Accumulation of Innate Amyloid Beta Peptide in Glioblastoma Tumors

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
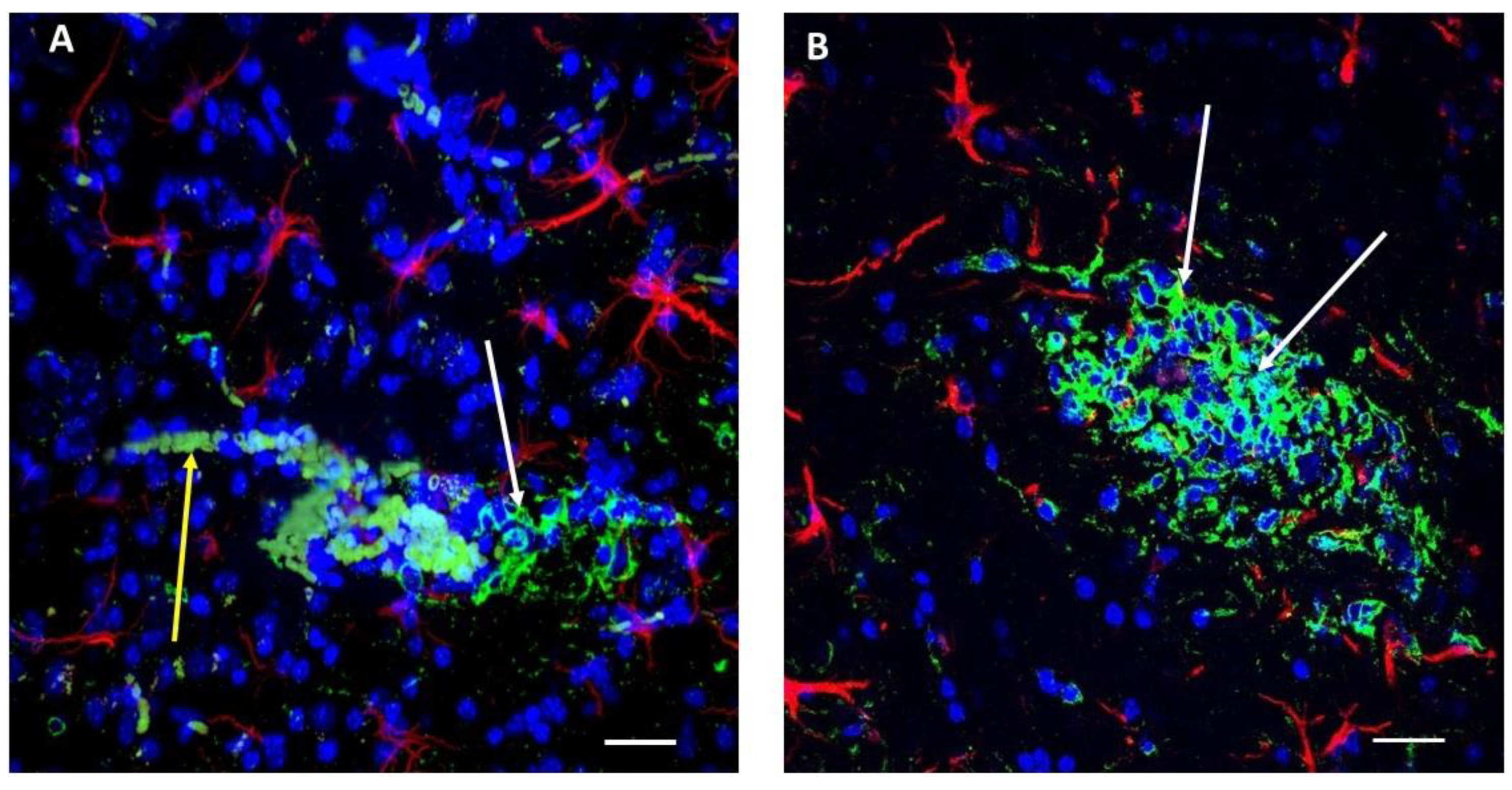
2.1. Immunoreactivity against Aβ Peptides Is Present in Glioma Cells in Primary and Secondary Tumors as Well as in Blood Vessels and Erythrocytes in the Near Vicinity, Indicating that the Aβ Level Is Elevated in the Tumor Zone
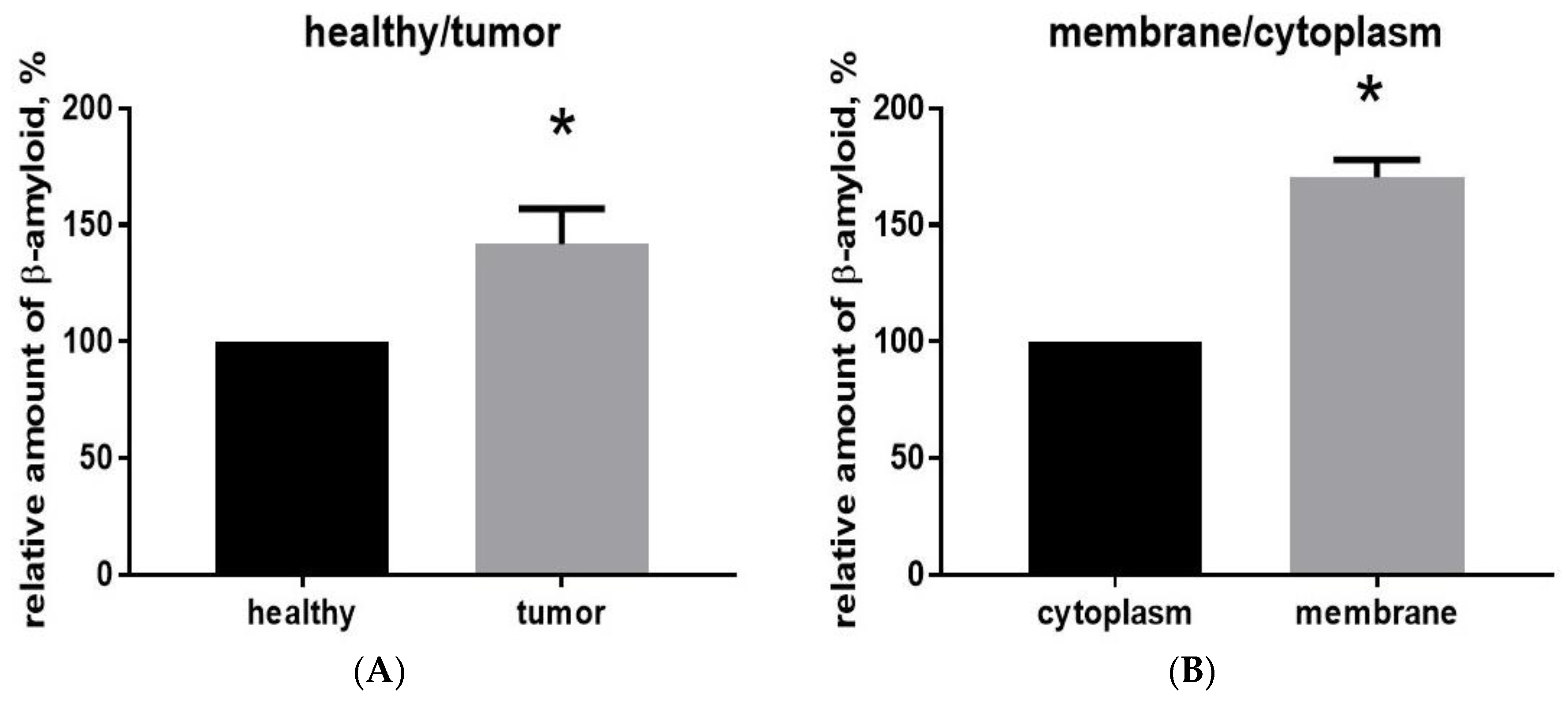
2.2. Aβ40 Is Concentrated in the Membrane Cell Fraction in Glioma Tumor Tissue
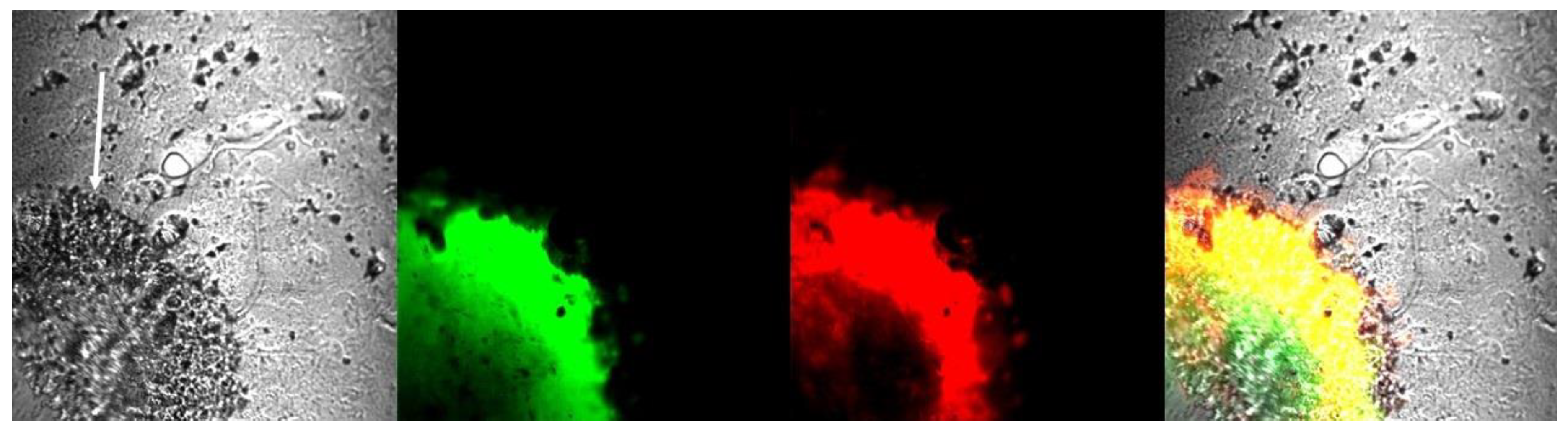
2.3. Glioma Tumor Tissue Contains Aggregated Amyloid
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Glioma Cell Culture
4.3. Intracranial Implantation of Glioma Cells
4.4. Percoll Purification of Blood Cells from Tissue Samples for Membrane Fraction Isolation
4.5. Isolation of Membrane and Cytoplasmic Proteins
4.6. Enzyme-Linked Immunosorbent Assay (ELISA) Measurements
4.7. Immunohistochemistry and Confocal Microscopy
4.8. Statistics and Measurements
5. Conclusions
- Aggregated amyloid is present inside glioma tumor borders;
- Aβ peptide immunofluorescence is present in glioma tumors, marking glioma cells and nearby ruptured blood vessels.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ELISA | enzyme-linked immunosorbent assay |
| ThS | Thioflavin S |
| ThT | Thioflavin T |
| PMSF | Phenylmethylsulfonyl fluoride |
| DTT | Dithiothreitol |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| Aβ | Amyloid beta peptide |
References
- Guerreiro, R.; Bras, J. The age factor in Alzheimer’s disease. Genome Med. 2015, 7, 106. [Google Scholar] [CrossRef] [PubMed]
- Young, J.S.; Chmura, S.J.; Wainwright, D.A.; Yamini, B.; Peters, K.B.; Lukas, R.V. Management of glioblastoma in elderly patients. J. Neurol. Sci. 2017, 380, 250–255. [Google Scholar] [CrossRef]
- Ou, S.M.; Lee, Y.J.; Hu, Y.W.; Liu, C.J.; Chen, T.J.; Fuh, J.L.; Wang, S.J. Does Alzheimer’s disease protect against cancers? A nationwide population-based study. Neuroepidemiology 2013, 40, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Musicco, M.; Adorni, F.; Di Santo, S.; Prinelli, F.; Pettenati, C.; Caltagirone, C.; Palmer, K.; Russo, A. Inverse occurrence of cancer and Alzheimer disease: A population-based incidence study. Neurology 2013, 81, 322–328. [Google Scholar] [CrossRef]
