Single-Cell RNA-Sequencing Identifies Activation of TP53 and STAT1 Pathways in Human T Lymphocyte Subpopulations in Response to Ex Vivo Radiation Exposure

 , ,
, ,
Abstract
1. Introduction
2. Results
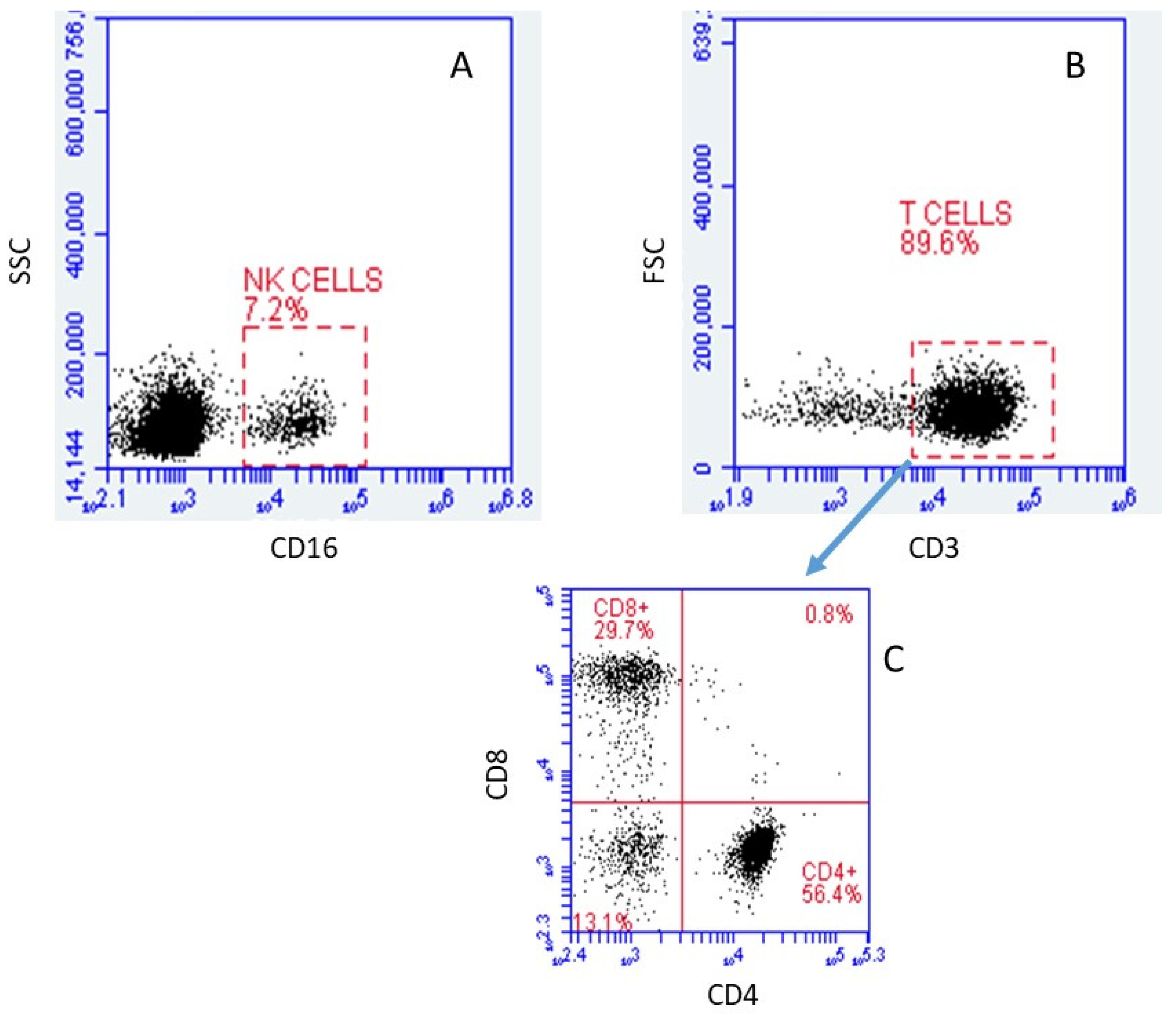
2.1. Verification and Identification of Isolated Cell Subpopulations by FACS
2.1.1. Cell Types
2.1.2. Quality of Single-Cell RNA Sequencing
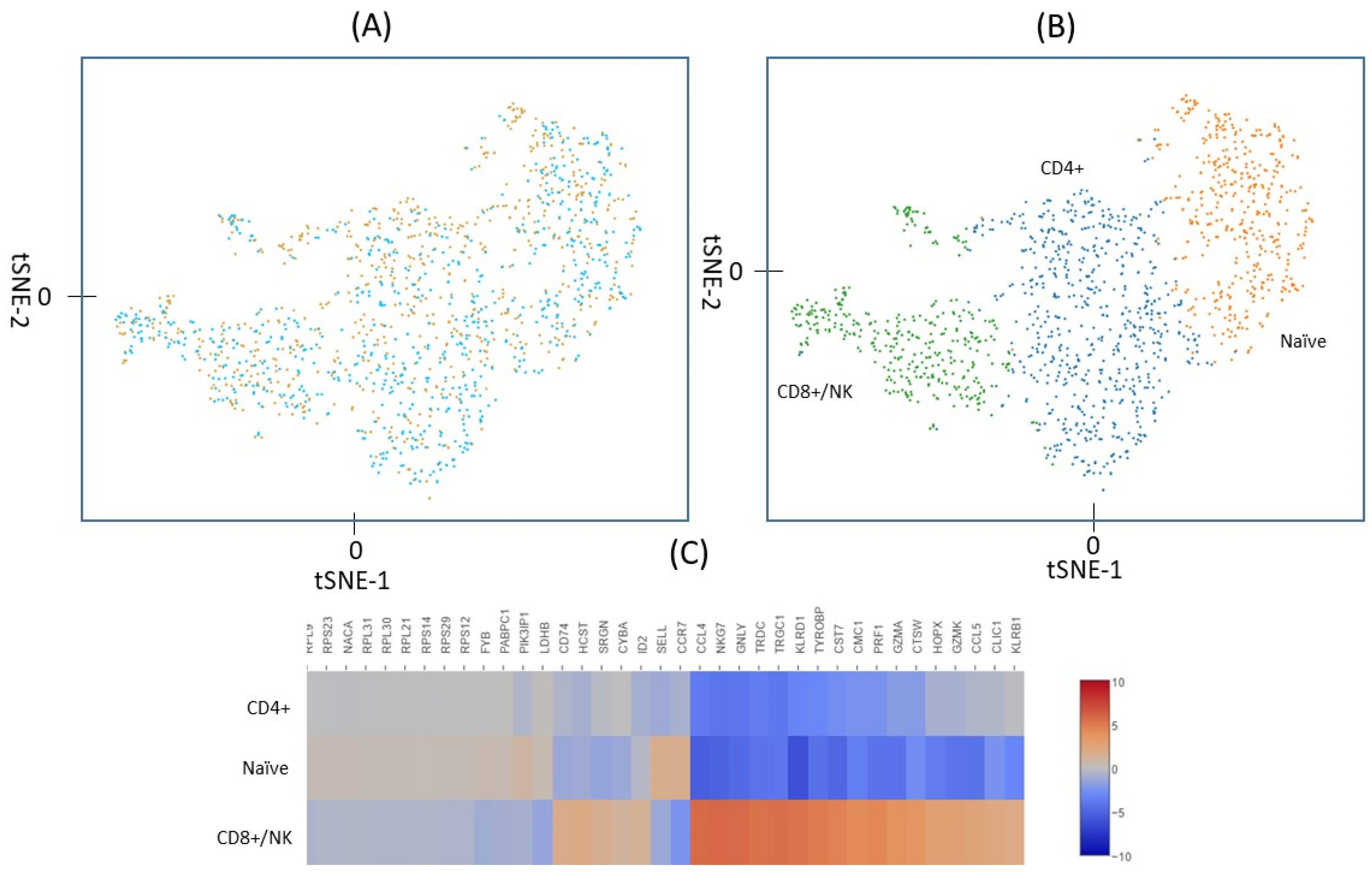
2.2. Identification of Lymphocyte Subpopulations Based on scRNA-seq
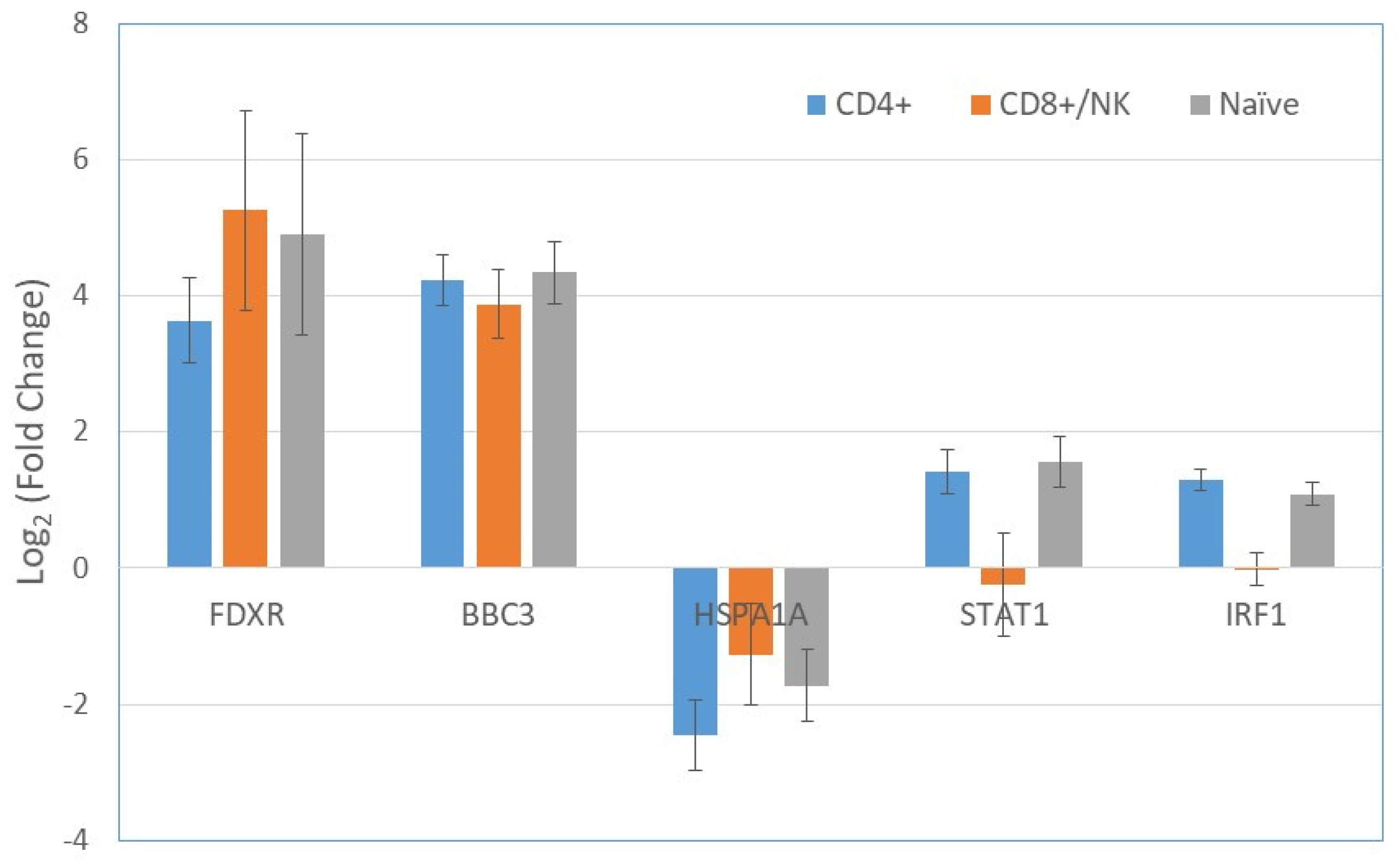
2.3. Gene Expression in T cells Subpopulations
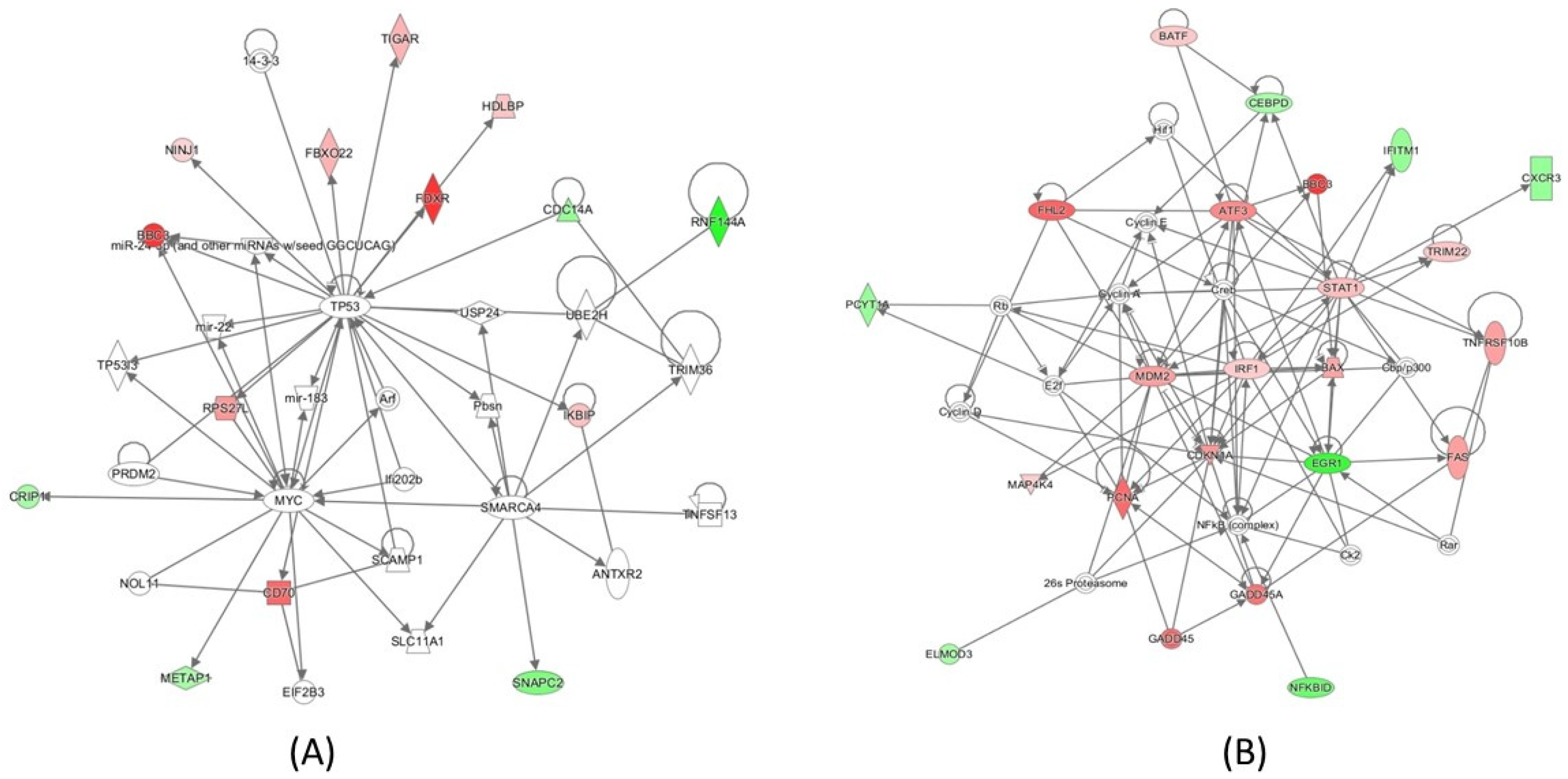
2.4. Pathway Analysis
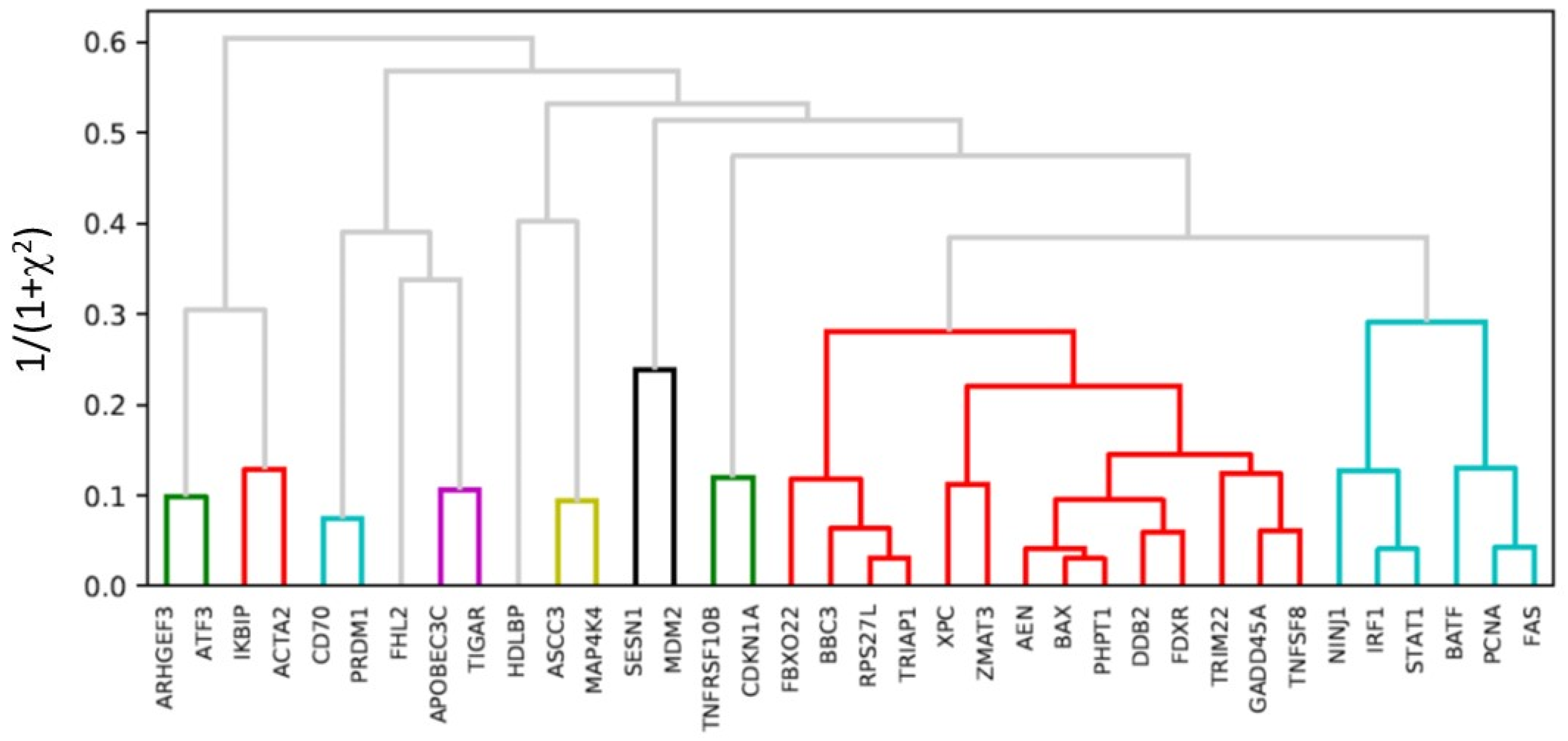
2.5. Hierarchical Clustering
3. Discussion
4. Materials and Methods
4.1. Cells and Radiation Exposure
4.2. Single Cell RNA-Seq Library Preparation and Sequencing
4.3. Transcriptome Analysis
4.4. Gene Expression Analysis in Response to Radiation
4.5. Pathway Analysis
4.6. Hierarchical Clustering of Genes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cucinotta, F.A.; Durante, M. Cancer risk from exposure to galactic cosmic rays: Implications for space exploration by human beings. Lancet Oncol. 2006, 7, 431–435. [Google Scholar] [CrossRef]
- Moreno-Villanueva, M.; Wong, M.; Lu, T.; Zhang, Y.; Wu, H. Interplay of space radiation and microgravity in DNA damage and DNA damage response. NPJ Microgravity 2017, 3, 14. [Google Scholar] [CrossRef]
- Hada, M.; Cucinotta, F.A.; Gonda, S.R.; Wu, H. mBAND analysis of chromosomal aberrations in human epithelial cells exposed to low- and high-LET radiation. Radiat. Res. 2007, 168, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Nakano-Aoki, M.; Matsumoto, Y.; Hirayama, R.; Kobayashi, A.; Konishi, T. Equivalency of the quality of sublethal lesions after photons and high-linear energy transfer ion beams. J. Radiat. Res. 2017, 58, 58803–58808. [Google Scholar]
- De Toledo, S.M.; Buonanno, M.; Li, M.; Asaad, N.; Qin, Y.; Gonon, G.; Shim, G.; Galdass, M.; Boateng, Y.; Zhang, J.; et al. The impact of adaptive and non-targeted effects in the biological responses to low dose/low fluence ionizing radiation: The modulating effect of linear energy transfer. Health Phys. 2011, 100, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Hei, T.K.; Zhou, H.; Chai, Y.; Ponnaiya, B.; Ivanov, V.N. Radiation induced non-targeted response: Mechanism and potential clinical implications. Curr Mol. Pharmacol. 2011, 4, 96–105. [Google Scholar] [CrossRef]
- Desai, N.; Davis, E.; O’Neill, P.; Durante, M.; Cucinotta, F.A.; Wu, H. Immunofluorescence detection of clustered gamma-H2AX foci induced by HZE-particle radiation. Radiat. Res. 2005, 164, 518–522. [Google Scholar] [CrossRef]
