Opin. Biotechnol. 2017, 48, 1–7. [Google Scholar] [CrossRef]
- Exner, M.P.; Kuenzl, T.; To, T.M.T.; Ouyang, Z.; Schwagerus, S.; Hoesl, M.G.; Hackenberger, C.P.R.; Lensen, M.C.; Panke, S.; Budisa, N. Design of S-Allylcysteine in Situ Production and Incorporation Based on a Novel Pyrrolysyl-tRNA Synthetase Variant. ChemBioChem 2017, 18, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N.; Pipitone, O.; Siwanowicz, I.; Rubini, M.; Pal, P.; Holak, T.; Gelmi, M. Efforts towards the Design of “Teflon” Proteins:In vivo Translation with Trifluorinated Leucine and Methionine Analogues. Chem. Biodivers. 2004, 1, 1465–1475. [Google Scholar] [CrossRef]
- Yoo, T.H.; Tirrell, D.A. High-Throughput Screening for Methionyl-tRNA Synthetases That Enable Residue-Specific Incorporation of Noncanonical Amino Acids into Recombinant Proteins in Bacterial Cells. Angew. Chem. Int. Ed. Engl. 2007, 46, 5340–5343. [Google Scholar] [CrossRef] [PubMed]
- Montclare, J.K.; Tirrell, D.A. Evolving Proteins of Novel Composition. Angew. Chem. Int. Ed. Engl. 2006, 45, 4518–4521. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.H.; Link, A.J.; Tirrell, D.A. Evolution of a fluorinated green fluorescent protein. Proc. Natl. Acad. Sci. USA 2007, 104, 13887–13890. [Google Scholar] [CrossRef]
- Nagasundarapandian, S.; Merkel, L.; Budisa, N.; Govindan, R.; Ayyadurai, N.; Sriram, S.; Yun, H.; Lee, S.-G. Engineering Protein Sequence Composition for Folding Robustness Renders Efficient Noncanonical Amino acid Incorporations. ChemBioChem 2010, 11, 2521–2524. [Google Scholar] [CrossRef]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P.R. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed. Engl. 2015, 54, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Völler, J.-S.; Koksch, B.; Acevedo-Rocha, C.G.; Kubyshkin, V.; Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew. Chem. Int. Ed. Engl. 2017, 56, 9680–9703. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y. Metabolic Engineering of O-acetyl-L-homoserine Sulfhydrylase and Met-Biosynthetic Pathway in Escherichia coli. Ph.D. Thesis, Technische Universität Berlin, Berlin, Germany, 2016. [Google Scholar]
- Ma, Y.; Di Salvo, M.L.; Budisa, N. Self-Directed in Cell Production of Methionine Analogue Azidohomoalanine by Synthetic Metabolism and Its Incorporation into Model Proteins. Methods. Mol. Biol. 2018, 1728, 127–135. [Google Scholar]
- Di Salvo, M.L.; Budisa, N.; Contestabile, R. PLP-dependent Enzymes: A Powerful Tool for Metabolic Synthesis of Non-canonical Amino Acids. In Proceedings of the Beilstein Bozen Symposium on Molecular Engineering and Control, Prien, Bavaria, Germany, 14–18 May 2012; Hicks, M.G., Kettner, C., Eds.; pp. 27–66. [Google Scholar]
- Budisa, N. Engineering the Genetic Code; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Baden-Wuerttemberg, Germany, 2005; ISBN 9783527607181. [Google Scholar]
- Ma, Y.; Biava, H.; Contestabile, R.; Budisa, N.; Di Salvo, M. Coupling Bioorthogonal Chemistries with Artificial Metabolism: Intracellular Biosynthesis of Azidohomoalanine and Its Incorporation into Recombinant Proteins. Molecules 2014, 19, 1004–1022. [Google Scholar] [CrossRef]