- Yarchoan, M.; James, B.D.; Shah, R.C.; Arvanitakis, Z.; Wilson, R.S.; Schneider, J.; Bennett, D.A.; Arnold, S.E. Association of Cancer History with Alzheimer’s Disease Dementia and Neuropathology. J. Alzheimers Dis. 2017, 56, 699–706. [Google Scholar] [CrossRef]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S. Glioblastoma and dementia may share a common cause. Med. Hypotheses 2010, 75, 67–68. [Google Scholar] [CrossRef]
- Lehrer, S. Glioma and Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2018, 2, 213–218. [Google Scholar] [CrossRef]
- Behrens, M.I.; Lendon, C.; Roe, C.M. A common biological mechanism in cancer and Alzheimer’s disease? Curr. Alzheimer Res. 2009, 6, 196–204. [Google Scholar] [CrossRef]
- Sánchez-Valle, J.; Tejero, H.; Ibáñez, K.; Portero, J.L.; Krallinger, M.; Al-Shahrour, F.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. A molecular hypothesis to explain direct and inverse co-morbidities between Alzheimer’s Disease, Glioblastoma and Lung cancer. Sci. Rep. 2017, 7, 4474. [Google Scholar] [CrossRef]
- Hansel, D.E.; Rahman, A.; Wehner, S.; Herzog, V.; Yeo, C.J.; Maitra, A. Increased expression and processing of the Alzheimer amyloid precursor protein in pancreatic cancer may influence cellular proliferation. Cancer Res. 2003, 63, 7032–7037. [Google Scholar] [PubMed]
- Tsang, J.Y.S.; Lee, M.A.; Ni, Y.B.; Chan, S.K.; Cheung, S.Y.; Chan, W.W.; Lau, K.F.; Tse, G.M.K. Amyloid Precursor Protein Is Associated with Aggressive Behavior in Nonluminal Breast Cancers. Oncologist 2018, 23, 1273–1281. [Google Scholar] [CrossRef] [PubMed]
- Tsang, J.Y.S.; Lee, M.A.; Chan, T.H.; Li, J.; Ni, Y.B.; Shao, Y.; Chan, S.K.; Cheungc, S.Y.; Lau, K.F.; Tse, G.M.K. Proteolytic cleavage of amyloid precursor protein by ADAM10 mediates proliferation and migration in breast cancer. EBioMedicine 2018, 38, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.S.; Bu, X.L.; Liu, Y.H.; Shen, L.L.; Zhuang, Z.Q.; Jiao, S.S.; Zhu, C.; Wang, Q.H.; Zhou, H.D.; Zhang, T.; et al. Plasma Amyloid-Beta Levels in Patients with Different Types of Cancer. Neurotox. Res. 2017, 31, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Morato, E.; Mayor, F., Jr. Production of the Alzheimer’s beta-amyloid peptide by C6 glioma cells. FEBS Lett. 1993, 336, 275–278. [Google Scholar] [CrossRef]