- Asaithamby, A.; Chen, D.J. Cellular responses to DNA double-strand breaks after low-dose gamma-irradiation. Nucleic Acids Res. 2009, 37, 3912–3923. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Durante, M.; Furusawa, Y.; George, K.; Kawata, T.; Cucinotta, F.A. Truly incomplete and complex exchanges in prematurely condensed chromosomes of human fibroblasts exposed in vitro to energetic heavy ions. Radiat. Res. 2003, 160, 418–424. [Google Scholar] [CrossRef]
- Lu, T.; Zhang, Y.; Wong, M.; Feiveson, A.; Gaza, R.; Stoffle, N.; Wang, H.; Wilson, B.; Rohde, L.; Stodieck, L.; Karouia, F.; Wu, H. Detection of DNA damage by space radiation in human fibroblasts flown on the International Space Station. Life Sci. Space Res. 2017, 12, 24–31. [Google Scholar] [CrossRef]
- Hwang, B.; Lee, J.H.; Bang, D. Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp. Mol. Med. 2018, 50, 96. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Novak, R.; Shuga, J.; Smith, M.T.; Mathies, R.A. High-performance single cell genetic analysis using microfluidic emulsion generator arrays. Anal. Chem. 2010, 82, 3183–3190. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356. [Google Scholar] [CrossRef]
- Kumar, M.P.; Du, J.; Lagoudas, G.; Jiao, Y.; Sawyer, A.; Drummond, D.C.; Lauffenburger, D.A.; Raue, A. Analysis of Single-Cell RNA-Seq Identifies Cell-Cell Communication Associated with Tumor Characteristics. Cell Rep. 2018, 25, 1458–1468. [Google Scholar] [CrossRef]
- Li, N.; van Unen, V.; Abdelaal, T.; Guo, N.; Kasatskaya, S.A.; Ladell, K.; McLaren, J.E.; Egorov, E.S.; Izraelson, M.; Chuva de Sousa Lopes, S.M.; et al. Memory CD4+ T cells are generated in the human fetal intestine. Nat. Immunol. 2019, 20, 301. [Google Scholar] [CrossRef]
- Gupta, I.; Collier, P.G.; Haase, B.; Mahfouz, A.; Joglekar, A.; Floyd, T.; Koopmans, F.; Barres, B.; Smit, A.B.; Sloan, S.A.; et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018, 36, 1197. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Gonzalo, R.; Baatout, S.; Moreels, M. Impact of Particle Irradiation on the Immune System: From the Clinic to Mars. Front. Immunol. 2017, 8, 177. [Google Scholar] [CrossRef]
- Amundson, S.A.; Do, K.T.; Shahab, S.; Bittner, M.; Meltzer, P.; Trent, J.; Fornace, A.J. Identification of potential mRNA biomarkers in peripheral blood lymphocytes for human exposure to ionizing radiation. Radiat. Res. 2000, 154, 342–346. [Google Scholar] [CrossRef]
- Tucker, J.D.; Joiner, M.C.; Thomas, R.A.; Grever, W.E.; Bakhmutsky, M.V.; Chinkhota, C.N.; Smolinski, J.M.; Divine, G.W.; Auner, G.W. Accurate gene expression-based biodosimetry using a minimal set of human gene transcripts. Int. J. Radiat. Oncol. Biol. Phys. 2014, 88, 933–939. [Google Scholar] [CrossRef]
- Macaeva, E.; Mysara, M.; De Vos, W.H.; Baatout, S.; Quintens, R. Gene expression-based biodosimetry for radiological incidents: Assessment of dose and time after radiation exposure. Int. J. Radiat. Biol. 2019, 95, 64–75. [Google Scholar] [CrossRef]
