Hidden Aggregation Hot-Spots on Human Apolipoprotein E: A Structural Study

 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
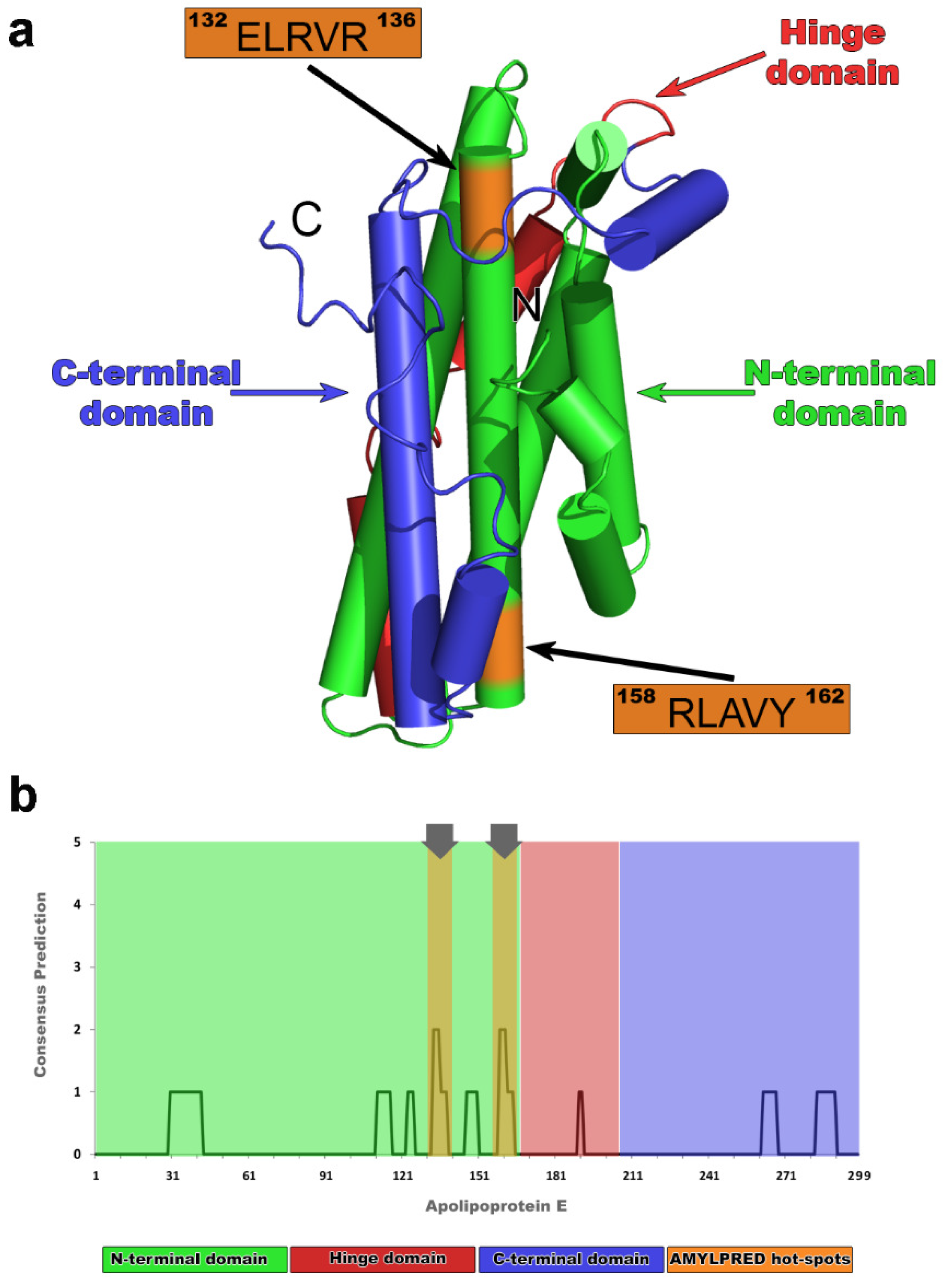
2.1. Computational Identification of apoE Hot-Spots
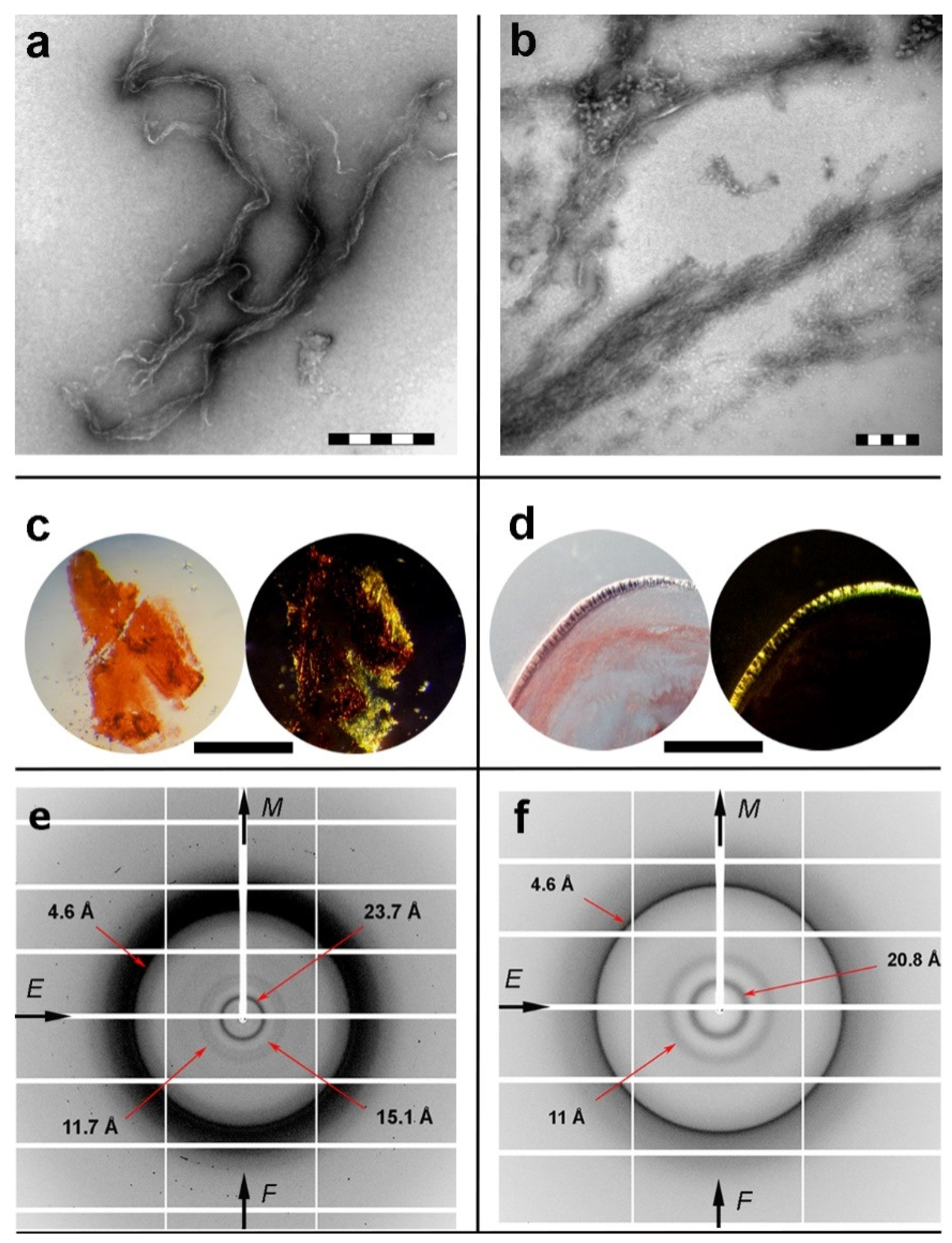
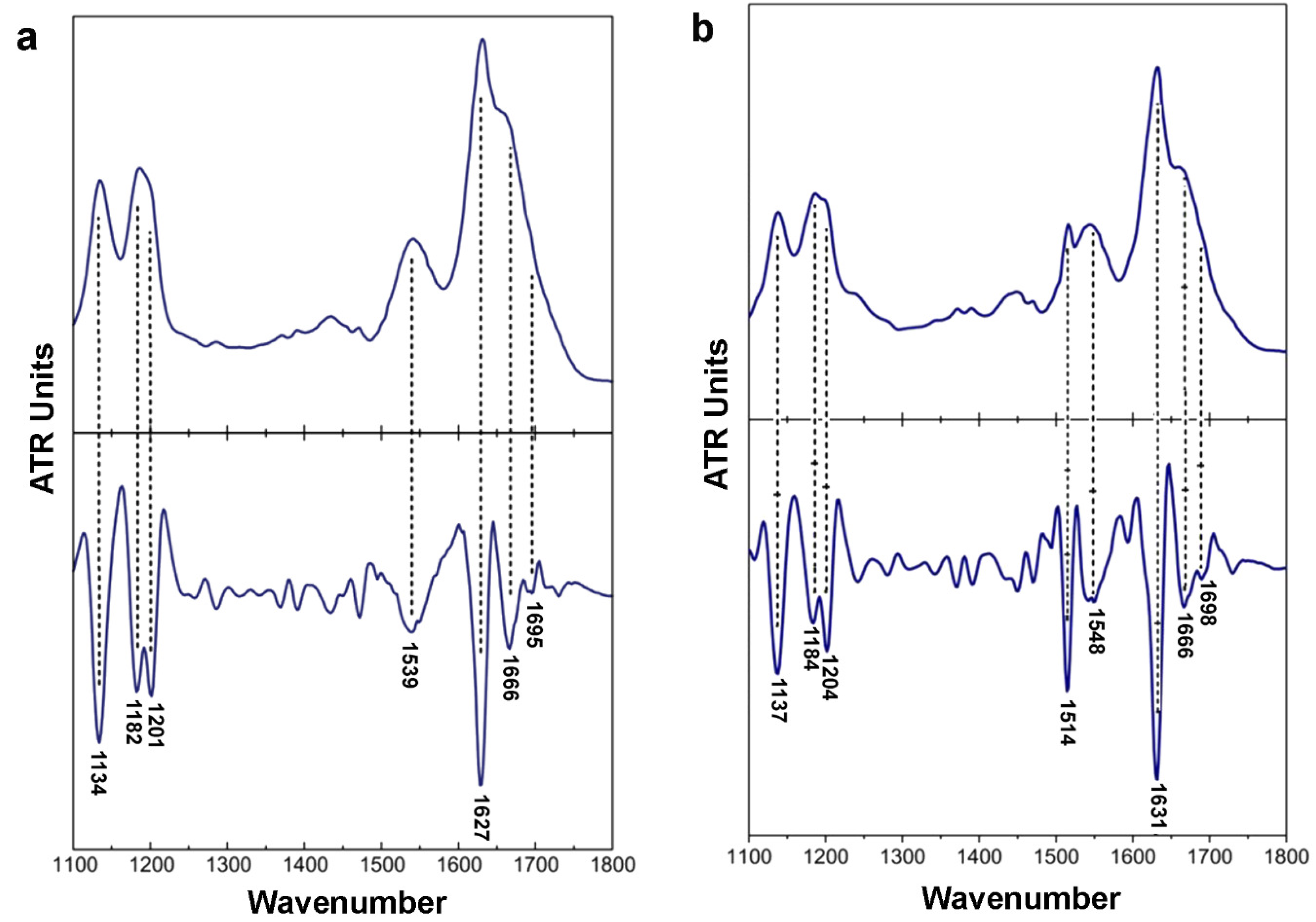
2.2. Isolated apoE Peptide–Analogues Fulfill All Basic Amyloid Criteria
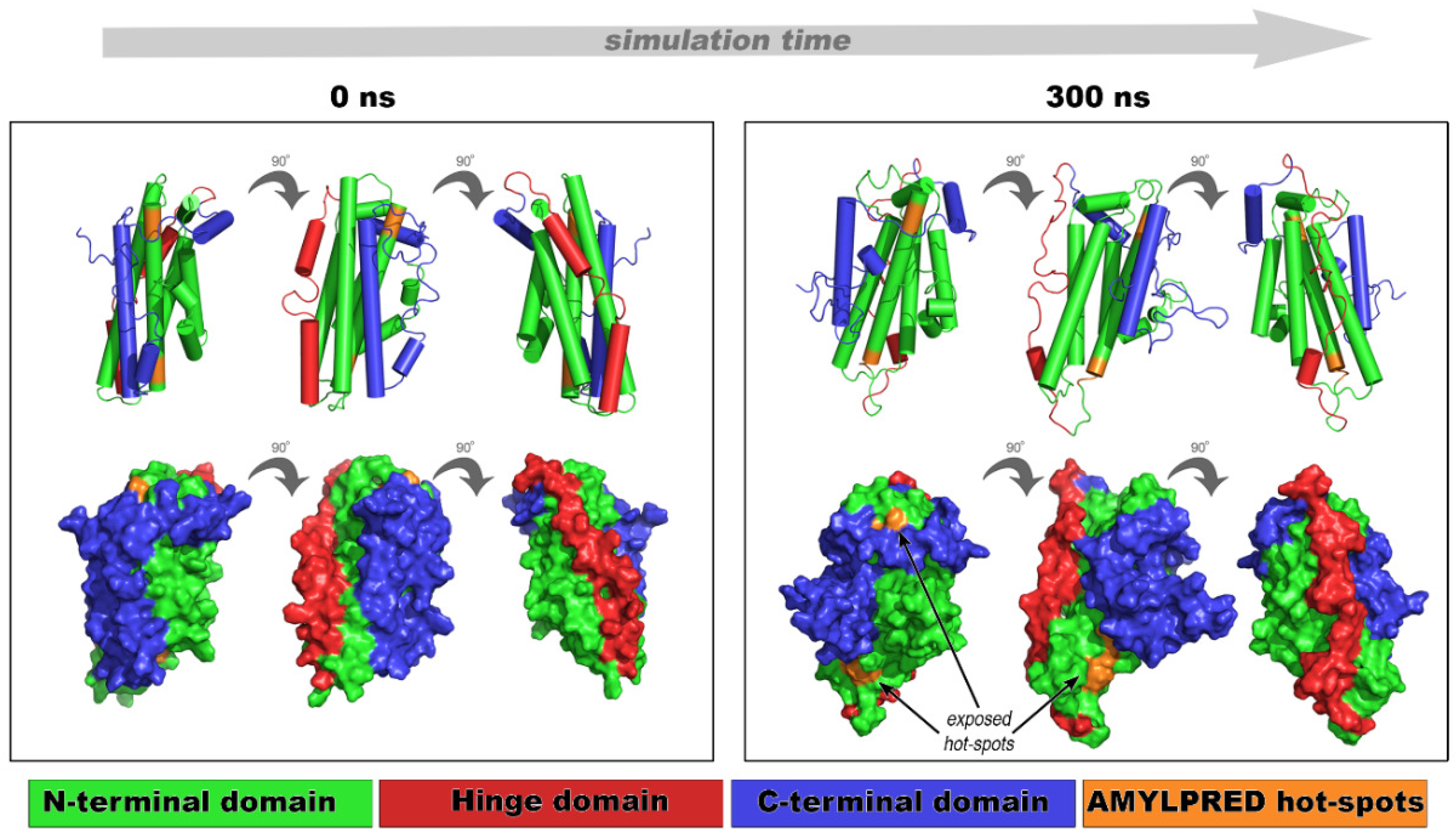
2.3. Implication of apoE Peptide–Analogues in the Tertiary Structural Stability of apoE
2.4. An apoE Aggregation Hot-Spot Anchors Oligomeric Aβ
3. Materials and Methods
3.1. Identification of Aggregation-Prone Peptides in apoE and Αβ
3.2. Peptide Design, Synthesis,and Preparation of Peptide Samples
3.3. X-ray Diffraction
3.4. Negative Staining and Transmission Electron Microscopy
3.5. Attenuated Total Reflectance Fourier-Transform Infrared Spectroscopy (ATR FTIR) and Post-Run Computations of the Spectra
3.6. Congo Red Staining and Polarized Light Microscopy
3.7. Molecular Docking and Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| APP | Amyloid-Beta Precursor Protein |
| apoE | Apolipoprotein E |
| CSF | Cerebrospinal Fluid |
| HDL | High-Density Lipoproteins |
| LDLR | Low-Density Lipoprotein Receptor |
| MD | Molecular Dynamics |
| NMR | Nuclear Magnetic Resonance |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
| TEM | Transmission Electron Microscopy |
| VLDL | Very Low-Density Lipoproteins |
References
- Rall, S.C.; Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E. The complete amino acid sequence. J. Biol. Chem. 1982, 257, 4171–4178. [Google Scholar]
- McLean, J.W.; Elshourbagy, N.A.; Chang, D.J.; Mahley, R.W.; Taylor, J.M. Human apolipoprotein E mRNA. cDNA cloning and nucleotide sequencing of a new variant. J. Biol. Chem. 1984, 259, 6498–6504. [Google Scholar]
- Mahley, R. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 1988, 240, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.-C.; Li, W.-H.; Moore, M.N.; Chan, L. Structure and evolution of the apolipoprotein multigene family. J. Mol. Biol. 1986, 187, 325–340. [Google Scholar] [CrossRef]
- Li, W.H.; Tanimura, M.; Luo, C.C.; Datta, S.; Chan, L. The apolipoprotein multigene family: Biosynthesis, structure, structure-function relationships, and evolution. J. Lipid Res. 1988, 29, 245–271. [Google Scholar] [PubMed]
- Mahley, R.W.; Innerarity, T.L.; Rall, S.C.; Weisgraber, K.H. Plasma lipoproteins: apolipoprotein structure and function. J. Lipid Res. 1984, 25, 1277–1294. [Google Scholar] [PubMed]
- Elshourbagy, N.A.; Liao, W.S.; Mahley, R.W.; Taylor, J.M. Apolipoprotein E mRNA is abundant in the brain and adrenals, as well as in the liver, and is present in other peripheral tissues of rats and marmosets. Proc. Natl. Acad. Sci. USA 1985, 82, 203–207. [Google Scholar] [CrossRef]
- Beisiegel, U.; Weber, W.; Ihrke, G.; Herz, J.; Stanley, K.K. The LDL–receptor–related protein, LRP, is an apolipoprotein E-binding protein. Nat. Cell Biol. 1989, 341, 162–164. [Google Scholar] [CrossRef]