- Murphy, S.F.; Banasiak, M.; Yee, G.-T.; Wotoczek-Obadia, M.; Tran, Y.; Prak, A.; Albright, R.; Mullan, M.; Paris, D.; Brem, S. A synthetic fragment of beta-amyloid peptide suppresses glioma proliferation, angiogenesis, and invasiveness in vivo and in vitro. Neuro-Oncol. 2010, 12, iv5. [Google Scholar] [CrossRef]
- Paris, D. Modulation of Angiogenesis by a-Beta Peptide Fragments. Patent US20080031954A1, 7 February 2005. [Google Scholar]
- Paris, D.; Gan ey, N.; Banasiak, M.; Laporte, V.; Patel, N.; Mullan, M.; Murphy, S.F.; Yee, G.T.; Bachmeier, C.; Ganey, C.; et al. Impaired orthotopic glioma growth and vascularization in transgenic mouse models of Alzheimer’s disease. J. Neurosci. 2010, 30, 11251–11258. [Google Scholar] [CrossRef]
- Inyushin, M.Y.; Sanabria, P.; Rojas, L.; Kucheryavykh, Y.; Kucheryavykh, L. Aβ Peptide Originated from Platelets Promises New Strategy in Anti-Alzheimer’s Drug Development. Biomed. Res. Int. 2017, 2017, 3948360. [Google Scholar] [CrossRef]
- Inyushin, M.; Zayas-Santiago, A.; Rojas, L.; Kucheryavykh, Y.; Kucheryavykh, L. Platelet-generated amyloid beta peptides in Alzheimer’s disease and glaucoma. Histol. Histopathol. 2019, 18111. [Google Scholar] [CrossRef]
- Kucheryavykh, L.Y.; Dávila-Rodríguez, J.; Rivera-Aponte, D.E.; Zueva, L.V.; Washington, A.V.; Sanabria, P.; Inyushin, M.Y. Platelets are responsible for the accumulation of β-amyloid in blood clots inside and around blood vessels in mouse brain after thrombosis. Brain Res. Bull. 2017, 128, 98–105. [Google Scholar] [CrossRef]
- Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Washington, A.V.; Inyushin, M.Y. Amyloid Beta Peptide Is Released during Thrombosis in the Skin. Int. J. Mol. Sci. 2018, 19, 1705. [Google Scholar] [CrossRef]
- Jurasz, P.; Alonso-Escolano, D.; Radomski, M.W. Platelet–cancer interactions: Mechanisms and pharmacology of tumour cell-induced platelet aggregation. Br. J. Pharm. 2004, 143, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Goubran, H.A.; Burnouf, T.; Radosevic, M.; El-Ekiaby, M. The platelet-cancer loop. Eur. J. Intern. Med. 2013, 24, 393–400. [Google Scholar] [CrossRef]
