Human Liver Regeneration: An Etiology Dependent Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Regeneration after Resection
3. Liver Regeneration in the Diseased Liver
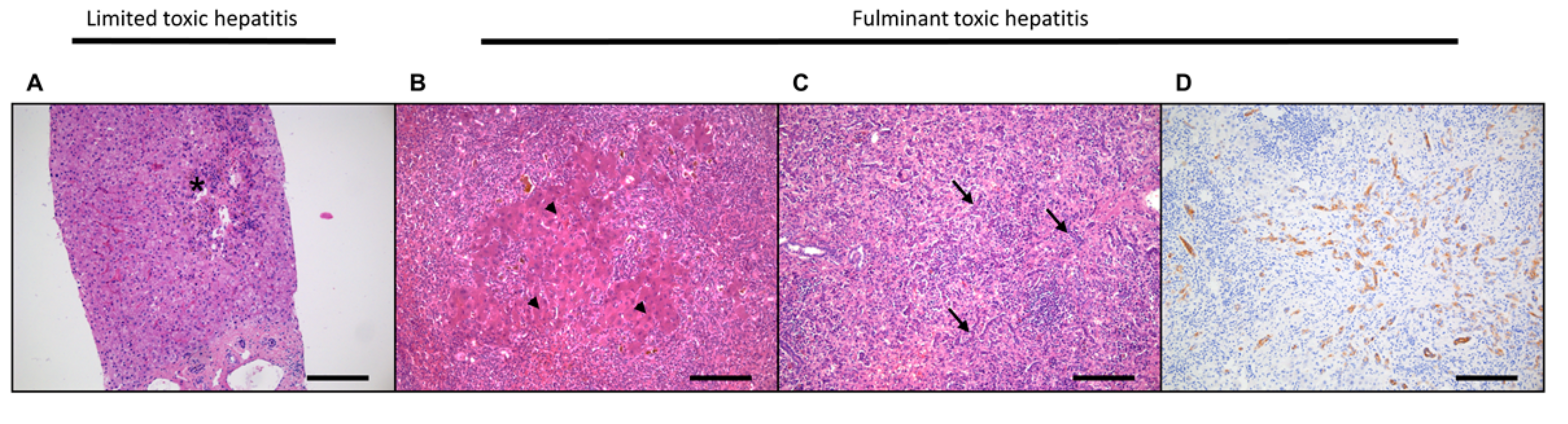
3.1. Acute Liver Damage and Regeneration
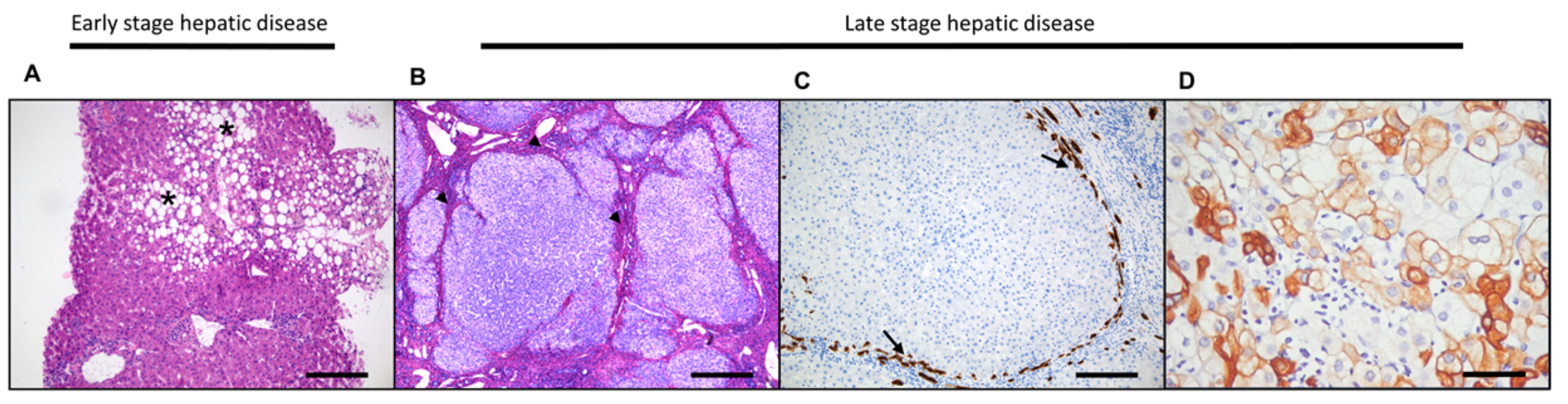
3.2. Hepatocellular Damage and Regeneration
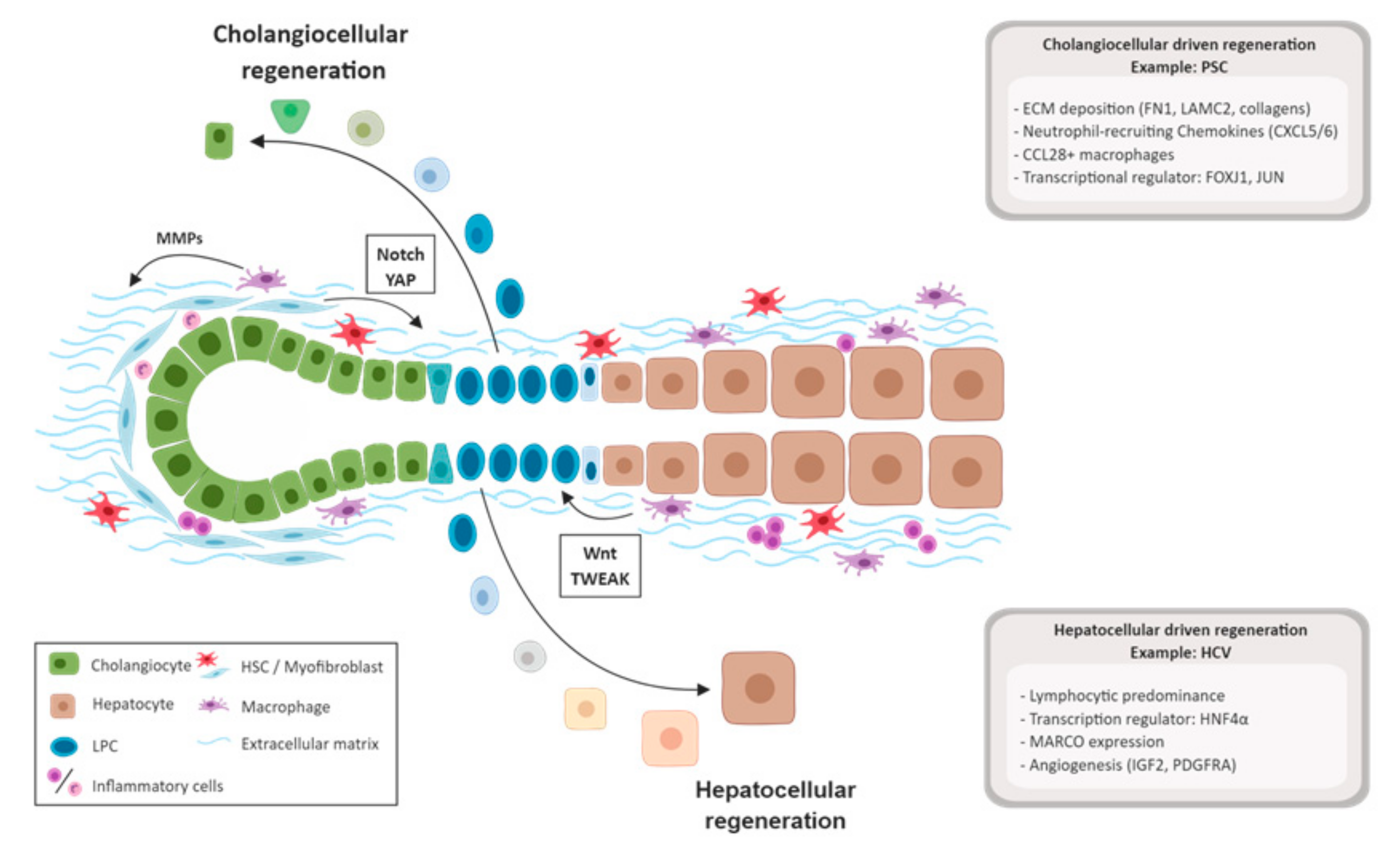
3.3. Biliary Damage and Regeneration
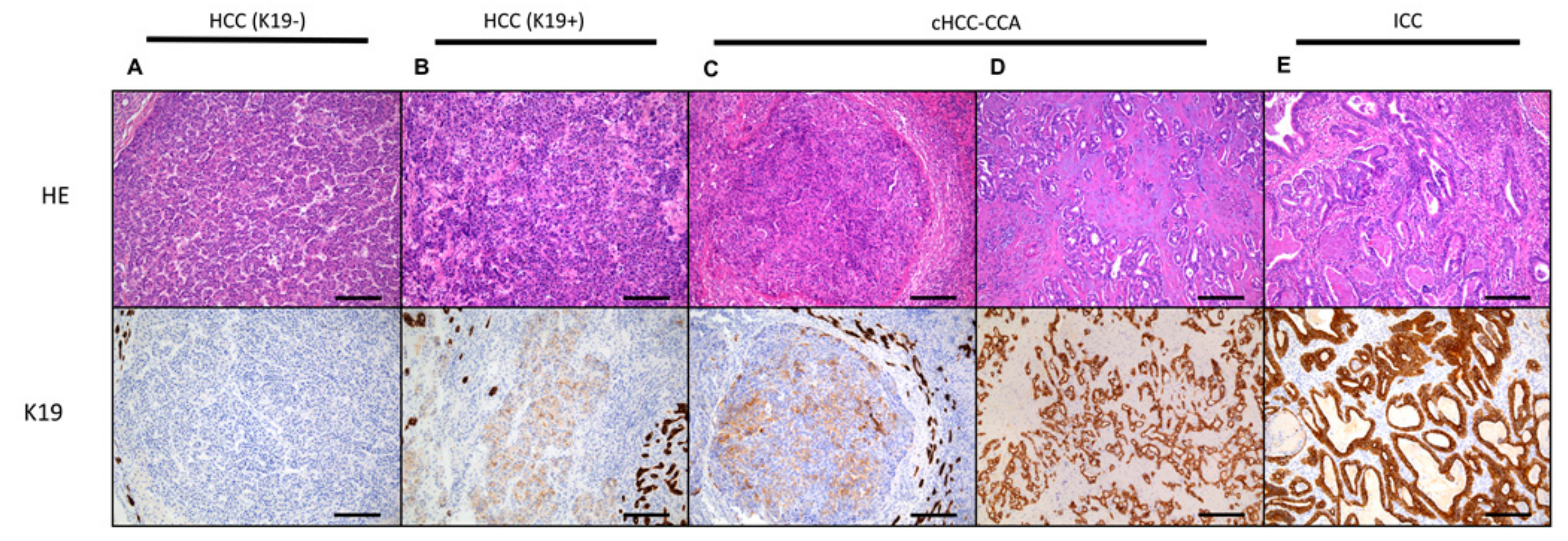
4. Carcinogenesis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| LPCs | Liver progenitor cells |
| LSECs | Liver sinusoidal endothelial cells |
| HSCs | Hepatic stellate cells |
| PLC | Primary liver cancer |
| HCC | Hepatocellular carcinoma |
| ICC | Intrahepatic cholangiocarcinoma |
| HNF4α | hepatocyte nuclear factor 4 alfa |
| TWEAK | TNF-related weak inducer of apoptosis |
| IGF2 | insulin-like growth factor 2 |
| APAP | N-acetyl-para-aminophenol |
| PDGFRA | Platelet-derived growth factor receptor A |
| FN1 | Fibronectin 1 |
| LAMC2 | Laminin subunit gamma-2 |
| CXCL5 | C-X-C motif chemokine ligand 5 |
| CXCL6 | C-X-C motif chemokine ligand 6 |
| FOXJ1 | Forkhead box protein J1 |
| JUN | Jun proto-oncogene |
| AIH | Autoimmune hepatitis |
| TGFβ1 | Transforming growth factor–β1 |
| CSF1 | Colony-stimulating factor |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| NAFLD | Non-alcoholic fatty liver disease |
| NAFL | Non-alcoholic fatty liver |
| NASH | Non-alcoholic steatohepatitis |
| MARCO | Macrophage receptor with collagenous structure |
| CCL28 | Chemokine (C-C motif) ligand 28 |
| PSC | Primary sclerosing cholangitis |
| PBC | primary biliary cholangitis |
| AMA | Antimitochondrial antibodies |
| ECM | Extracellular matrix |
| K7 | Kertin-7 |
| K19 | Keratin-19 |
| YAP | Yes-associated protein 1 |
| WWTR1 | WW domain-containing transcription regulator protein 1 |
| SOX9 | Express Sex-Determining Region Y-Box 9 |
| NCAM | Neural cell adhesion molecule |
| EpCAM | Epithelial cell adhesion molecule |
| PBG | Peribiliary gland |
| HGF | Hepatocyte growth factor |
| HSP70 | Beat shock protein 70 |
| TERT | Telomerase reverse transcriptase |
| cHCC-CCA | Combined hepatocellular-cholangiocarcinoma |
References
- Sutherland, F.; Harris, J. Claude couinaud: A passion for the liver. Arch. Surh. 2002, 137, 1305–1310. [Google Scholar] [CrossRef]
- Felmlee, D.J.; Grun, D.; Baumert, T.F. Zooming in on liver zonation. Hepatology 2017, 67, 784–787. [Google Scholar] [CrossRef]
- Roskams, T.A.; Theise, N.D.; Balabaud, C.; Bhagat, G.; Bhathal, P.S.; Bioulac-Sage, P.; Brunt, E.M.; Crawford, J.M.; Crosby, H.A.; Desmet, V.; et al. Nomenclature of the finer branches of the biliary tree: Canals, ductules, and ductular reactions in human livers. Hepatology 2004, 39, 1739–1745. [Google Scholar] [CrossRef]
- Theise, N.D.; Saxena, R.; Portmann, B.C.; Thung, S.N.; Yee, H.; Chiriboga, L.; Kumar, A.; Crawford, J.M. The canals of hering and hepatic stem cells in humans. Hepatology 1999, 30, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver regeneration. J. Cell Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology 2017, 65, 1384–1392. [Google Scholar] [CrossRef]
- Taub, R. Liver regeneration: From myth to mechanism. Nat. Rev. Mol. Cell Biol. 2004, 5, 836–847. [Google Scholar] [CrossRef]