- Takahashi, S.; Kawarabayasi, Y.; Nakai, T.; Sakai, J.; Yamamoto, T. Rabbit very low density lipoprotein receptor: a low density lipoprotein receptor-like protein with distinct ligand specificity. Proc. Natl. Acad. Sci. USA 1992, 89, 9252–9256. [Google Scholar] [CrossRef]
- Willnow, T.E.; Goldstein, J.L.; Orth, K.; Brown, M.S.; Herz, J. Low density lipoprotein receptor-related protein and gp330 bind similar ligands, including plasminogen activator-inhibitor complexes and lactoferrin, an inhibitor of chylomicron remnant clearance. J. Biol. Chem. 1992, 267, 26172–26180. [Google Scholar] [PubMed]
- Kim, D.-H.; Iijima, H.; Goto, K.; Sakai, J.; Ishii, H.; Kim, H.-J.; Suzuki, H.; Kondo, H.; Saeki, S.; Yamamoto, T. Human Apolipoprotein E Receptor 2: A Novel Lipoprotein Receptor of the Low Density Lipoprotein Receptor Family Predominantly Expressed in Brain. J. Biol. Chem. 1996, 271, 8373–8380. [Google Scholar] [CrossRef]
- Das, H.K.; McPherson, J.; Bruns, G.A.; Karathanasis, S.K.; Breslow, J.L. Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J. Biol. Chem. 1985, 260, 6240–6247. [Google Scholar]
- Davison, P.; Norton, P.; Wallis, S.; Gill, L.; Cook, M.; Williamson, R.; Humphries, S. There are two gene sequences for human apolipoprotein CI (apo CI) on chromosome 19, one of which is 4 KB from the gene for apo E. Biochem. Biophys. Commun. 1986, 136, 876–884. [Google Scholar] [CrossRef]
- Scott, J.; Knott, T.J.; Shaw, D.J.; Brook, J.D. Localization of genes encoding apolipoproteins CI, CII, and E to the p13?cen region of human chromosome 19. Hum. Genet. 1985, 71, 144–146. [Google Scholar] [CrossRef]
- Humphries, S.E.; Berg, K.; Gill, L.; Cumming, A.M.; Robertson, F.W.; Stalenhoef, A.F.; Williamson, R.; Børresen, A.L. The gene for apolipoprotein C-ll is closely linked to the gene for apolipoprotein E on chromosome 19. Clin. Genet. 1984, 26, 389–396. [Google Scholar] [CrossRef]
- Myklebost, O.; Rogne, S. A physical map of the apolipoprotein gene cluster on human chromosome 19. Hum. Genet. 1988, 78, 244–247. [Google Scholar] [CrossRef]
- Utermann, G.; Langenbeck, U.; Beisiegel, U.; Weber, W. Genetics of the apolipoprotein E-system in man. Am. J. Hum. Genet. 1980, 32, 339–347. [Google Scholar] [PubMed]
- Zannis, V.I.; Breslow, J.L. Human very low density lipoprotein apolipoprotein E isoprotein polymorphism is explained by genetic variation and posttranslational modification. Biochemistry 1981, 20, 1033–1041. [Google Scholar] [CrossRef]
- Hatters, D.M.; Peters-Libeu, C.A.; Weisgraber, K.H. Apolipoprotein E structure: insights into function. Trends Biochem. Sci. 2006, 31, 445–454. [Google Scholar] [CrossRef]
- Poirier, J.; Bertrand, P.; Poirier, J.; Kogan, S.; Gauthier, S.; Poirier, J.; Gauthier, S.; Davignon, J.; Bouthillier, D.; Davignon, J. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 1993, 342, 697–699. [Google Scholar] [CrossRef]