- Hermanson, M.; Funa, K.; Hartman, M.; Claesson-Welsh, L.; Heldin, C.H.; Westermark, B.; Nistér, M. Platelet-derived growth factor and its receptors in human glioma tissue: Expression of messenger RNA and protein suggests the presence of autocrine and paracrine loops. Cancer Res. 1992, 52, 3213–3219. [Google Scholar]
- Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Rolón-Reyes, K.; Skatchkov, S.N.; Eaton, M.J.; Cubano, L.A.; Inyushin, M.Y. Visualization of implanted GL261 glioma cells in living mouse brain slices using fluorescent 4-(4-(dimethylamino)-styryl)-N-methylpyridinium iodide (ASP+). Biotechniques 2012. [Google Scholar] [CrossRef] [PubMed]
- Rolón-Reyes, K.; Kucheryavykh, Y.V.; Cubano, L.A.; Inyushin, M.; Skatchkov, S.N.; Eaton, M.J.; Harrison, J.K.; Kucheryavykh, L.Y. Microglia Activate Migration of Glioma Cells through a Pyk2 Intracellular Pathway. PLoS ONE 2015, 10, e0131059. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Liu, J.; Zhao, Z.; Xue, R.; Zhang, N.; Zhang, P.; Zhao, P.; Zheng, F.; Sun, X. The peripheral blood of Aβ binding RBC as a biomarker for diagnosis of Alzheimer’s disease. Age Ageing 2015, 44, 458–464. [Google Scholar] [CrossRef]
- Mohanty, J.G.; Eckley, D.M.; Williamson, J.D.; Launer, L.J.; Rifkind, J.M. Do red blood cell-β-amyloid interactions alter oxygen delivery in Alzheimer’s disease? Adv. Exp. Med. Biol. 2008, 614, 29–35. [Google Scholar]
- Kelényi, G. Thioflavin S fluorescent and Congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol. 1967, 7, 336–348. [Google Scholar] [CrossRef]
- Biancalana, M.; Koide, S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 2010, 1804, 1405–1412. [Google Scholar] [CrossRef]
- Wu, Y.; Du, S.; Johnson, J.L.; Tung, H.Y.; Landers, C.T.; Liu, Y.; Seman, B.G.; Wheeler, R.T.; Costa-Mattioli, M.; Kheradmand, F.; et al. Microglia and amyloid precursor protein coordinate control of transient Candida cerebritis with memory deficits. Nat. Commun. 2019, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Brody, A.H.; Strittmatter, S.M. Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease Through Prion Protein and mGluR5. Adv. Pharm. 2018, 82, 293–323. [Google Scholar] [CrossRef]
