Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development

 , , , and
, , , and 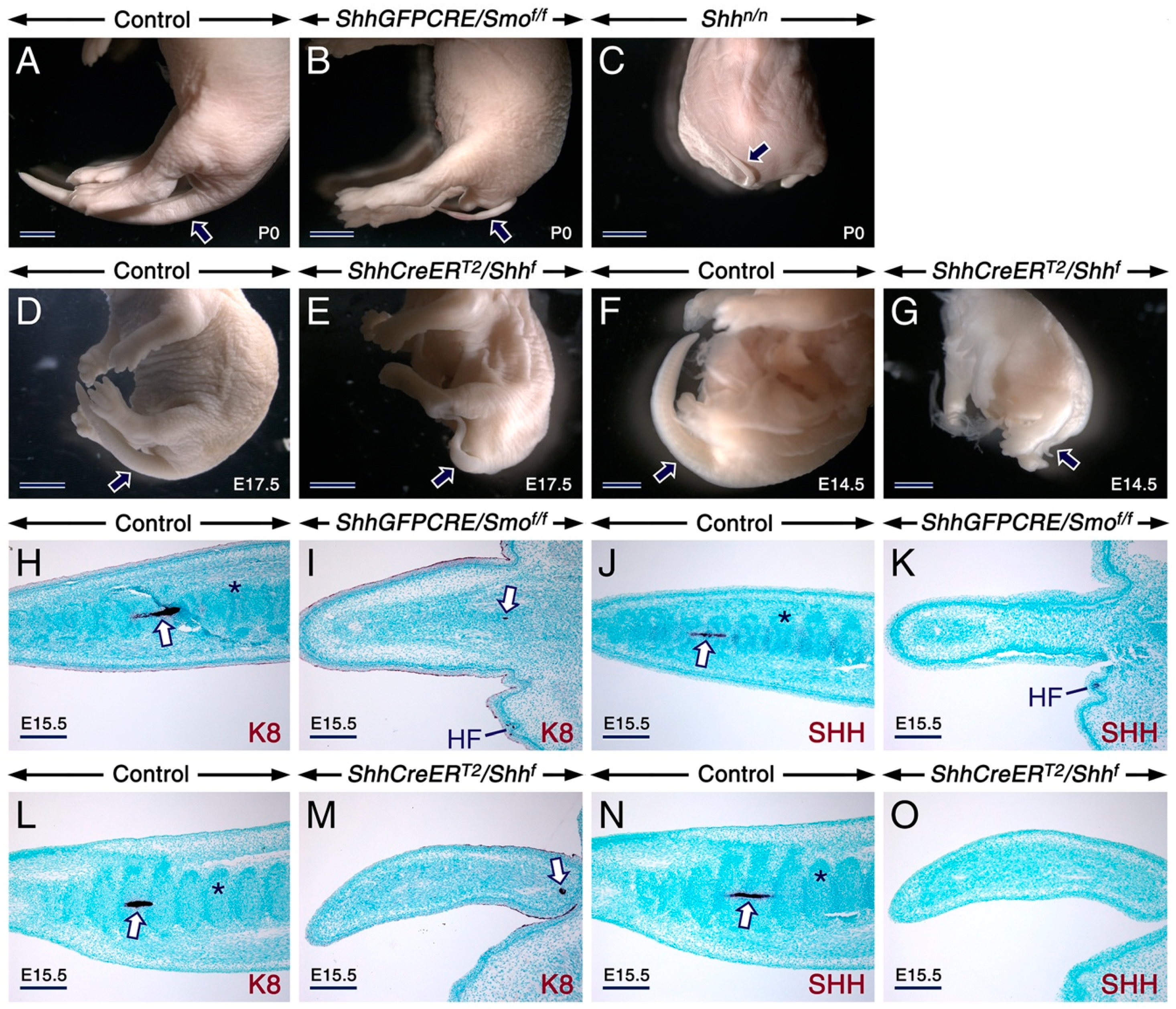{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results and Discussion
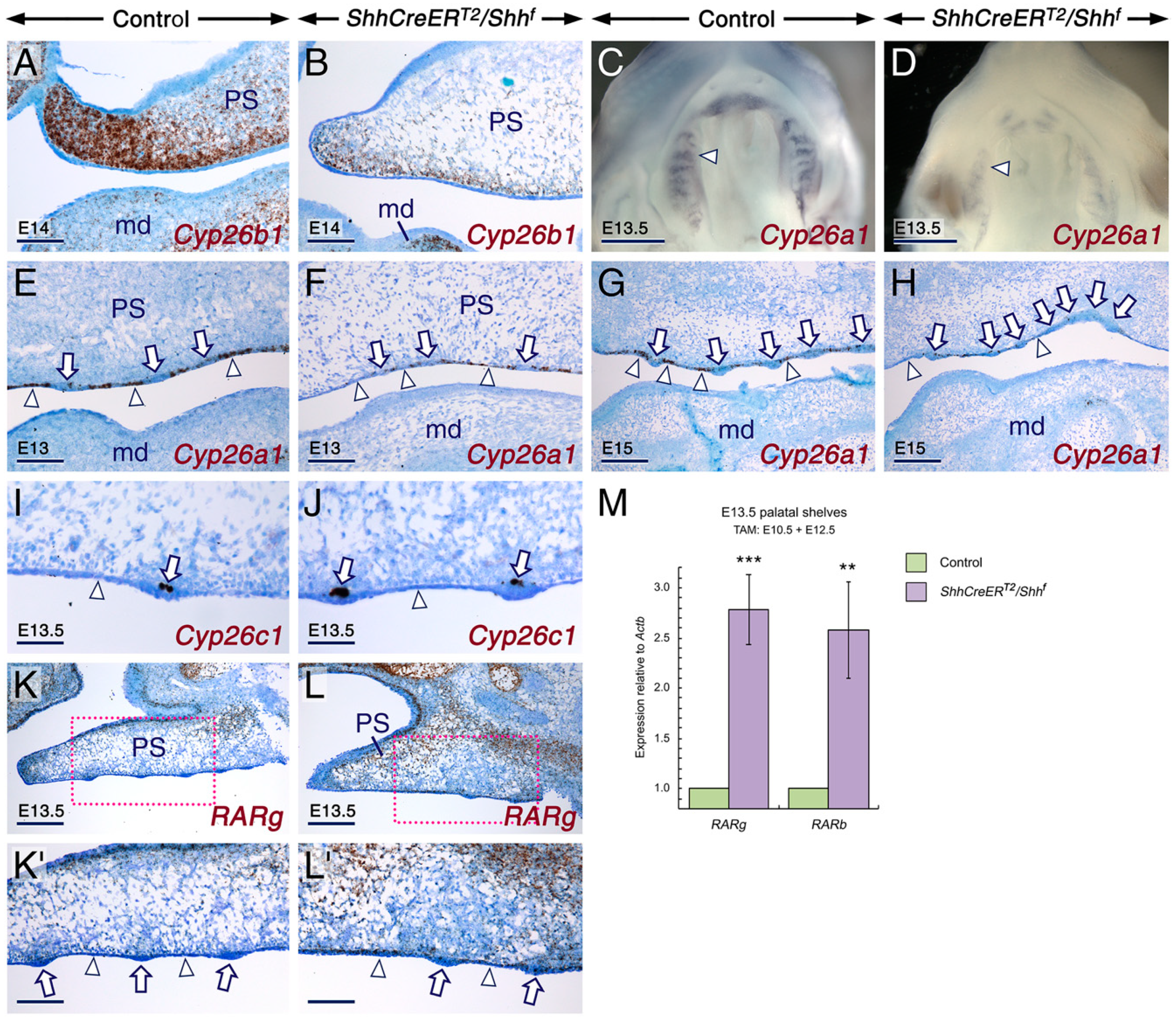
2.1. SHH Signaling Antagonizes RA Activity through CYP26A1 to Ensure Proper Development of the Tail
2.2. SHH Signaling in the Developing Secondary Palate Is Required to Prevent Enhancement of RA Activity
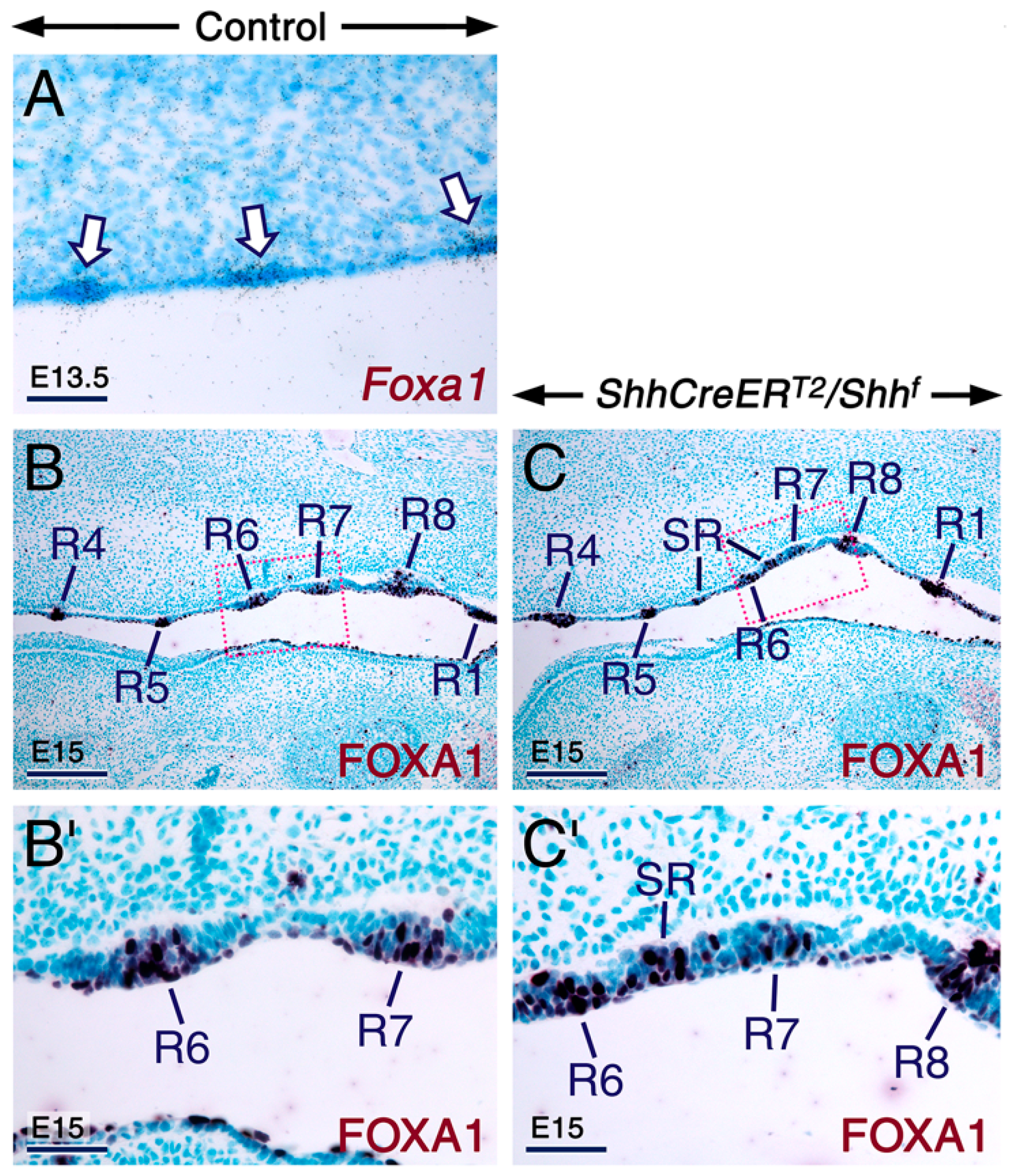
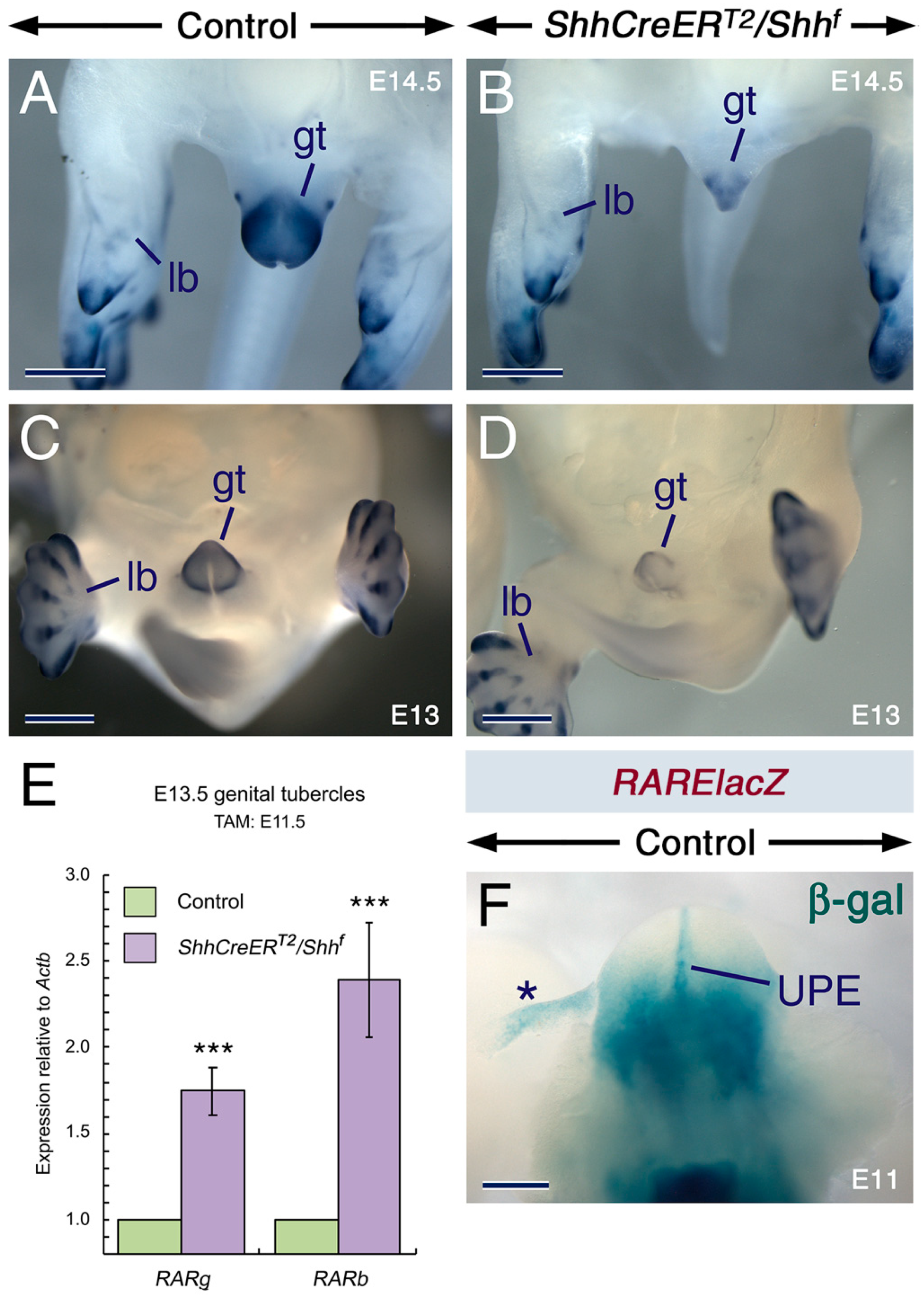
2.3. SHH Signaling Is Required for Cyp26 Expression in Other Developing Structures
2.4. Conclusions
3. Materials and Methods
3.1. Ethics Statement
3.2. Mouse Lines
3.3. Histology, Immunohistochemistry, In Situ Hybridization, β-Galactosidase Histochemistry and RT-qPCR
3.4. In Vitro Explant Cultures and Quantification of Apoptosis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BMS493 | Pan Retinoic acid receptor antagonist |
| CRE | (Cyclization recombination) DNA recombinase |
| CYP26A1,B1,C1 | Cytochrome P450 isoenzymes A1, B1 and C1 |
| f | Floxed allele |
| FGF | Fibroblast growth factor |
| FNP | Frontonasal process |
| FOXA1/Foxa1 | Forkhead box protein A1 protein/gene |