- Farrer, L.A.; Cupples, L.A.; Haines, J.L.; Hyman, B.; Kukull, W.A.; Mayeux, R.; Myers, R.H.; Pericak-Vance, M.A.; Risch, N.; van Duijn, C.M. Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and alzheimer disease: A meta-analysis. JAMA 1997, 278, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Hauser, P.S.; Ryan, R.O. Impact of apolipoprotein E on Alzheimer’s disease. Curr. Res. 2013, 10, 809–817. [Google Scholar] [CrossRef]
- Mahley, R.W.; Weisgraber, K.H.; Huang, Y. Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5644–5651. [Google Scholar] [CrossRef]
- Blennow, K.; de Leon, M.J.; Zetterberg, H. Alzheimer’s disease. Lancet 2006, 368, 387–403. [Google Scholar] [CrossRef]
- Weisgraber, K.H.; Rall, S.C.; Mahley, R.W. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J. Biol. Chem. 1981, 256, 9077–9083. [Google Scholar] [PubMed]
- Innerarity, T.L.; Pitas, R.E.; Mahley, R.W. Binding of arginine-rich (E) apoprotein after recombination with phospholipid vesicles to the low density lipoprotein receptors of fibroblasts. J. Biol. Chem. 1979, 254, 4186–4190. [Google Scholar] [PubMed]
- Peters-Libeu, C.A.; Newhouse, Y.; Hatters, D.M.; Weisgraber, K.H. Model of Biologically Active Apolipoprotein E Bound to Dipalmitoylphosphatidylcholine. J. Biol. Chem. 2006, 281, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M.; Budamagunta, M.S.; Voss, J.C.; Weisgraber, K.H. Modulation of Apolipoprotein E Structure by Domain Interaction: Differences in Lipid-Bound and lipid-Free Forms. J. Biol. Chem. 2005, 280, 34288–34295. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Szeto, S.S.W.; Ryan, R.O. Lipid association-induced N- and C-terminal domain reorganization in human apolipoprotein E3. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Ryan, R.O. Molecular basis of exchangeable apolipoprotein function. Biochim. Biophys. Acta 2000, 1483, 15–36. [Google Scholar] [CrossRef]
- Peters-Libeu, C.A.; Newhouse, Y.; Hall, S.C.; Witkowska, H.E.; Weisgraber, K.H. Apolipoprotein E*dipalmitoylphosphatidylcholine particles are ellipsoidal in solution. J. Lipid Res. 2007, 48, 1035–1044. [Google Scholar] [CrossRef]
- Patel, A.B.; Khumsupan, P.; Narayanaswami, V. Pyrene Fluorescence Analysis Offers New Insights into the Conformation of the Lipoprotein-Binding Domain of Human Apolipoprotein, E. Biophys. J. 2010, 98, 23a. [Google Scholar] [CrossRef][Green Version]
- Chen, J.; Li, Q.; Wang, J. Topology of human apolipoprotein E3 uniquely regulates its diverse biological functions. Proc. Natl. Acad. Sci. USA 2011, 108, 14813–14818. [Google Scholar] [CrossRef]
- Wilson, C.; Wardell, M.; Weisgraber, K.; Mahley, R.; Agard, D. Three-dimensional structure of the LDL receptor-binding domain of human apolipoprotein E. Science 1991, 252, 1817–1822. [Google Scholar] [CrossRef]
- Wilson, C.; Mau, T.; Weisgraber, K.H.; Wardell, M.R.; Mahley, R.W.; Agard, D.A. Salt bridge relay triggers defective LDL receptor binding by a mutant apolipoprotein. Structure 1994, 2, 713–718. [Google Scholar] [CrossRef]