- Salazar, S.V.; Cox, T.O.; Lee, S.; Brody, A.H.; Chyung, A.S.; Haas, L.T.; Strittmatter, S.M. Alzheimer’s Disease Risk Factor Pyk2 Mediates Amyloid-β-Induced Synaptic Dysfunction and Loss. J. Neurosci. 2019, 39, 758–772. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Tran, N.L.; Menashi, E.; Rohl, C.; Kloss, J.; Bay, R.C.; Berens, M.E.; Loftus, J.C. The tyrosine kinase pyk2 promotes migration and invasion of glioma cells. Neoplasia 2005, 7, 435–445. [Google Scholar] [CrossRef]
- Fan, Q.W.; Weiss, W.A. Targeting the RTK-PI3K-mTOR axis in malignant glioma: Overcoming resistance. Curr. Top. Microbiol. Immunol. 2010, 347, 279–296. [Google Scholar] [CrossRef]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef] [PubMed]
- Klippel, A.; Reinhard, C.; Kavanaugh, W.M.; Apell, G.; Escobedo, M.A.; Williams, L.T. Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol. Cell. Biol. 1996, 16, 4117–4127. [Google Scholar] [CrossRef]
- Gao, X.; Lowry, P.R.; Zhou, X.; Depry, C.; Wei, Z.; Wong, G.W.; Zhang, J. PI3K/Akt signaling requires spatial compartmentalization in plasma membrane microdomains. Proc. Natl. Acad. Sci. USA 2011, 108, 14509–14514. [Google Scholar] [CrossRef]
- Chen, T.J.; Wang, D.C.; Chen, S.S. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J. Neurosci. Res. 2009, 87, 2297–2307. [Google Scholar] [CrossRef]
- Mruthinti, S.; Hill, W.D.; Swamy-Mruthinti, S.; Buccafusco, J.J. Relationship between the induction of RAGE cell-surface antigen and the expression of amyloid binding sites. J. Mol. Neurosci. 2003, 20, 223–232. [Google Scholar] [CrossRef]
- Deane, R.; Du Yan, S.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Fuentes, M.K.; Huang, E.H.; Arumugam, T. RAGE and RAGE ligands in cancer. Curr. Mol. Med. 2007, 7, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Olabarria, M.; Noristani, H.N.; Yeh, C.-Y.; Rodriguez, J.J. Astrocytes in Alzheimer’s Disease. Neurotherapeutics 2010, 7, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Veeraraghavalu, K.; Zhang, C.; Zhang, X.; Tanzi, R.E.; Sisodia, S.S. Age-dependent non-cell-autonomous deposition of amyloid from synthesis of β-amyloid by cells other than excitatory neurons. J. Neurosci. 2014, 34, 3668–3673. [Google Scholar] [CrossRef]
- Frost, G.R.; Li, Y.M. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017, 7, 170228. [Google Scholar] [CrossRef] [PubMed]