| GLI1-3 | Glioma-associated oncogene family members 1, 2 and 3 |
| IHH | Indian hedgehog |
| ISH | In situ Hybridization |
| K8 | Keratin 8 |
| K14 | Keratin 14 |
| LacZ | Gene encoding E. Coli β-galactosidase |
| n | Null allele |
| SAG | Smoothened agonist |
| SHH/Shh | Sonic Hedgehog protein/gene |
| SMO/Smo | Smoothened protein/gene |
| PS | Palatal shelf/shelves |
| Ptch1 | Patched 1 |
| RA | Retinoic acid |
| RALDH/Aldh1a | Retinaldehyde dehydrogenase protein/gene |
| RAR | Retinoic acid receptor |
| RARE | Retinoic acid response element |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| RXR | Retinoid X receptor |
| TAM | Tamoxifen |
| WMISH | Whole-mount in situ hybridizationWingless/integrated 3a |
| 4-OH-TAM | 4-hydroxytamoxifen |
References
- McMahon, A.P.; Ingham, P.W.; Tabin, T.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Dollé, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P. The mechanisms of hedgehog signaling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef]
- Mark, M.; Teletin, M.; Vernet, N.; Ghyselinck, N. Role of retinoic acid receptor (RAR) signaling in postnatal male germ cell differentiation. Biochim. Biophys. Acta 2015, 1849, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Xavier, M.G.; Seppala, M.; Barrell, W.; Birjandi, A.A.; Geoghegan, F.; Cobourne, M. Hedgehog receptor function during craniofacial development. Dev. Biol. 2016, 415, 198–215. [Google Scholar] [CrossRef]
- Cotton, J.L.; Li, Q.; Ma, L.; Wang, J.; Park, J.S.; Ou, J.; Zhu, L.J.; YT, I.P.; Johnson, R.L.; Mao, J. YAP/TAZ and Hedgehog coordinate growth and patterning in gastrointestinal mesenchyme. Dev. Cell 2017, 43, 35–47. [Google Scholar] [CrossRef]
- Hibsher, D.; Epshtein, A.; Oren, N.; Landsman, L. Pancreatic mesenchyme regulates islet cellular composition in a Patched/Hedgehog-dependent manner. Sci. Rep. 2016, 28, 38008. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, X.; Li, Y.; Zhang, X.; Zhang, Z. Supressor of fused restraint of Hedgehog activity level is critical for osteogenic proliferation and differentiation during calvarial bone development. J. Biol. Chem. 2017, 292, 1514–1525. [Google Scholar]
- Millington, G.; Elliott, K.H.; Chang, Y.T.; Chang, C.F.; Dlugosz, A.; Brugmann, S.A. Cilia-dependent GLI processing in neural crest cells is required for tongue development. Dev. Biol. 2017, 424, 124–137. [Google Scholar] [CrossRef]