- Forstner, M.; Peters-Libeu, C.; Contreras-Forrest, E.; Newhouse, Y.; Knapp, M.; Rupp, B.; Weisgraber, K.H. Carboxyl-Terminal Domain of Human Apolipoprotein E: Expression, Purification, and Crystallization. ProteinExpr. Purif. 1999, 17, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Frousios, K.K.; Iconomidou, V.A.; Karletidi, C.-M.; Hamodrakas, S.J. Amyloidogenic determinants are usually not buried. BMC Struct. Biol. 2009, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.I.; Merlini, G.; Saraiva, M.J.; Westermark, P. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef]
- Hatters, D.M.; Howlett, G.J. The structural basis for amyloid formation by plasma apolipoproteins: A review. Eur. Biophys. J. 2002, 31, 2–8. [Google Scholar] [CrossRef]
- Corder, E.; Saunders, A.; Strittmatter, W.; Schmechel, D.; Gaskell, P.; Small, G.; Roses, A.; Haines, J.; Pericak-Vance, M. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef]
- Roses, A.D. Apolipoprotein E genotyping in the differential diagnosis, not prediction, of Alzheimer’s disease. Ann. Neurol. 1995, 38, 6–14. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef]
- Han, S.H.; Einstein, G.; Weisgraber, K.H.; Strittmatter, W.J.; Saunders, A.M.; Pericak-Vance, M.; Roses, A.D.; Schmechel, D.E. Apolipoprotein E is localized to the cytoplasm of human cortical neurons: A light and electron microscopic study. J. Neuropathol. Exp. Neurol. 1994, 53, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Diedrich, J.F.; Minnigan, H.; Carp, R.I.; Whitaker, I.N.; Race, R.; Frey, W.; Hazse, A.T. Neuropathological changes in scrapIe and AlzheImer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J. Virol. 1991, 65, 4759–4768. [Google Scholar]
- Esteras-Chopo, A.; Serrano, L.; de la Paz, M.L. The amyloid stretch hypothesis: Recruiting proteins toward the dark side. Proc. Natl. Acad. Sci. USA 2005, 102, 16672–16677. [Google Scholar] [CrossRef]
- Iconomidou, V.A.; Pheida, D.; Hamodraka, E.S.; Antony, C.; Hoenger, A.; Hamodrakas, S.J. An amyloidogenic determinant in N-terminal pro-brain natriuretic peptide (NT-proBNP): Implications for cardiac amyloidoses. Biopolymers 2012, 98, 67–75. [Google Scholar] [CrossRef]
- Iconomidou, V.A.; Leontis, A.; Hoenger, A.; Hamodrakas, S.J. Identification of a novel ‘aggregation-prone’/‘amyloidogenic determinant’ peptide in the sequence of the highly amyloidogenic human calcitonin. FEBS Lett. 2013, 587, 569–574. [Google Scholar] [CrossRef]
- Louros, N.N.; Iconomidou, V.A.; Tsiolaki, P.L.; Chrysina, E.D.; Baltatzis, G.E.; Patsouris, E.S.; Hamodrakas, S.J. An N-terminal pro-atrial natriuretic peptide (NT-proANP) ’aggregation-prone’ segment involved in isolated atrial amyloidosis. FEBS Lett. 2014, 588, 52–57. [Google Scholar] [CrossRef]
- Teng, P.K.; Eisenberg, D. Short protein segments can drive a non-fibrillizing protein into the amyloid state. Protein Eng. Sel. 2009, 22, 531–536. [Google Scholar] [CrossRef]
- Tenidis, K.; Waldner, M.; Bernhagen, J.; Fischle, W.; Bergmann, M.; Weber, M.; Merkle, M.-L.; Voelter, W.; Brunner, H.; Kapurniotu, A. Identification of a penta- and hexapeptide of islet amyloid polypeptide (IAPP) with amyloidogenic and cytotoxic properties11Edited by R. Huber. J. Mol. Biol. 2000, 295, 1055–1071. [Google Scholar] [CrossRef]