- Macosko, E.Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas, A.R.; Kamitaki, N.; Martersteck, E.M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.; Diehn, M.; Alizadeh, A.A.; Brown, P.O. Cell-type specific gene expression profiles of leukocytes in human peripheral blood. BMC Genomics 2006, 7, 115. [Google Scholar]
- Berard, M.; Tough, D.F. Qualitative differences between naïve and memory T cells. Immunology 2002, 106, 127–138. [Google Scholar] [CrossRef]
- Unsoeld, H.; Pircher, H. Complex memory T-cell phenotypes revealed by coexpression of CD62L and CCR7. J Virol. 2005, 79, 4510–4513. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Shekhar, K. Single-Cell RNA Sequencing of Human T Cells. Methods Mol. Biol. 2017, 1514, 203–239. [Google Scholar] [PubMed]
- Zemmour, D.; Zilionis, R.; Kiner, E.; Klein, A.M.; Mathis, D.; Benoist, C. Single-cell gene expression reveals a landscape of regulatory T cell phenotypes shaped by the TCR. Nat. Immunol. 2018, 19, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Lacombe, J.; Sima, C.; Amundson, S.A.; Zenhausern, F. Candidate gene biodosimetry markers of exposure to external ionizing radiation in human blood: A systematic review. PLoS ONE 2018, 13, e0198851. [Google Scholar] [CrossRef]
- O’Brien, G.; Cruz-Garcia, L.; Majewski, M.; Grepl, J.; Abend, M.; Port, M.; Tichý, A.; Sirak, I.; Malkova, A.; Donovan, E.; et al. FDXR is a biomarker of radiation exposure in vivo. Sci Rep. 2018, 8, 684. [Google Scholar] [CrossRef] [PubMed]
- Riecke, A.; Rufa, C.G.; Cordes, M.; Hartmann, J.; Meineke, V.; Abend, M. Gene expression comparisons performed for biodosimetry purposes on in vitro peripheral blood cellular subsets and irradiated individuals. Radiat. Res. 2012, 178, 234–243. [Google Scholar] [CrossRef]
- Gruel, G.; Voisin, P.; Vaurijoux, A.; Roch-Lefevre, S.; Grégoire, E.; Maltere, P.; Petat, C.; Gidrol, X.; Voisin, P.; Roy, L. Broad modulation of gene expression in CD4+ lymphocyte subpopulations in response to low doses of ionizing radiation. Radiat. Res. 2008, 170, 335–344. [Google Scholar] [CrossRef]
- Gerber, S.A.; Sedlacek, A.L.; Cron, K.R.; Murphy, S.P.; Frelinger, J.G.; Lord, E.M. IFN-γ mediates the antitumor effects of radiation therapy in a murine colon tumor. Am. J. Pathol. 2013, 182, 2345–2354. [Google Scholar] [CrossRef]