- Miyaoka, Y.; Ebato, K.; Kato, H.; Arakawa, S.; Shimizu, S.; Miyajima, A. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr. Biol. 2012, 22, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Gilgenkrantz, H.; Collin de l’Hortet, A. Understanding liver regeneration: From mechanisms to regenerative medicine. Am. J. Pathol. 2018, 188, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Alkhalili, E.; Berber, E. Laparoscopic liver resection for malignancy: A review of the literature. World J. Gastroenterol. 2014, 20, 13599–13606. [Google Scholar] [CrossRef]
- Balzan, S.; Belghiti, J.; Farges, O.; Ogata, S.; Sauvanet, A.; Delefosse, D.; Durand, F. The “50-50 criteria” on postoperative day 5: An accurate predictor of liver failure and death after hepatectomy. Ann. Surg. 2005, 242, 824–829. [Google Scholar] [CrossRef] [PubMed]
- Engstrand, J.; Nilsson, H.; Stromberg, C.; Jonas, E.; Freedman, J. Colorectal cancer liver metastases—A population-based study on incidence, management and survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Abdalla, E.K.; Chun, Y.S.; Zorzi, D.; Madoff, D.C.; Wallace, M.J.; Curley, S.A.; Vauthey, J.N. Three hundred and one consecutive extended right hepatectomies: Evaluation of outcome based on systematic liver volumetry. Ann. Surh. 2009, 250, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Shimada, R.; Kubota, M.; Matsuyama, Y.; Nakayama, A.; Miyagawa, S.; Makuuchi, M.; Kawasaki, S. Preoperative portal vein embolization: An audit of 84 patients. Hepatology 1999, 29, 1099–1105. [Google Scholar] [CrossRef] [PubMed]
- Isfordink, C.J.; Samim, M.; Braat, M.; Almalki, A.M.; Hagendoorn, J.; Borel Rinkes, I.H.M.; Molenaar, I.Q. Portal vein ligation versus portal vein embolization for induction of hypertrophy of the future liver remnant: A systematic review and meta-analysis. Surh. Oncol 2017, 26, 257–267. [Google Scholar] [CrossRef]
- Goto, Y.; Nagino, M.; Nimura, Y. Doppler estimation of portal blood flow after percutaneous transhepatic portal vein embolization. Ann. Surh. 1998, 228, 209–213. [Google Scholar] [CrossRef]
- Liu, K.; Zhang, X.; Xu, W.; Chen, J.; Yu, J.; Gamble, J.R.; McCaughan, G.W. Targeting the vasculature in hepatocellular carcinoma treatment: Starving versus normalizing blood supply. Clin. Transl. Gastroenterol. 2017, 8, e98. [Google Scholar] [CrossRef]
- Tao, Y.; Wang, M.; Chen, E.; Tang, H. Liver regeneration: Analysis of the main relevant signaling molecules. Mediat. Inflamm. 2017, 2017, 4256352. [Google Scholar] [CrossRef]
- Kele, P.G.; van der Jagt, E.J.; Gouw, A.S.; Lisman, T.; Porte, R.J.; de Boer, M.T. The impact of hepatic steatosis on liver regeneration after partial hepatectomy. Liver Int. 2013, 33, 469–475. [Google Scholar] [CrossRef]
- Berg, C.L.; Gillespie, B.W.; Merion, R.M.; Brown, R.S., Jr.; Abecassis, M.M.; Trotter, J.F.; Fisher, R.A.; Freise, C.E.; Ghobrial, R.M.; Shaked, A.; et al. Improvement in survival associated with adult-to-adult living donor liver transplantation. Gastroenterology 2007, 133, 1806–1813. [Google Scholar] [CrossRef]
- Kawasaki, S.; Makuuchi, M.; Ishizone, S.; Matsunami, H.; Terada, M.; Kawarazaki, H. Liver regeneration in recipients and donors after transplantation. Lancet 1992, 339, 580–581. [Google Scholar] [CrossRef]