- Teich, A.F.; Patel, M.; Arancio, O. A reliable way to detect endogenous murine β-amyloid. PLoS ONE 2013, 8, e55647. [Google Scholar] [CrossRef]
- Okereke, O.I.; Xia, W.; Irizarry, M.C.; Sun, X.; Qiu, W.Q.; Fagan, A.M.; Mehta, P.D.; Hyman, B.T.; Selkoe, D.J.; Grodstein, F. Performance characteristics of plasma amyloid-beta 40 and 42 assays. J. Alzheimers Dis. 2009, 16, 277–285. [Google Scholar] [CrossRef]
- Aluise, C.D.; Sowell, R.A.; Butterfield, D.A. Peptides and proteins in plasma and cerebrospinal fluid as biomarkers for the prediction, diagnosis, and monitoring of therapeutic efficacy of Alzheimer’s disease. Biochim. Biophys. Acta 2008, 1782, 549–558. [Google Scholar] [CrossRef]
- Stewart, K.L.; Radford, S.E. Amyloid plaques beyond Aβ: A survey of the diverse modulators of amyloid aggregation. Biophys. Rev. 2017, 9, 405–419. [Google Scholar] [CrossRef]
- Ganesan, A.; Debulpaep, M.; Wilkinson, H.; Van Durme, J.; De Baets, G.; Jonckheere, W.; Ramakers, M.; Ivarsson, Y.; Zimmermann, P.; Van Eldere, J.; et al. Selectivity of aggregation-determining interactions. J. Mol. Biol. 2015, 427, 236–247. [Google Scholar] [CrossRef]
- Bolognesi, B.; Tartaglia, G.G. Physicochemical principles of protein aggregation. Prog. Mol. Biol. Transl. Sci. 2013, 117, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Valenta, L.J.; Michel-Bechet, M.; Mattson, J.C.; Singer, F.R. Microfollicular thyroid carcinoma with amyloid rich stroma, resembling the medullary carcinoma of the thyroid (MCT). Cancer 1977, 39, 1573–1586. [Google Scholar] [CrossRef]
- Khan, I.S.; Loh, K.S.; Petersson, F. Amyloid and hyaline globules in undifferentiated nasopharyngeal carcinoma. Ann. Diagn. Pathol. 2019, 40, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Franklin, C.D.; Martin, M.V.; Clark, A.; Smith, C.J.; Hindle, M.O. An investigation into the origin and nature of ‘amyloid’ in a calcifying epithelial odontogenic tumour. J. Oral Pathol. 1981, 10, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Delaney, M.A.; Singh, K.; Murphy, C.L.; Solomon, A.; Nel, S.; Boy, S.C. Immunohistochemical and biochemical evidence of ameloblastic origin of amyloid-producing odontogenic tumors in cats. Vet. Pathol. 2013, 50, 238–242. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hirayama, K.; Endoh, C.; Kagawa, Y.; Ohmachi, T.; Yamagami, T.; Nomura, K.; Matsuda, K.; Okamoto, M.; Taniyama, H. Amyloid-Producing Odontogenic Tumors of the Facial Skin in Three Cats. Vet. Pathol. 2017, 54, 218–221. [Google Scholar] [CrossRef]