- Ferla, M.P.; Patrick, W.M. Bacterial methionine biosynthesis. Microbiology 2014, 160, 1571–1584. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Al-Shameri, A.; Schipp, C.J.; Contestabile, R.; Budisa, N.; Di Salvo, M.L. Metabolically reconfigured Escherichia coli with self-directed in-cell production of synthetic amino acids as substrates for reprogrammed protein translation. 2019; Manuscript in preparation. [Google Scholar]
- Greene, R.C. Biosynthesis of Methionine. In Escherichia coli and Salmonella typhimurium, Cellular and Molecular Biology; Neidhardt, F., Ed.; ASM Press: Washington, DC, USA, 1996; pp. 542–560. [Google Scholar]
- Wiltschi, B.; Merkel, L.; Budisa, N. Fine tuning the N-terminal residue excision with methionine analogues. ChemBioChem 2009, 10, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Yesildag, C.; Ouyang, Z.; Zhang, Z.; Lensen, M.C. Micro-Patterning of PEG-Based Hydrogels With Gold Nanoparticles Using a Reactive Micro-Contact-Printing Approach. Front. Chem. 2019, 6, 667. [Google Scholar] [CrossRef] [PubMed]
- Torres-Kolbus, J.; Chou, C.; Liu, J.; Deiters, A. Synthesis of Non-linear Protein Dimers through a Genetically Encoded Thiol-ene Reaction. PLoS ONE 2014, 9, e105467. [Google Scholar] [CrossRef]
- Kharkar, P.M.; Rehmann, M.S.; Skeens, K.M.; Maverakis, E.; Kloxin, A.M. Thiol-ene click hydrogels for therapeutic delivery. ACS Biomater. Sci. Eng. 2016, 2, 165–179. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Ivarez, M.; Alberiicio, F. Amino acid-protecting groups. Chem. Rev. 2009, 109, 2455–2504. [Google Scholar] [CrossRef]
- Li, J.; Yu, J.; Zhao, J.; Wang, J.; Zheng, S.; Lin, S.; Chen, L.; Yang, M.; Jia, S.; Zhang, X.; et al. Palladium-triggered deprotection chemistry for protein activation in living cells. Nat. Chem. 2014, 6, 352–361. [Google Scholar] [CrossRef]
- Heyrovský, M.; Vavřička, S. Electrochemical reactivity of homocysteine at mercury electrodes as compared with cysteine. Bioelectrochem. Bioenerg. 1999, 48, 43–51. [Google Scholar] [CrossRef]
- Kasper, M.-A.; Glanz, M.; Stengl, A.; Penkert, M.; Klenk, S.; Sauer, T.; Schumacher, D.; Helma, J.; Krause, E.; Cardoso, M.C.; et al. Cysteine-selective phosphonamidate electrophiles for modular protein bioconjugations. Angew. Chem. Int. Ed. Engl. 2019. [Google Scholar] [CrossRef] [PubMed]
- Siebertz, K.D.; Hackenberger, C.P.R. Chemoselective triazole-phosphonamidate conjugates suitable for photorelease. Chem. Commun. 2018, 54, 763–766. [Google Scholar] [CrossRef]
- Mark, S.S. (Ed.) Bioconjugation Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2011; Volume 751, ISBN 978-1-61779-150-5. [Google Scholar]
- Schumacher, D.; Hackenberger, C.P.R. More than add-on: Chemoselective reactions for the synthesis of functional peptides and proteins. Curr. Opin. Chem. Biol. 2014, 22, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N.; Karnbrock, W.; Steinbacher, S.; Humm, A.; Prade, L.; Neuefeind, T.; Moroder, L.; Huber, R. Bioincorporation of telluromethionine into proteins: A promising new approach for X-ray structure analysis of proteins. J. Mol. Biol. 1997, 270, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Schultz, P.G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [PubMed]