- Han, S.K.; Song, J.Y.; Yun, Y.S.; Yi, S.Y. Gamma irradiation-reduced IFN-gamma expression, STAT1 signals, and cell-mediated immunity. J. Biochem. Mol. Biol. 2002, 35, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Ozsahin, M.; Crompton, N.E.; Gourgou, S.; Kramar, A.; Li, L.; Shi, Y.; Sozzi, W.J.; Zouhair, A.; Mirimanoff, R.O.; Azria, D. CD4 and CD8 T-lymphocyte apoptosis can predict radiation-induced late toxicity: a prospective study in 399 patients. Clin. Cancer Res. 2005, 11, 7426–7433. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ishihara, M.; Lamphier, M.S.; Tanaka, N.; Oishi, I.; Aizawa, S.; Matsuyama, T.; Mak, T.W.; Taki, S.; Taniguchi, T. An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen-activated T lymphocytes. Nature 1995, 376, 596–599. [Google Scholar] [CrossRef]
- El Jamal, S.M.; Taylor, E.B.; Abd Elmageed, Z.Y.; Alamodi, A.A.; Selimovic, D.; Alkhateeb, A.; Hannig, M.; Hassan, S.Y.; Santourlidis, S.; Friedlander, P.L.; et al. Interferon gamma-induced apoptosis of head and neck squamous cell carcinoma is connected to indoleamine-2,3-dioxygenase via mitochondrial and ER stress-associated pathways. Cell Div. 2016, 11, 11. [Google Scholar] [CrossRef]
- Brzostek-Racine, S.; Gordon, C.; Van Scoy, S.; Reich, N.C. The DNA damage response induces IFN. J. Immunol. 2011, 187, 5336–5345. [Google Scholar] [CrossRef]
- Kim, M.K.; Song, J.Y.; Koh, D.I.; Kim, J.Y.; Hatano, M.; Jeon, B.N.; Kim, M.Y.; Cho, S.Y.; Kim, K.S.; Hur, M.W. Reciprocal negative regulation between the tumor suppressor protein p53 and B cell CLL/lymphoma 6 (BCL6) via control of caspase-1 expression. J. Biol. Chem. 2019, 294, 299–313. [Google Scholar] [CrossRef]
- Youlyouz-Marfak, I.; Gachard, N.; Le Clorennec, C.; Najjar, I.; Baran-Marszak, F.; Reminieras, L.; May, E.; Bornkamm, G.W.; Fagard, R.; Feuillard, J. Identification of a novel p53-dependent activation pathway of STAT1 by antitumour genotoxic agents. Cell Death Differ. 2008, 15, 376–385. [Google Scholar] [CrossRef]
- Chapgier, A.; Kong, X.F.; Boisson-Dupuis, S.; Jouanguy, E.; Averbuch, D.; Feinberg, J.; Zhang, S.Y.; Bustamante, J.; Vogt, G.; Lejeune, J.; et al. A partial form of recessive STAT1 deficiency in humans. J. Clin. Invest. 2009, 119, 1502–1514. [Google Scholar]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef]
- Kernbauer, E.; Maier, V.; Stoiber, D.; Strobl, B.; Schneckenleithner, C.; Sexl, V.; Reichart, U.; Reizis, B.; Kalinke, U.; Jamieson, A.; Müller, M.; Decker, T. Conditional Stat1 ablation reveals the importance of interferon signaling for immunity to Listeria monocytogenes infection. PLoS Pathog. 2012, 8, e1002763. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhao, J.; Chen, L.; Dong, N.; Ying, Z.; Cai, Z.; Ji, D.; Zhang, Y.; Dong, L.; Li, Y.; et al. STAT1 modification improves therapeutic effects of interferons on lung cancer cells. J. Transl. Med. 2015, 13, 293. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Mandinova, A.; Aaronson, S.A.; Lee, S.W. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat. Rev. Immunol. 2016, 16, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Jyothi Prasanna, S.; Chandrasekar, B.; Nandi, D.C. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine 2010, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zenke, K.; Muroi, M.; Tanamoto, K.I. IRF1 supports DNA binding of STAT1 by promoting its phosphorylation. Immunol. Cell. Biol. 2018, 96, 1095–1103. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Sun, Y. Ribosomal protein S27L is a direct p53 target that regulates apoptosis. Oncogene 2007, 26, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Park, W.R.; Nakamura, Y. p53CSV, a novel p53-inducible gene involved in the p53-dependent cell-survival pathway. Cancer Res. 2005, 65, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Andrysik, Z.; Kim, J.; Tan, A.C.; Espinosa, J.M. A genetic screen identifies TCF3/E2A and TRIAP1 as pathway-specific regulators of the cellular response to p53 activation. Cell Rep. 2013, 3, 1346–1354. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Villanueva, M.; Wu, H. Radiation and microgravity – Associated stress factors and carcinogenesis. REACH 2019, 13, 100027. [Google Scholar] [CrossRef]
- Cekanaviciute, E.; Rosi, S.; Costes, S.V. Central Nervous System Responses to Simulated Galactic Cosmic Rays. Int. J. Mol. Sci. 2018, 19, 3669. [Google Scholar] [CrossRef]
- Kabacik, M.G.; Raffy, C.; Bouffler, S.; Badie, C. Time, dose and ataxia telangiectasia mutated (ATM) status dependency of coding and noncoding RNA expression after ionizing radiation exposure. Radiat. Res. 2015, 183, 325–337. [Google Scholar] [CrossRef]
- Rràs-Fresneda, M.; Barquinero, J.F.; Gomolka, M.; Hornhardt, S.; Rössler, U.; Armengol, G.; Barrios, L. Differences in DNA Repair Capacity, Cell Death and Transcriptional Response after Irradiation between a Radiosensitive and a Radioresistant Cell Line. Sci. Rep. 2016, 6, 27043. [Google Scholar] [CrossRef]
- Chen, J.; Cheung, F.; Shi, R.; Zhou, H.; Lu, W.; CHI Consortium. PBMC fixation and processing for Chromium single-cell RNA sequencing. J. Transl. Med. 2018, 16, 198. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Van der Maaten, L. Accelerating t-SNE using Tree-Based Algorithms. J. Mach. Learn. Res. 2018, 15, 3221–3245. [Google Scholar]
- Yu, D.; Huber, W.; Vitek, O. Shrinkage estimation of dispersion in Negative Binomial models for RNA-seq experiments with small sample size. Bioinformatics 2013, 29, 1275–1282. [Google Scholar] [CrossRef]
- Famoye, F. A new bivariate generalized Poisson distribution. Stat. Neerl. 2010, 64, 112–124. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Parameters | Control | Irradiated |
|---|---|---|
| Cell Number | 690 | 733 |
| Total Reads | 40 Million | 31 Million |
| Genes per Cell | 1131 | 1067 |
| Valid Barcodes | 97.8% | 97.7% |
| Sequencing Saturation | 87.1% | 83.6% |
| Barcode Q30 | 96.4% | 96.3% |
| Gene | All Cells log2 FC | p | CD4+ log2 FC | p | CD8+/NK log2 FC | p | Naïve log2 FC | p |