- Silverman, J.F.; Dabbs, D.J.; Norris, H.T.; Pories, W.J.; Legier, J.; Kay, S. Localized primary (AL) amyloid tumor of the breast. Cytologic, histologic, immunocytochemical and ultrastructural observations. Am. J. Surg. Pathol. 1986, 10, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Kotani, H.; Sawaki, M.; Hattori, M.; Yoshimura, A.; Gondo, N.; Adachi, Y.; Kataoka, A.; Sugino, K.; Horisawa, N.; et al. Amyloid tumor of the breast. Surg. Case Rep. 2019, 5, 31. [Google Scholar] [CrossRef]
- Rosenzweig, M.; Landau, H. Light chain (AL) amyloidosis: Update on diagnosis and management. J. Hematol. Oncol. 2011, 4, 47. [Google Scholar] [CrossRef]
- Zayas-Santiago, A.; Ríos, D.S.; Zueva, L.V.; Inyushin, M.Y. Localization of αA-Crystallin in Rat Retinal Müller Glial Cells and Photoreceptors. Microsc. Microanal. 2018, 24, 545–552. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kucheryavykh, L.Y.; Ortiz-Rivera, J.; Kucheryavykh, Y.V.; Zayas-Santiago, A.; Diaz-Garcia, A.; Inyushin, M.Y. Accumulation of Innate Amyloid Beta Peptide in Glioblastoma Tumors. Int. J. Mol. Sci. 2019, 20, 2482. https://doi.org/10.3390/ijms20102482
Kucheryavykh LY, Ortiz-Rivera J, Kucheryavykh YV, Zayas-Santiago A, Diaz-Garcia A, Inyushin MY. Accumulation of Innate Amyloid Beta Peptide in Glioblastoma Tumors. International Journal of Molecular Sciences. 2019; 20(10):2482. https://doi.org/10.3390/ijms20102482
Chicago/Turabian StyleKucheryavykh, Lilia Y., Jescelica Ortiz-Rivera, Yuriy V. Kucheryavykh, Astrid Zayas-Santiago, Amanda Diaz-Garcia, and Mikhail Y. Inyushin. 2019. "Accumulation of Innate Amyloid Beta Peptide in Glioblastoma Tumors" International Journal of Molecular Sciences 20, no. 10: 2482. https://doi.org/10.3390/ijms20102482
APA StyleKucheryavykh, L. Y., Ortiz-Rivera, J., Kucheryavykh, Y. V., Zayas-Santiago, A., Diaz-Garcia, A., & Inyushin, M. Y. (2019). Accumulation of Innate Amyloid Beta Peptide in Glioblastoma Tumors. International Journal of Molecular Sciences, 20(10), 2482. https://doi.org/10.3390/ijms20102482
{kind=link}