- Haga, J.; Shimazu, M.; Wakabayashi, G.; Tanabe, M.; Kawachi, S.; Fuchimoto, Y.; Hoshino, K.; Morikawa, Y.; Kitajima, M.; Kitagawa, Y. Liver regeneration in donors and adult recipients after living donor liver transplantation. Liver Transplant. 2008, 14, 1718–1724. [Google Scholar] [CrossRef]
- Bernal, W.; Wendon, J. Acute liver failure. N. Engl. J. Med. 2013, 369, 2525–2534. [Google Scholar] [CrossRef]
- Lefkowitch, J.H. The pathology of acute liver failure. Adv. Anat. Pathol. 2016, 23, 144–158. [Google Scholar] [CrossRef]
- Possamai, L.A.; Antoniades, C.G.; Anstee, Q.M.; Quaglia, A.; Vergani, D.; Thursz, M.; Wendon, J. Role of monocytes and macrophages in experimental and human acute liver failure. World J. Gastroenterol. 2010, 16, 1811–1819. [Google Scholar] [CrossRef]
- Samuel, D.; Ichai, P. Prognosis indicator in acute liver failure: Is there a place for cell death markers? J. Hepatol. 2010, 53, 593–595. [Google Scholar] [CrossRef][Green Version]
- Katoonizadeh, A.; Nevens, F.; Verslype, C.; Pirenne, J.; Roskams, T. Liver regeneration in acute severe liver impairment: A clinicopathological correlation study. Liver Int. 2006, 26, 1225–1233. [Google Scholar] [CrossRef]
- Libbrecht, L. Hepatic progenitor cells in human liver tumor development. World J. Gastroenterol. 2006, 12, 6261–6265. [Google Scholar] [CrossRef]
- Spee, B.; Carpino, G.; Schotanus, B.A.; Katoonizadeh, A.; Vander Borght, S.; Gaudio, E.; Roskams, T. Characterisation of the liver progenitor cell niche in liver diseases: Potential involvement of wnt and notch signalling. Gut 2010, 59, 247–257. [Google Scholar] [CrossRef]
- Bhushan, B.; Walesky, C.; Manley, M.; Gallagher, T.; Borude, P.; Edwards, G.; Monga, S.P.S.; Apte, U. Pro-regenerative signaling after acetaminophen-induced acute liver injury in mice identified using a novel incremental dose model. Am. J. Pathol. 2014, 184, 3013–3025. [Google Scholar] [CrossRef]
- Apte, U.; Singh, S.; Zeng, G.; Cieply, B.; Virji, M.A.; Wu, T.; Monga, S.P. Beta-catenin activation promotes liver regeneration after acetaminophen-induced injury. Am. J. Pathol. 2009, 175, 1056–1065. [Google Scholar] [CrossRef]
- Bird, T.G.; Muller, M.; Boulter, L.; Vincent, D.F.; Ridgway, R.A.; Lopez-Guadamillas, E.; Lu, W.Y.; Jamieson, T.; Govaere, O.; Campbell, A.D.; et al. Tgfbeta inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci. Transl. Med. 2018, 10, eaan1230. [Google Scholar] [CrossRef]
- Antoniades, C.G.; Quaglia, A.; Taams, L.S.; Mitry, R.R.; Hussain, M.; Abeles, R.; Possamai, L.A.; Bruce, M.; McPhail, M.; Starling, C.; et al. Source and characterization of hepatic macrophages in acetaminophen-induced acute liver failure in humans. Hepatology 2012, 56, 735–746. [Google Scholar] [CrossRef]
- Stutchfield, B.M.; Antoine, D.J.; Mackinnon, A.C.; Gow, D.J.; Bain, C.C.; Hawley, C.A.; Hughes, M.J.; Francis, B.; Wojtacha, D.; Man, T.Y.; et al. Csf1 restores innate immunity after liver injury in mice and serum levels indicate outcomes of patients with acute liver failure. Gastroenterology 2015, 149, 1896–1909. [Google Scholar] [CrossRef]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis c virus infection: An up-date of the distribution and circulation of hepatitis c virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef]
- Polaris Observatory Collaborators. Global prevalence, treatment, and prevention of hepatitis b virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar]