|---|---|---|---|---|---|---|---|---|
| FDXR | 4.05 | 5.9 × 10−16 | 3.63 | 5.2 × 10−9 | 4.52 | 1.7 × 10−5 | 4.90 | 8.9 × 10−4 |
| BBC3 | 4.00 | 0.0 × 10+00 | 4.23 | 4.2 × 10−29 | 3.44 | 1.6 × 10−18 | 4.34 | 2.3 × 10−20 |
| CD70 | 2.99 | 2.6 × 10−9 | 4.17 | 2.8 × 10−6 | 1.75 | 1.1 × 10−2 | 2.58 | 1.1 × 10−1 |
| AEN | 3.15 | 6.1 × 10−30 | 3.31 | 2.3 × 10−17 | 2.83 | 1.8 × 10−6 | 3.06 | 1.2 × 10−8 |
| FHL2 | 3.08 | 5.3 × 10−4 | 2.95 | 7.9 × 10−3 | 2.29 | 1.5 × 10−1 | ||
| PCNA | 2.94 | 0.0 × 10+00 | 3.20 | 6.1 × 10−25 | 3.53 | 2.8 × 10−14 | 2.04 | 2.9 × 10−7 |
| DDB2 | 2.87 | 0.0 × 10+00 | 3.17 | 1.5 × 10−21 | 2.82 | 7.3 × 10−11 | 2.49 | 4.0 × 10−12 |
| GADD45A | 2.79 | 1.5 × 10−15 | 2.66 | 6.0 × 10−9 | 5.64 | 1.3 × 10−4 | 1.47 | 1.9 × 10−2 |
| ATF3 | 2.33 | 4.8 × 10−4 | 4.20 | 5.6 × 10−3 | 1.78 | 1.2 × 10−1 | 0.77 | 4.5 × 10−1 |
| CDKN1A | 2.08 | 3.6 × 10−4 | 3.65 | 6.0 × 10−4 | 0.23 | 8.1 × 10−1 | 0.77 | 6.6 × 10−1 |
| TNFSF8 | 2.39 | 1.4 × 10−12 | 2.22 | 2.9 × 10−8 | 2.72 | 4.3 × 10−4 | 3.02 | 5.6 × 10−3 |
| PHPT1 | 2.07 | 0.0 × 10+00 | 2.46 | 7.2 × 10−30 | 1.57 | 6.8 × 10−9 | 1.97 | 3.6 × 10−14 |
| ACTA2 | 2.08 | 6.1 × 10−6 | 1.95 | 1.4 × 10−3 | 2.29 | 3.8 × 10−3 | ||
| RPS27L | 2.06 | 0.0 × 10+00 | 2.14 | 0.0 × 10+00 | 2.04 | 1.6 × 10−33 | 1.93 | 0.0 × 10+00 |
| MDM2 | 1.95 | 7.0 × 10−8 | 1.46 | 1.7 × 10−3 | 4.67 | 1.6 × 10−3 | 1.70 | 1.9 × 10−2 |
| TNFRSF10B | 1.89 | 1.3 × 10−5 | 2.00 | 9.8 × 10−4 | 1.97 | 9.8 × 10−2 | 1.54 | 3.6 × 10−2 |
| FAS | 1.87 | 3.9 × 10−12 | 2.06 | 7.4 × 10−9 | 1.60 | 1.4 × 10−3 | 1.58 | 2.5 × 10−2 |
| BAX | 1.85 | 0.0 × 10+00 | 1.93 | 7.1 × 10−37 | 1.68 | 8.2 × 10−16 | 2.03 | 3.1 × 10−21 |
| ASCC3 | 1.67 | 2.5 × 10−9 | 1.68 | 4.5 × 10−5 | 1.60 | 2.3 × 10−3 | 1.69 | 2.5 × 10−03 |
| PRDM1 | 1.66 | 8.0 × 10−4 | 1.69 | 1.2 × 10−2 | 1.11 | 1.6 × 10−1 | ||
| TRIAP1 | 1.62 | 1.2 × 10−17 | 1.85 | 1.3 × 10−10 | 1.66 | 3.3 × 10−5 | 1.19 | 2.7 × 10−4 |
| FBXO22 | 1.52 | 1.5 × 10−7 | 1.95 | 3.6 × 10−5 | 1.55 | 2.2 × 10−2 | 1.02 | 2.3 × 10−2 |
| ARHGEF3 | 1.50 | 6.5 × 10−9 | 0.92 | 1.1 × 10−2 | 2.34 | 3.5 × 10−4 | 1.90 | 8.4 × 10−5 |
| TIGAR | 1.50 | 3.6 × 10−5 | 1.30 | 1.2 × 10−2 | 1.25 | 5.4 × 10−2 | 2.36 | 1.1 × 10−2 |
| STAT1 | 1.32 | 1.2 × 10−8 | 1.42 | 1.3 × 10−5 | −0.27 | 6.9 × 10−1 | 1.55 | 3.3 × 10−5 |
| APOBEC3C | 1.39 | 4.2 × 10−7 | 1.41 | 1.3 × 10−3 | 1.65 | 1.4 × 10−4 | 0.99 | 1.1 × 10−1 |
| XPC | 1.33 | 3.8 × 10−14 | 1.22 | 9.0 × 10−7 | 0.92 | 6.7 × 10−3 | 1.99 | 1.9 × 10−7 |
| SESN1 | 1.30 | 6.6 × 10−10 | 1.66 | 2.1 × 10−6 | 1.11 | 3.3 × 10−3 | 0.95 | 9.7 × 10−3 |
| IKBIP | 1.26 | 3.2 × 10−5 | 1.58 | 1.1 × 10−3 | 1.23 | 3.6 × 10−02 | 0.97 | 7.5 × 10−2 |
| HDLBP | 1.18 | 9.2 × 10−4 | 1.25 | 1.7 × 10−2 | 1.55 | 3.9 × 10−2 | 0.77 | 2.4 × 10−1 |
| ZMAT3 | 1.15 | 1.8 × 10-04 | 1.45 | 1.2 × 10−03 | 0.80 | 1.8 × 10−1 | 0.77 | 2.1 × 10−1 |