- López de la Paz, M.; Serrano, L. Sequence determinants of amyloid fibril formation. Proc. Natl. Acad. Sci. USA 2004, 101, 87–92. [Google Scholar] [CrossRef]
- Tsiolaki, P.L.; Louros, N.N.; Hamodrakas, S.J.; Iconomidou, V.A. Exploring the ‘aggregation-prone’ core of human Cystatin C: A structural study. J. Struct. Biol. 2015, 191, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Louros, N.; Tsiolaki, P.L.; Zompra, A.A.; Pappa, E.V.; Magafa, V.; Pairas, G.; Cordopatis, P.; Cheimonidou, C.; Trougakos, I.P.; Iconomidou, V.A.; et al. Structural studies and cytotoxicity assays of “aggregation-prone” IAPP 8-16 and its non-amyloidogenic variants suggest its important role in fibrillogenesis and cytotoxicity of human amylin. Biopolymers 2015, 104, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Conchillo-Solé, O.; De Groot, N.S.; Aviles, F.X.; Vendrell, J.; Daura, X.; Ventura, S. AGGRESCAN: A server for the prediction and evaluation of "hot spots" of aggregation in polypeptides. BMC Bioinform. 2007, 8, 65. [Google Scholar] [CrossRef]
- Das, M.; Gursky, O. Amyloid-Forming Properties of Human Apolipoproteins: Sequence Analyses and Structural Insights. Adv. Exp. Med. Biol. 2015, 855, 175–211. [Google Scholar] [PubMed]
- Louros, N.N.; Tsiolaki, P.L.; Griffin, M.D.; Howlett, G.J.; Hamodrakas, S.J.; Iconomidou, V.A. Chameleon ‘aggregation-prone’ segments of apoA-I: A model of amyloid fibrils formed in apoA-I amyloidosis. Int. J. Biol. Macromol. 2015, 79, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Scharnagl, H.; Tisljar, U.; Hoschützky, H.; Friedrich, I.; Hoffmann, M.M.; Hüttinger, M.; Wieland, H.; März, W. Competition of Abeta amyloid peptide and apolipoprotein E for receptor-mediated endocytosis. J. Lipid Res. 1999, 40, 447–455. [Google Scholar] [PubMed]
- Wisniewski, T.; Lalowski, M.; Golabek, A.; Frangione, B.; Vogel, T. Is Alzheimer’s disease an apolipoprotein E amyloidosis? Lancet 1995, 345, 956–958. [Google Scholar] [CrossRef]
- Weisgraber, K.H. Apolipoprotein E distribution among human plasma lipoproteins: Role of the cysteine-arginine interchange at residue 112. J. Lipid Res. 1990, 31, 1503–1511. [Google Scholar]
- Azriel, R.; Gazit, E. Analysis of the Minimal Amyloid-forming Fragment of the Islet Amyloid Polypeptide: An Experimental Support for The Key Role of The Phenylalanine Residue in Amyloid Formation. J. Biol. Chem. 2001, 276, 34156–34161. [Google Scholar] [CrossRef]
- Close, W.; Neumann, M.; Schmidt, A.; Hora, M.; Annamalai, K.; Schmidt, M.; Reif, B.; Schmidt, V.; Grigorieff, N.; Fändrich, M. Physical basis of amyloid fibril polymorphism. Nat. Commun. 2018, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, M.R.; Sambashivan, S.; Nelson, R.; Ivanova, M.I.; Sievers, S.A.; Apostol, M.I.; Thompson, M.J.; Balbirnie, M.; Wiltzius, J.J.; McFarlane, H.T.; et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature 2007, 447, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Hatters, D.M.; Zhong, N.; Rutenber, E.; Weisgraber, K.H. Amino-terminal Domain Stability Mediates Apolipoprotein E Aggregation into Neurotoxic Fibrils. J. Mol. Biol. 2006, 361, 932–944. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Wisniewski, T.; Frangione, B. Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 1992, 135, 235–238. [Google Scholar] [CrossRef]