- Trivedi, P.J.; Hubscher, S.G.; Heneghan, M.; Gleeson, D.; Hirschfield, G.M. Grand round: Autoimmune hepatitis. J. Hepatol. 2019, 70, 773–784. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of nafld and nash: Trends, predictions, risk factors and prevention. Nat. Rev. Gastro. Hepat. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the american association for the study of liver diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Sakhuja, P. Pathology of alcoholic liver disease, can it be differentiated from nonalcoholic steatohepatitis? World J. Gastroenterol. 2014, 20, 16474–16479. [Google Scholar] [CrossRef]
- Font-Burgada, J.; Shalapour, S.; Ramaswamy, S.; Hsueh, B.; Rossell, D.; Umemura, A.; Taniguchi, K.; Nakagawa, H.; Valasek, M.A.; Ye, L.; et al. Hybrid periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015, 162, 766–779. [Google Scholar] [CrossRef]
- Lin, W.R.; Lim, S.N.; McDonald, S.A.; Graham, T.; Wright, V.L.; Peplow, C.L.; Humphries, A.; Kocher, H.M.; Wright, N.A.; Dhillon, A.P.; et al. The histogenesis of regenerative nodules in human liver cirrhosis. Hepatology 2010, 51, 1017–1026. [Google Scholar] [CrossRef]
- Van Haele, M.; Roskams, T. Hepatic progenitor cells: An update. Gastroenterol. Clin. North Am. 2017, 46, 409–420. [Google Scholar] [CrossRef]
- Lu, W.Y.; Bird, T.G.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 2015, 17, 971–983. [Google Scholar] [CrossRef]
- Manco, R.; Leclercq, I.A.; Clerbaux, L.A. Liver regeneration: Different sub-populations of parenchymal cells at play choreographed by an injury-specific microenvironment. Int. J. Mol. Sci. 2018, 19, E4115. [Google Scholar] [CrossRef]
- Russell, J.O.; Lu, W.Y.; Okabe, H.; Abrams, M.; Oertel, M.; Poddar, M.; Singh, S.; Forbes, S.J.; Monga, S.P. Hepatocyte-specific beta-catenin deletion during severe liver injury provokes cholangiocytes to differentiate into hepatocytes. Hepatology 2019, 69, 742–759. [Google Scholar] [CrossRef]
- Raven, A.; Lu, W.Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwyer, B.J.; Thomson, J.P.; Meehan, R.R.; Bogorad, R.; Koteliansky, V.; et al. Cholangiocytes act as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350. [Google Scholar] [CrossRef]
- Yoon, S.M.; Gerasimidou, D.; Kuwahara, R.; Hytiroglou, P.; Yoo, J.E.; Park, Y.N.; Theise, N.D. Epithelial cell adhesion molecule (epcam) marks hepatocytes newly derived from stem/progenitor cells in humans. Hepatology 2011, 53, 964–973. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, X.; Li, W.P.; Feng, R.X.; Li, L.; Yi, G.R.; Zhang, X.N.; Yin, C.; Yu, H.Y.; Zhang, J.P.; et al. Chronic liver injury induces conversion of biliary epithelial cells into hepatocytes. Cell Stem Cell 2018, 23, 114–122. [Google Scholar] [CrossRef]
- Zhao, L.; Westerhoff, M.; Pai, R.K.; Choi, W.T.; Gao, Z.H.; Hart, J. Centrilobular ductular reaction correlates with fibrosis stage and fibrosis progression in non-alcoholic steatohepatitis. Mod. Pathol. 2018, 31, 150–159. [Google Scholar] [CrossRef]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef]
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.W.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol. 2003, 163, 1301–1311. [Google Scholar] [CrossRef]