| TRIM22 | 1.15 | 1.5 × 10−10 | 1.04 | 1.4 × 10−5 | 1.52 | 5.2 × 10−4 | 1.07 | 2.0 × 10−3 |
| BATF | 1.08 | 2.7 × 10−4 | 1.44 | 6.7 × 10−4 | 0.05 | 9.3 × 10−1 | 1.77 | 1.1 × 10−2 |
| IRF1 | 1.03 | 1.3 × 10−23 | 1.29 | 2.5 × 10−17 | 0.04 | 8.5 × 10−1 | 1.09 | 1.9 × 10−10 |
| MAP4K4 | 1.01 | 1.8 × 10−4 | 0.73 | 4.7 × 10−2 | 1.19 | 9.1 × 10−2 | 1.44 | 3.9 × 10−3 |
| NINJ1 | 0.91 | 2.7 × 10−06 | 1.22 | 7.3 × 10−5 | 0.71 | 5.2 × 10−2 | 0.67 | 5.8 × 10−2 |
| Gene | All Cells log2 FC | p | CD4+ log2 FC | p | CD8+/NK log2 FC | p | Naïve log2 FC | p |
|---|---|---|---|---|---|---|---|---|
| CRIP1 | −1.02 | 2.6 × 10−9 | −1.03 | 1.1 × 10−6 | −1.16 | 2.4 × 10−3 | −0.64 | 1.1 × 10−1 |
| METAP1 | −1.05 | 2.2 × 10−4 | −1.39 | 4.1 × 10−2 | −1.01 | 3.8 × 10−2 | ||
| ISG20 | −1.02 | 5.2 × 10−13 | −1.28 | 3.1 × 10−9 | −0.84 | 4.5 × 10−3 | −0.79 | 1.6 × 10−3 |
| DNAJB1 | −1.03 | 2.7 × 10−14 | −0.61 | 1.0 × 10−3 | −0.77 | 4.3 × 10−3 | −1.46 | 9.7 × 10−10 |
| CEBPD | −1.03 | 9.8 × 10−4 | −1.69 | 1.4 × 10−2 | −0.84 | 1.2 × 10−2 | −2.23 | 1.7 × 10−1 |
| DBF4 | −1.04 | 9.8 × 10−5 | −0.62 | 8.4 × 10−2 | −1.96 | 8.2 × 10−3 | −1.39 | 5.1 × 10−3 |
| ELMOD3 | −1.04 | 8.4 × 10−4 | −0.91 | 3.9 × 10−2 | −2.53 | 5.2 × 10−3 | −0.53 | 3.3 × 10−1 |
| NDUFA7 | −1.05 | 6.7 × 10−4 | −1.31 | 1.8 × 10−2 | −0.69 | 1.9 × 10−1 | −1.04 | 4.7 × 10−2 |
| LINC00954 | −1.07 | 7.7 × 10-04 | −1.05 | 3.1 × 10−2 | −1.77 | 6.3 × 10−2 | −1.09 | 2.5 × 10−2 |
| SHOC2 | −1.10 | 1.8 × 10−5 | −1.05 | 5.4 × 10−3 | −1.56 | 8.6 × 10−03 | −0.79 | 8.3 × 10−2 |
| TSEN54 | −1.09 | 9.8 × 10−5 | −0.86 | 5.9 × 10−2 | −1.80 | 5.3 × 10−4 | −0.59 | 2.5 × 10−1 |
| THEMIS2 | −1.20 | 6.7 × 10-04 | −1.70 | 6.6 × 10−3 | −1.28 | 4.7 × 10−2 | −0.37 | 5.6 × 10-01 |
| WDR20 | −1.13 | 3.3 × 10−4 | −0.86 | 6.0 × 10−2 | −1.94 | 1.7 × 10−2 | −1.09 | 4.4 × 10−2 |
| CXCR3 | −1.17 | 2.0 × 10−5 | −1.63 | 3.2 × 10−4 | −0.65 | 6.2 × 10−2 | ||
| PCYT1A | −1.16 | 4.0 × 10−4 | −1.14 | 1.0 × 10−2 | −0.67 | 3.2 × 10−1 | −1.81 | 1.5 × 10−2 |
| CCDC65 | −1.18 | 2.6 × 10−4 | −1.15 | 1.4 × 10−2 | −0.52 | 5.4 × 10−1 | −1.55 | 4.2 × 10−3 |
| IFITM1 | −1.18 | 4.6 × 10−7 | −1.42 | 8.2 × 10−5 | −1.57 | 4.6 × 10−3 | −0.70 | 6.5 × 10−2 |
| SF3A2 | −1.20 | 1.8 × 10−4 | −1.00 | 3.0 × 10−2 | −1.80 | 1.5 × 10−2 | −1.15 | 5.4 × 10−2 |
| DOK2 | −1.23 | 1.5 × 10−4 | −1.56 | 1.3 × 10−3 | −0.72 | 2.3 × 10−1 | −1.23 | 9.5 × 10−2 |
| CDC14A | −1.23 | 5.8 × 10−4 | −1.21 | 1.5 × 10−2 | −0.55 | 4.6 × 10−1 | −1.81 | 1.6 × 10−2 |
| ABHD3 | −1.22 | 6.9 × 10−6 | −1.52 | 1.4 × 10−4 | −0.15 | 8.0 × 10−1 | −1.41 | 6.7 × 10−3 |
| APBB1 | −1.29 | 8.7 × 10−4 | −1.97 | 6.9 × 10−3 | −0.77 | 3.0 × 10−1 | −1.15 | 5.4 × 10−2 |
| YPEL2 | −1.39 | 4.4 × 10−4 | −2.63 | 7.3 × 10−4 | −0.64 | 2.0 × 10−1 | ||
| RP11284N83 | −1.41 | 9.5 × 10−4 | −3.11 | 4.2 × 10−4 | −0.25 | 7.5 × 10−1 | −0.23 | 7.7 × 10−1 |