- Choi-Miura, N.H.; Takahashi, Y.; Nakano, Y.; Tobe, T.; Tomita, M. Identification of the Disulfide Bonds in Human Plasma Protein SP-40,40 (Apolipoprotein-J)1. J. Econ. Èntomol. 1992, 112, 557–561. [Google Scholar] [CrossRef]
- Ma, J.; Yee, A.; Brewer, H.B.; Das, S.; Potter, H. Amyloid-associated proteins [alpha]1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer [beta]-protein into filaments. Nature 1994, 372, 92–94. [Google Scholar] [CrossRef]
- Sanan, D.A.; Weisgraber, K.H.; Russell, S.J.; Mahley, R.W.; Huang, D.; Saunders, A.; Schmechel, D.; Wisniewski, T.; Frangione, B.; Roses, A.D. Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Investig. 1994, 94, 860–869. [Google Scholar] [CrossRef]
- Wisniewski, T.; Castaño, E.M.; Golabek, A.; Vogel, T.; Frangione, B. Acceleration of Alzheimer’s fibril formation by apolipoprotein E in vitro. Am. J. Pathol. 1994, 145, 1030–1035. [Google Scholar]
- Evans, K.C.; Berger, E.P.; Cho, C.G.; Weisgraber, K.H.; Lansbury, P.T. Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: Implications for the pathogenesis and treatment of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1995, 92, 763–767. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef]
- Luhrs, T.; Ritter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Breydo, L.; Uversky, V.N. Structural, morphological, and functional diversity of amyloid oligomers. FEBS Lett. 2015, 589, 2640–2648. [Google Scholar] [CrossRef]
- Pechmann, S.; Tartaglia, G.G.; Vendruscolo, M.; Levy, E.D. Physicochemical principles that regulate the competition between functional and dysfunctional association of proteins. Proc. Natl. Acad. Sci. USA 2009, 106, 10159–10164. [Google Scholar] [CrossRef]
- Castillo, V.; Ventura, S. Amyloidogenic Regions and Interaction Surfaces Overlap in Globular Proteins Related to Conformational Diseases. PLoS Comput. Biol. 2009, 5, e1000476. [Google Scholar] [CrossRef]
- CrysAlisPRO, Agilent Technologies. Software System; Agilent Technologies UK Ltd.: Oxford, UK, 2012. [Google Scholar]
- CrysAlisPRO Agilent Technologies. Version 1.171.37.31; Agilent Technologies UK Ltd.: Oxford, UK, 2014. [Google Scholar]
- Leslie, A.G.W.; Powell, H.R. Processing Diffraction Data with Mosflm; Springer: Dordrecht, The Netherlands, 2007; pp. 41–51. [Google Scholar]
- Savitzky, A.; Golay, M.J.E. Smoothing and Differentiation of Data by Simplified Least Squares Procedures. Anal. Chem. 1964, 36, 1627–1639. [Google Scholar] [CrossRef]
- Van Zundert, G.; Rodrigues, J.; Trellet, M.; Schmitz, C.; Kastritis, P.; Karaca, E.; Melquiond, A.; Van Dijk, M.; De Vries, S.; Bonvin, A.M.; et al. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef]
- Chandrasekhar, J.; Impey, R.W.; Jorgensen, W.L.; Madura, J.D.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 19–25. [Google Scholar] [CrossRef]
- Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E.; Lindorff-Larsen, K.; Lindorff-Larsen, K.; Lindorff-Larsen, K. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins: Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; DiNola, A.; Haak, J.R.; Van Gunsteren, W.F. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Physical A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Gr. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; DeLano Scientific: San Carlos, CA, USA, 2015. [Google Scholar]
- Wisniewski, T.; Golabek, A.A.; Kida, E.; Wisniewski, K.E.; Frangione, B. Conformational mimicry in Alzheimer’s disease. Role of apolipoproteins in amyloidogenesis. Am. J. Pathol. 1995, 147, 238–244. [Google Scholar]
- Nichols, W.C.; Dwulet, F.E.; Liepnieks, J.; Benson, M.D. Variant apolipoprotein AI as a major constituent of a human hereditary amyloid. Biochem. Biophys. Commun. 1988, 156, 762–768. [Google Scholar] [CrossRef]
- Murphy, C.L.; Wang, S.; Weaver, K.; Gertz, M.A.; Weiss, D.T.; Solomon, A. Renal apolipoprotein A-I amyloidosis associated with a novel mutant Leu64Pro. Am. J. Kidney Dis. 2004, 44, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Soutar, A.K.; Hawkins, P.N.; Vigushin, D.M.; Tennent, G.A.; Booth, S.E.; Hutton, T.; Nguyen, O.; Totty, N.F.; Feest, T.G.; Hsuan, J.J. Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis. Proc. Natl. Acad. Sci. USA 1992, 89, 7389–7393. [Google Scholar] [CrossRef]
- Johnson, K.H.; Sletten, K.; Hayden, D.W.; O’Brien, T.D.; Roertgen, K.E.; Westermark, P. Pulmonary vascular amyloidosis in aged dogs. A new form of spontaneously occurring amyloidosis derived from apolipoprotein AI. Am. J. Pathol. 1992, 141, 1013–1019. [Google Scholar]
- Benson, M.D.; Liepnieks, J.J.; Yazaki, M.; Yamashita, T.; Asl, K.H.; Guenther, B.; Kluve-Beckerman, B. A New Human Hereditary Amyloidosis: The Result of a Stop-Codon Mutation in the Apolipoprotein AII Gene. Genomics 2001, 72, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, K.; Kitagawa, K.; Naiki, H.; Hanada, K.; Hosokawa, M.; Takeda, T. Polymorphism of apolipoprotein A-II (apoA-II) among inbred strains of mice. Relationship between the molecular type of apoA-II and mouse senile amyloidosis. Biochem. J. 1991, 279, 427–433. [Google Scholar] [CrossRef]
- Bergstrom, J.; Murphy, C.L.; Weiss, D.T.; Solomon, A.; Sletten, K.; Hellman, U.; Westermark, P. Two different types of amyloid deposits[mdash]apolipoprotein A-IV and transthyretin[mdash]in a patient with systemic amyloidosis. Lab Investig. 2004, 84, 981–988. [Google Scholar] [CrossRef]
- Sethi, S.; Theis, J.D.; Shiller, S.M.; Nast, C.C.; Harrison, D.; Rennke, H.G.; Vrana, J.A.; Dogan, A. Medullary amyloidosis associated with apolipoprotein A-IV deposition. Kidney Int. 2012, 81, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Bois, M.C.; Dasari, S.; Mills, J.R.; Theis, J.; Highsmith, W.E.; Vrana, J.A.; Grogan, M.; Dispenzieri, A.; Kurtin, P.J.; Maleszewski, J.J. Apolipoprotein A-IV–Associated Cardiac Amyloidosis. J. Am. Cardiol. 2017, 69, 2248–2249. [Google Scholar] [CrossRef]