- Prakoso, E.; Tirnitz-Parker, J.E.E.; Clouston, A.D.; Kayali, Z.; Lee, A.; Gan, E.K.; Ramm, G.A.; Kench, J.G.; Bowen, D.G.; Olynyk, J.K.; et al. Analysis of the intrahepatic ductular reaction and progenitor cell responses in hepatitis c virus recurrence after liver transplantation. Liver Transpl. 2014, 20, 1508–1519. [Google Scholar] [PubMed]
- Lanthier, N.; Rubbia-Brandt, L.; Lin-Marq, N.; Clement, S.; Frossard, J.L.; Goossens, N.; Hadengue, A.; Spahr, L. Hepatic cell proliferation plays a pivotal role in the prognosis of alcoholic hepatitis. J. Hepatol. 2015, 63, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Yuan, W.G.; He, P.; Lei, J.H.; Wang, C.X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Clouston, A.D.; Forbes, S.J. Links between hepatic fibrosis, ductular reaction, and progenitor cell expansion. Gastroenterology 2014, 146, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Factor, V.M.; Marquardt, J.U.; Raggi, C.; Seo, D.; Kitade, M.; Conner, E.A.; Thorgeirsson, S.S. Hepatocyte growth factor/c-met signaling is required for stem-cell-mediated liver regeneration in mice. Hepatology 2012, 55, 1215–1226. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Evarts, R.P.; Fujio, K.; Marsden, E.R.; Thorgeirsson, S.S. Expression of hepatocyte growth-factor and c-met genes during hepatic differentiation and liver development in the rat. Am. J. Pathol. 1993, 142, 1823–1830. [Google Scholar]
- Kaur, S.; Siddiqui, H.; Bhat, M.H. Hepatic progenitor cells in action liver regeneration or fibrosis? Am. J. Pathol. 2015, 185, 2342–2350. [Google Scholar] [CrossRef]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Van Rooijen, N.; et al. Macrophage-derived wnt opposes notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 2012, 18, 572–579. [Google Scholar] [CrossRef]
- Kisseleva, T. The origin of fibrogenic myofibroblasts in fibrotic liver. Hepatology 2017, 65, 1039–1043. [Google Scholar] [CrossRef]
- Cassiman, D.; Libbrecht, L.; Desmet, V.; Denef, C.; Roskams, T. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J. Hepatol. 2002, 36, 200–209. [Google Scholar] [CrossRef]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Kwon, H.; Song, K.; Han, C.; Chen, W.; Wang, Y.; Dash, S.; Lim, K.; Wu, T. Inhibition of hedgehog signaling ameliorates hepatic inflammation in mice with nonalcoholic fatty liver disease. Hepatology 2016, 63, 1155–1169. [Google Scholar] [CrossRef]
- Jakubowski, A.; Ambrose, C.; Parr, M.; Lincecum, J.M.; Wang, M.Z.; Zheng, T.S.; Browning, B.; Michaelson, J.S.; Baetscher, M.; Wang, B.; et al. Tweak induces liver progenitor cell proliferation. J. Clin. Invest. 2005, 115, 2330–2340. [Google Scholar] [CrossRef]
- Cassiman, D.; Sinelli, N.; Bockx, I.; Vander Borght, S.; Petersen, B.; De Vos, R.; van Pelt, J.; Nevens, F.; Libbrecht, L.; Roskams, T. Human hepatic progenitor cells express vasoactive intestinal peptide receptor type 2 and receive nerve endings. Liver Int. 2007, 27, 323–328. [Google Scholar] [CrossRef]
- Oben, J.A.; Roskams, T.; Yang, S.; Lin, H.; Sinelli, N.; Li, Z.; Torbenson, M.; Huang, J.; Guarino, P.; Kafrouni, M.; et al. Sympathetic nervous system inhibition increases hepatic progenitors and reduces liver injury. Hepatology 2003, 38, 664–673. [Google Scholar] [CrossRef]
- Oben, J.A.; Roskams, T.; Yang, S.; Lin, H.; Sinelli, N.; Torbenson, M.; Smedh, U.; Moran, T.H.; Li, Z.; Huang, J.; et al. Hepatic fibrogenesis requires sympathetic neurotransmitters. Gut 2004, 53, 438–445. [Google Scholar] [CrossRef]