- McKay, C.S.; Finn, M.G. Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem. Biol. 2014, 21, 1075–1101. [Google Scholar] [CrossRef] [PubMed]
- Hoyle, C.E.; Bowman, C.N. Thiol-Ene Click Chemistry. Angew. Chem. Int. Ed. Engl. 2010, 49, 1540–1573. [Google Scholar] [CrossRef]
- Exner, M.; Köhling, S.; Rivollier, J.; Gosling, S.; Srivastava, P.; Palyancheva, Z.; Herdewijn, P.; Heck, M.-P.; Rademann, J.; Budisa, N. Incorporation of Amino Acids with Long-Chain Terminal Olefins into Proteins. Molecules 2016, 21, 287. [Google Scholar] [CrossRef]
- Teramoto, H.; Kojima, K. Incorporation of Methionine Analogues Into Bombyx mori Silk Fibroin for Click Modifications. Macromol. Biosci. 2015, 15, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, X.; Jiang, Y.; Li, Z. Influence of α-methylation in constructing stapled peptides with olefin metathesis. Tetrahedron 2014, 70, 7621–7626. [Google Scholar] [CrossRef]
- Köhling, S.; Exner, M.P.; Nojoumi, S.; Schiller, J.; Budisa, N.; Rademann, J. One-Pot Synthesis of Unprotected Anomeric Glycosyl Thiols in Water for Glycan Ligation Reactions with Highly Functionalized Sugars. Angew. Chem. Int. Ed. Engl. 2016, 55, 15510–15514. [Google Scholar] [CrossRef] [PubMed]
- Lowe, A.B. Thiol-ene “click” reactions and recent applications in polymer and materials synthesis. Polym. Chem. 2010, 1, 17–36. [Google Scholar] [CrossRef]
- Chen, Y.X.; Triola, G.; Waldmann, H. Bioorthogonal chemistry for site-specific labeling and surface immobilization of proteins. Acc. Chem. Res. 2011, 44, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Buhl, M.; Vonhören, B.; Ravoo, B.J. Immobilization of Enzymes via Microcontact Printing and Thiol–Ene Click Chemistry. Bioconjug. Chem. 2015, 26, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Fallacara, A.; Baldini, E.; Manfredini, S.; Vertuani, S. Hyaluronic Acid in the Third Millennium. Polymers (Basel) 2018, 10, 701. [Google Scholar] [CrossRef] [PubMed]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef]
- Sekowska, A.; Dénervaud, V.; Ashida, H.; Michoud, K.; Haas, D.; Yokota, A.; Danchin, A. Bacterial variations on the methionine salvage pathway. BMC Microbiol. 2004, 4, 9. [Google Scholar] [CrossRef]
- Krömer, J.O.; Wittmann, C.; Schröder, H.; Heinzle, E. Metabolic pathway analysis for rational design of L-methionine production by Escherichia coli and Corynebacterium glutamicum. Metab. Eng. 2006, 8, 353–369. [Google Scholar] [CrossRef]
- Weissbach, H.; Brot, N. Regulation of methionine synthesis in Escherichia coli. Mol. Microbiol. 1991, 5, 1593–1597. [Google Scholar] [CrossRef] [PubMed]
- Su, C.-H.; Greene, R.C. Regulation of Methionine Biosynthesis in Escherichia coli: Mapping of the metJ Locus and Properties of a metJ+/metJ- Diploid. Proc. Natl. Acad. Sci. USA 1970, 68, 367–371. [Google Scholar] [CrossRef]
- Huang, J.-F.; Liu, Z.-Q.; Jin, L.-Q.; Tang, X.-L.; Shen, Z.-Y.; Yin, H.-H.; Zheng, Y.-G. Metabolic engineering of Escherichia coli for microbial production of L-methionine. Biotechnol. Bioeng. 2017, 114, 843–851. [Google Scholar] [CrossRef]