- Hatters, D.M.; Macphee, C.E.; Lawrence, L.J.; Sawyer, W.H.; Howlett, G.J. Human Apolipoprotein C-II Forms Twisted Amyloid Ribbons and Closed Loops. Biochemistry 2000, 39, 8276–8283. [Google Scholar] [CrossRef] [PubMed]
- De Messieres, M.; Huang, R.K.; He, Y.; Lee, J.C. Amyloid Triangles, Squares, and Loops of Apolipoprotein C-III. Biochemistry 2014, 53, 3261–3263. [Google Scholar] [CrossRef] [PubMed]
- Nasr, S.H.; Dasari, S.; Hasadsri, L.; Theis, J.D.; Vrana, J.A.; Gertz, M.A.; Muppa, P.; Zimmermann, M.T.; Grogg, K.L.; Dispenzieri, A.; et al. Novel Type of Renal Amyloidosis Derived from Apolipoprotein-CII. J. Am. Soc. Nephrol. 2017, 28, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Valleix, S.; Verona, G.; Jourde-Chiche, N.; Nedelec, B.; Mangione, P.P.; Bridoux, F.; Mangé, A.; Doğan, A.; Goujon, J.-M.; Lhomme, M.; et al. D25V apolipoprotein C-III variant causes dominant hereditary systemic amyloidosis and confers cardiovascular protective lipoprotein profile. Nat. Commun. 2016, 7, 10353. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Blake, C. The Structure of Amyloid Fibrils by Electron Microscopy and X-ray Diffraction. In Advances in Protein Chemistry; Elsevier BV: Amsterdam, The Netherlands, 1997; Volume 50, pp. 123–159. [Google Scholar]
- Sunde, M.; Serpell, L.C.; Bartlam, M.; Fraser, P.E.; Pepys, M.B.; Blake, C.C.F. Common core structure of amyloid fibrils by synchrotron X-ray diffraction11Edited by F. E. Cohen. J. Mol. Biol. 1997, 273, 729–739. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber (cm−1) | Assignment | |
|---|---|---|
| 132ERLVR136 | 158RLAVY162 | |
| 1134 | 1137 | TFA |
| 1182 | 1184 | TFA |
| 1201 | 1201 | TFA |
| - | 1514 | Tyrosine |
| 1539 | 1548 | β-sheet (Amide II) |
| 1627 | 1631 | β-sheet (Amide I) |
| 1666 | 1666 | TFA |
| 1695 | 1689 | Antiparallel β-sheets |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsiolaki, P.L.; Katsafana, A.D.; Baltoumas, F.A.; Louros, N.N.; Iconomidou, V.A. Hidden Aggregation Hot-Spots on Human Apolipoprotein E: A Structural Study. Int. J. Mol. Sci. 2019, 20, 2274. https://doi.org/10.3390/ijms20092274
Tsiolaki PL, Katsafana AD, Baltoumas FA, Louros NN, Iconomidou VA. Hidden Aggregation Hot-Spots on Human Apolipoprotein E: A Structural Study. International Journal of Molecular Sciences. 2019; 20(9):2274. https://doi.org/10.3390/ijms20092274
Chicago/Turabian StyleTsiolaki, Paraskevi L., Aikaterini D. Katsafana, Fotis A. Baltoumas, Nikolaos N. Louros, and Vassiliki A. Iconomidou. 2019. "Hidden Aggregation Hot-Spots on Human Apolipoprotein E: A Structural Study" International Journal of Molecular Sciences 20, no. 9: 2274. https://doi.org/10.3390/ijms20092274
APA StyleTsiolaki, P. L., Katsafana, A. D., Baltoumas, F. A., Louros, N. N., & Iconomidou, V. A. (2019). Hidden Aggregation Hot-Spots on Human Apolipoprotein E: A Structural Study. International Journal of Molecular Sciences, 20(9), 2274. https://doi.org/10.3390/ijms20092274