- McKee, C.; Sigala, B.; Soeda, J.; Mouralidarane, A.; Morgan, M.; Mazzoccoli, G.; Rappa, F.; Cappello, F.; Cabibi, D.; Pazienza, V.; et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci. Rep. 2015, 5, 8812. [Google Scholar] [CrossRef]
- Govaere, O.; Cockell, S.; Van Haele, M.; Wouters, J.; Van Delm, W.; Van den Eynde, K.; Bianchi, A.; van Eijsden, R.; Van Steenbergen, W.; Monbaliu, D.; et al. High-throughput sequencing identifies aetiology-dependent differences in ductular reaction in human chronic liver disease. J. Pathol. 2018, 248, 66–76. [Google Scholar] [CrossRef]
- Lee, Y.M.; Kaplan, M.M. Primary sclerosing cholangitis. N. Engl. J. Med. 1995, 332, 924–933. [Google Scholar] [CrossRef]
- Bjornsson, E.; Olsson, R.; Bergquist, A.; Lindgren, S.; Braden, B.; Chapman, R.W.; Boberg, K.M.; Angulo, P. The natural history of small-duct primary sclerosing cholangitis. Gastroenterology 2008, 134, 975–980. [Google Scholar] [CrossRef]
- Ruemmele, P.; Hofstaedter, F.; Gelbmann, C.M. Secondary sclerosing cholangitis. Nat. Rev. Gastroenterol. Hepatol 2009, 6, 287–295. [Google Scholar] [CrossRef]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary biliary cholangitis: 2018 practice guidance from the american association for the study of liver diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef]
- Weiler-Normann, C.; Lohse, A.W. Variant syndromes of autoimmune liver diseases: Classification, diagnosis and management. Dig. Dis. 2016, 34, 334–339. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Ellard, S. Alagille syndrome: Pathogenesis, diagnosis and management. Eur. J. Hum. Genet. 2012, 20, 251–257. [Google Scholar] [CrossRef]
- Andersson, E.R.; Chivukula, I.V.; Hankeova, S.; Sjoqvist, M.; Tsoi, Y.L.; Ramskold, D.; Masek, J.; Elmansuri, A.; Hoogendoorn, A.; Vazquez, E.; et al. Mouse model of alagille syndrome and mechanisms of jagged1 missense mutations. Gastroenterology 2018, 154, 1080–1095. [Google Scholar] [CrossRef]
- Ryan, M.J.; Bales, C.; Nelson, A.; Gonzalez, D.M.; Underkoffler, L.; Segalov, M.; Wilson-Rawls, J.; Cole, S.E.; Moran, J.L.; Russo, P.; et al. Bile duct proliferation in jag1/fringe heterozygous mice identifies candidate modifiers of the alagille syndrome hepatic phenotype. Hepatology 2008, 48, 1989–1997. [Google Scholar] [CrossRef]
- Overi, D.; Carpino, G.; Cardinale, V.; Franchitto, A.; Safarikia, S.; Onori, P.; Alvaro, D.; Gaudio, E. Contribution of resident stem cells to liver and biliary tree regeneration in human diseases. Int. J. Mol. Sci. 2018, 19, 2917. [Google Scholar] [CrossRef]
- Van Haele, M.; Moya, I.M.; Karaman, R.; Rens, G.; Snoeck, J.; Govaere, O.; Nevens, F.; Verslype, C.; Topal, B.; Monbaliu, D.; et al. Yap and taz heterogeneity in primary liver cancer: An analysis of its prognostic and diagnostic role. Int. J. Mol. Sci. 2019, 20, 2917. [Google Scholar] [CrossRef]
- De Jong, I.E.M.; Matton, A.P.M.; van Praagh, J.B.; van Haaften, W.T.; Wiersema-Buist, J.; van Wijk, L.A.; Oosterhuis, D.; Iswandana, R.; Suriguga, S.; Overi, D.; et al. Peribiliary glands are key in regeneration of the human biliary epithelium after severe bile duct injury. Hepatology 2019, 69, 1719–1734. [Google Scholar] [CrossRef]
- Roskams, T.A.; Libbrecht, L.; Desmet, V.J. Progenitor cells in diseased human liver. Semin Liver Dis. 2003, 23, 385–396. [Google Scholar]