- Li, H.; Wang, B.S.; Li, Y.R.; Zhang, L.; Ding, Z.Y.; Gu, Z.H.; Shi, G.Y. Metabolic engineering of Escherichia coli W3110 for the production of l-methionine. J. Ind. Microbiol. Biotechnol. 2017, 44, 75–88. [Google Scholar] [CrossRef]
- Nielsen, J.; Keasling, J.D. Engineering Cellular Metabolism. Cell 2016, 164, 1185–1197. [Google Scholar] [CrossRef]
- Giese, C.; Lepthien, S.; Metzner, L.; Brandsch, M.; Budisa, N.; Lilie, H. Intracellular uptake and inhibitory activity of aromatic fluorinated amino acids in human breast cancer cells. ChemMedChem 2008, 3, 1449–1456. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Rocha, C.G.; Geiermann, A.-S.; Budisa, N.; Merkel, L. Design of protein congeners containing β-cyclopropylalanine. Mol. Biosyst. 2012, 8, 2719. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, M.G.; Oehm, S.; Durkin, P.; Darmon, E.; Peil, L.; Aerni, H.-R.; Rappsilber, J.; Rinehart, J.; Leach, D.; Söll, D.; et al. Chemical Evolution of a Bacterial Proteome. Angew. Chem. Int. Ed. Engl. 2015, 54, 10030–10034. [Google Scholar] [CrossRef]
- Budisa, N.; Steipe, B.; Demange, P.; Eckerskorn, C.; Kellermann, J.; Huber, R. High-level Biosynthetic Substitution of Methionine in Proteins by its Analogs 2-Aminohexanoic Acid, Selenomethionine, Telluromethionine and Ethionine in Escherichia coli. Eur. J. Biochem. 1995, 230, 788–796. [Google Scholar] [CrossRef]
- Wiltschi, B.; Wenger, W.; Nehring, S.; Budisa, N. Expanding the genetic code of Saccharomyces cerevisiae with methionine analogues. Yeast 2008, 25, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Lepthien, S.; Merkel, L.; Budisa, N. In vivo double and triple labeling of proteins using synthetic amino acids. Angew. Chem. Int. Ed. Engl. 2010, 49, 5446–5450. [Google Scholar] [CrossRef]
- Bohlke, N. Protein Engineering and Bioorthogonal Chemistry for Multivalent Scaffold Design Global Reassignment of Rare Codons in the Genetic Code of Escherichia coli. Ph.D. Thesis, Technische Universität Berlin, Berlin, Germany, 2014. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nojoumi, S.; Ma, Y.; Schwagerus, S.; Hackenberger, C.P.R.; Budisa, N. In-Cell Synthesis of Bioorthogonal Alkene Tag S-Allyl-Homocysteine and Its Coupling with Reprogrammed Translation. Int. J. Mol. Sci. 2019, 20, 2299. https://doi.org/10.3390/ijms20092299
Nojoumi S, Ma Y, Schwagerus S, Hackenberger CPR, Budisa N. In-Cell Synthesis of Bioorthogonal Alkene Tag S-Allyl-Homocysteine and Its Coupling with Reprogrammed Translation. International Journal of Molecular Sciences. 2019; 20(9):2299. https://doi.org/10.3390/ijms20092299
Chicago/Turabian StyleNojoumi, Saba, Ying Ma, Sergej Schwagerus, Christian P. R. Hackenberger, and Nediljko Budisa. 2019. "In-Cell Synthesis of Bioorthogonal Alkene Tag S-Allyl-Homocysteine and Its Coupling with Reprogrammed Translation" International Journal of Molecular Sciences 20, no. 9: 2299. https://doi.org/10.3390/ijms20092299
APA StyleNojoumi, S., Ma, Y., Schwagerus, S., Hackenberger, C. P. R., & Budisa, N. (2019). In-Cell Synthesis of Bioorthogonal Alkene Tag S-Allyl-Homocysteine and Its Coupling with Reprogrammed Translation. International Journal of Molecular Sciences, 20(9), 2299. https://doi.org/10.3390/ijms20092299