| SNAPC2 | −1.48 | 1.4 × 10−4 | −1.80 | 1.9 × 10−3 | −1.73 | 3.9 × 10−2 | −0.97 | 1.9 × 10−1 |
| HSPA1B | −1.52 | 1.4X10−4 | −1.63 | 2.3 × 10−2 | −1.79 | 5.4 × 10−2 | −1.54 | 3.7 × 10−3 |
| NFKBID | −1.70 | 5.0 × 10−5 | −1.51 | 8.4 × 10−2 | −0.84 | 1.9 × 10−1 | −3.23 | 2.6 × 10−4 |
| HSPA1A | −1.76 | 1.4 × 10−7 | −2.46 | 3.0 × 10−6 | −1.42 | 5.2 × 10−2 | −1.72 | 9.1 × 10−4 |
| ERI1 | −1.83 | 9.2 × 10−4 | −2.63 | 3.7 × 10−3 | −0.35 | 7.1 × 10−1 | −2.04 | 7.8 × 10−2 |
| HAUS7 | −2.05 | 1.8 × 10−4 | −1.99 | 3.2 × 10−3 | −1.62 | 3.3 × 10−1 | −2.40 | 3.3 × 10−2 |
| LZTS2 | −2.20 | 7.5 × 10−4 | −0.53 | 5.3 × 10−1 | −3.35 | 2.7 × 10−2 | ||
| EGR1 | −2.29 | 2.4 × 10− | −1.63 | 1.0 × 10−1 | −2.84 | 4.9 × 10−2 | −2.48 | 9.5 × 10−3 |
| GZMH | −2.60 | 3.2 × 10−4 | −2.03 | 5.4 × 10−3 |
| CD4+ | CD8+/NK | Naïve | |
|---|---|---|---|
| TP53 | 4.51 | 3.8 | 3.58 |
| TP63 | 2.96 | 2.44 | 2.58 |
| NFATC2 | 2.35 | 1.11 | 1.91 |
| STAT1 | 2.28 | 0.54 | 2.74 |
| IRF1 | 2.19 | 1.09 | 2.36 |
| TP73 | 2.15 | 2.57 | 1.51 |
| STAT5B | −2.61 | −2.19 | −2.22 |
| STAT5A | −2.14 | −2.19 | −2.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Villanueva, M.; Zhang, Y.; Feiveson, A.; Mistretta, B.; Pan, Y.; Chatterjee, S.; Wu, W.; Clanton, R.; Nelman-Gonzalez, M.; Krieger, S.; et al. Single-Cell RNA-Sequencing Identifies Activation of TP53 and STAT1 Pathways in Human T Lymphocyte Subpopulations in Response to Ex Vivo Radiation Exposure. Int. J. Mol. Sci. 2019, 20, 2316. https://doi.org/10.3390/ijms20092316
Moreno-Villanueva M, Zhang Y, Feiveson A, Mistretta B, Pan Y, Chatterjee S, Wu W, Clanton R, Nelman-Gonzalez M, Krieger S, et al. Single-Cell RNA-Sequencing Identifies Activation of TP53 and STAT1 Pathways in Human T Lymphocyte Subpopulations in Response to Ex Vivo Radiation Exposure. International Journal of Molecular Sciences. 2019; 20(9):2316. https://doi.org/10.3390/ijms20092316
Chicago/Turabian StyleMoreno-Villanueva, Maria, Ye Zhang, Alan Feiveson, Brandon Mistretta, Yinghong Pan, Sujash Chatterjee, Winston Wu, Ryan Clanton, Mayra Nelman-Gonzalez, Stephanie Krieger, and et al. 2019. "Single-Cell RNA-Sequencing Identifies Activation of TP53 and STAT1 Pathways in Human T Lymphocyte Subpopulations in Response to Ex Vivo Radiation Exposure" International Journal of Molecular Sciences 20, no. 9: 2316. https://doi.org/10.3390/ijms20092316
APA StyleMoreno-Villanueva, M., Zhang, Y., Feiveson, A., Mistretta, B., Pan, Y., Chatterjee, S., Wu, W., Clanton, R., Nelman-Gonzalez, M., Krieger, S., Gunaratne, P., Crucian, B., & Wu, H. (2019). Single-Cell RNA-Sequencing Identifies Activation of TP53 and STAT1 Pathways in Human T Lymphocyte Subpopulations in Response to Ex Vivo Radiation Exposure. International Journal of Molecular Sciences, 20(9), 2316. https://doi.org/10.3390/ijms20092316