- Arriazu, E.; Ruiz de Galarreta, M.; Cubero, F.J.; Varela-Rey, M.; Perez de Obanos, M.P.; Leung, T.M.; Lopategi, A.; Benedicto, A.; Abraham-Enachescu, I.; Nieto, N. Extracellular matrix and liver disease. Antioxid Redox Signal. 2014, 21, 1078–1097. [Google Scholar] [CrossRef]
- Milani, S.; Herbst, H.; Schuppan, D.; Surrenti, C.; Riecken, E.O.; Stein, H. Cellular localization of type i iii and iv procollagen gene transcripts in normal and fibrotic human liver. Am. J. Pathol. 1990, 137, 59–70. [Google Scholar]
- Desmet, V.J.; Roskams, T. Cirrhosis reversal: A duel between dogma and myth. J. Hepatol 2004, 40, 860–867. [Google Scholar] [CrossRef]
- Li, M.K.; Crawford, J.M. The pathology of cholestasis. Semin Liver Dis. 2004, 24, 21–42. [Google Scholar]
- Libbrecht, L.; Cassiman, D.; Desmet, V.; Roskams, T. Expression of neural cell adhesion molecule in human liver development and in congenital and acquired liver diseases. Histochem Cell Biol. 2001, 116, 233–239. [Google Scholar] [CrossRef]
- Desmet, V.J. Ductal plates in hepatic ductular reactions. Hypothesis and implications. I. Types of ductular reaction reconsidered. Virchows Arch. 2011, 458, 251–259. [Google Scholar] [CrossRef]
- Carpino, G.; Cardinale, V.; Folseraas, T.; Overi, D.; Floreani, A.; Franchitto, A.; Onori, P.; Cazzagon, N.; Berloco, P.B.; Karlsen, T.H.; et al. Hepatic stem/progenitor cell activation differs between primary sclerosing and primary biliary cholangitis. Am. J. Pathol. 2018, 188, 627–639. [Google Scholar] [CrossRef]
- Kallis, Y.N.; Robson, A.J.; Fallowfield, J.A.; Thomas, H.C.; Alison, M.R.; Wright, N.A.; Goldin, R.D.; Iredale, J.P.; Forbes, S.J. Remodelling of extracellular matrix is a requirement for the hepatic progenitor cell response. Gut 2011, 60, 525–533. [Google Scholar] [CrossRef]
- Vogel, A.; Cervantes, A.; Chau, I.; Daniele, B.; Llovet, J.; Meyer, T.; Nault, J.C.; Neumann, U.; Ricke, J.; Sangro, B.; et al. Hepatocellular carcinoma: Esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv238–iv255. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef]
- Desai, A.; Sandhu, S.; Lai, J.P.; Sandhu, D.S. Hepatocellular carcinoma in non-cirrhotic liver: A comprehensive review. World J. Hepatol. 2019, 11, 1–18. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Borzio, M.; Fargion, S.; Borzio, F.; Fracanzani, A.L.; Croce, A.M.; Stroffolini, T.; Oldani, S.; Cotichini, R.; Roncalli, M. Impact of large regenerative, low grade and high grade dysplastic nodules in hepatocellular carcinoma development. J. Hepatol. 2003, 39, 208–214. [Google Scholar] [CrossRef]
- Di Tommaso, L.; Franchi, G.; Park, Y.N.; Fiamengo, B.; Destro, A.; Morenghi, E.; Montorsi, M.; Torzilli, G.; Tommasini, M.; Terracciano, L.; et al. Diagnostic value of hsp70, glypican 3, and glutamine synthetase in hepatocellular nodules in cirrhosis. Hepatology 2007, 45, 725–734. [Google Scholar] [CrossRef]
- Libbrecht, L.; Severi, T.; Cassiman, D.; Borght, S.V.; Pirenne, J.; Nevens, F.; Verslype, C.; van Pelt, J.; Roskams, T. Glypican-3 expression distinguishes small hepatocellular carcinomas from cirrhosis, dysplastic nodules, and focal nodular hyperplasia-like nodules. Am. J. Surh. Pathol. 2006, 30, 1405–1411. [Google Scholar] [CrossRef]
- Ahn, S.M.; Jang, S.J.; Shim, J.H.; Kim, D.; Hong, S.M.; Sung, C.O.; Baek, D.; Haq, F.; Ansari, A.A.; Lee, S.Y.; et al. Genomic portrait of resectable hepatocellular carcinomas: Implications of rb1 and fgf19 aberrations for patient stratification. Hepatology 2014, 60, 1972–1982. [Google Scholar] [CrossRef]
- Totoki, Y.; Tatsuno, K.; Covington, K.R.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouze, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumour classification. J. Hepatol. 2017, 67, 727–738. [Google Scholar] [CrossRef]
- Schlageter, M.; Terracciano, L.M.; D’Angelo, S.; Sorrentino, P. Histopathology of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 15955–15964. [Google Scholar] [CrossRef]
- Brunt, E.; Aishima, S.; Clavien, P.A.; Fowler, K.; Goodman, Z.; Gores, G.; Gouw, A.; Kagen, A.; Klimstra, D.; Komuta, M.; et al. Chcc-cca: Consensus terminology for primary liver carcinomas with both hepatocytic and cholangiocytic differentation. Hepatology 2018, 68, 113–126. [Google Scholar] [CrossRef]
- Komuta, M.; Spee, B.; Borght, S.V.; De Vos, R.; Verslype, C.; Aerts, R.; Yano, H.; Suzuki, T.; Matsuda, M.; Fujii, H.; et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology 2008, 47, 1544–1556. [Google Scholar] [CrossRef]
- Moeini, A.; Sia, D.; Zhang, Z.Y.; Camprecios, G.; Stueck, A.; Dong, H.; Montal, R.; Torrens, L.; Martinez-Quetglas, I.; Fiel, M.I.; et al. Mixed hepatocellular cholangiocarcinoma tumors: Cholangiolocellular carcinoma is a distinct molecular entity. J. Hepatol. 2017, 66, 952–961. [Google Scholar] [CrossRef]
- Govaere, O.; Komuta, M.; Berkers, J.; Spee, B.; Janssen, C.; de Luca, F.; Katoonizadeh, A.; Wouters, J.; van Kempen, L.C.; Durnez, A.; et al. Keratin 19: A key role player in the invasion of human hepatocellular carcinomas. Gut 2014, 63, 674–685. [Google Scholar] [CrossRef]
- Govaere, O.; Petz, M.; Wouters, J.; Vandewynckel, Y.P.; Scott, E.J.; Topal, B.; Nevens, F.; Verslype, C.; Anstee, Q.M.; Van Vlierberghe, H.; et al. The pdgfr alpha-laminin b1-keratin 19 cascade drives tumor progression at the invasive front of human hepatocellular carcinoma. Oncogene 2017, 36, 6605–6616. [Google Scholar] [CrossRef]
- Couri, T.; Pillai, A. Goals and targets for personalized therapy for hcc. Hepatol. Int. 2019, 13, 125–137. [Google Scholar] [CrossRef]
- Kurebayashi, Y.; Ojima, H.; Tsujikawa, H.; Kubota, N.; Maehara, J.; Abe, Y.; Kitago, M.; Shinoda, M.; Kitagawa, Y.; Sakamoto, M. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology 2018, 68, 1025–1041. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Haele, M.; Snoeck, J.; Roskams, T. Human Liver Regeneration: An Etiology Dependent Process. Int. J. Mol. Sci. 2019, 20, 2332. https://doi.org/10.3390/ijms20092332
Van Haele M, Snoeck J, Roskams T. Human Liver Regeneration: An Etiology Dependent Process. International Journal of Molecular Sciences. 2019; 20(9):2332. https://doi.org/10.3390/ijms20092332
Chicago/Turabian StyleVan Haele, Matthias, Janne Snoeck, and Tania Roskams. 2019. "Human Liver Regeneration: An Etiology Dependent Process" International Journal of Molecular Sciences 20, no. 9: 2332. https://doi.org/10.3390/ijms20092332
APA StyleVan Haele, M., Snoeck, J., & Roskams, T. (2019). Human Liver Regeneration: An Etiology Dependent Process. International Journal of Molecular Sciences, 20(9), 2332. https://doi.org/10.3390/ijms20092332