- Shimo, T.; Koyama, E.; Okui, T.; Kunisada, Y.; Ibaragi, S.; Yoshioka, N.; Yoshida, S.; Sasaki, A.; Masui, M.; Kurio, N.; et al. Retinoic receptor signaling regulates hypertrophic chondrocyte-specific gene expression. In Vivo 2019, 33, 85–91. [Google Scholar] [CrossRef]
- Smith, J.N.; Walker, H.M.; Thompson, H.; Collinson, J.M.; Vargesson, N.; Erskine, L. Lens-regulated retinoic acid signalling controls expansion of the developing eye. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Marchwicka, A.; Macinkowska, E. Regulation of expression of CEBP genes by variably expressed vitamin D and retinoic acid receptor in human acute myeloid leukemia cell lines. Int. J. Mol. Sci. 2018, 19, 918. [Google Scholar] [CrossRef]
- Akhavan-Sigari, R.; Schulz-Schaeffer, W.; Harcej, A.; Rohde, V. The importance of the hedgehog signaling pathway in tumorigenesis of spinal and cranial chordoma. J. Clin. Med. 2019, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelloto, L.G.; Peifer, M.; Sos, M.L.; Karnesis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Han, M.E.; Lee, Y.S.; Baek, S.Y.; Kim, B.S.; Kim, J.B.; Oh, S.O. Hedgehog signaling regulates the survival of gastric cancer cells by regulating the expression of Bcl-2. Int. J. Mol. Sci. 2009, 6, 3033–3043. [Google Scholar] [CrossRef]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Ann. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef]
- Bitgood, M.J.; McMahon, A.P. Hedgehog and Bmp genes are coexpressed at many sites of cell-cell interaction in the mouse embryo. Dev. Biol. 1995, 172, 126–138. [Google Scholar] [CrossRef]
- Corbit, K.C.; Aanstad, P.; Norman, A.R.; Stainier, D.Y.; Reiter, J.F. Vertebrate Smoothened functions at the primary cilium. Nature 2005, 437, 1018–1021. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M. Patched regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef]
- Rhinn, M.; Dollé, P. Retinoic acid signaling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar] [CrossRef]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Beckett, B.; Chambon, P.; Petkovitch, M. The retinoic acid-metabolizing enzyme, Cyp26A1, is essential for normal hindbrain patterning, vertebral identity, and development of posterior structures. Genes Dev. 2001, 15, 226–240. [Google Scholar] [CrossRef]
- Yashiro, K.; Zhao, X.; Uehara, M.; Yamashita, J.; Nishijima, M.; Nishino, J.; Saijoh, Y.; Sakai, Y.; Hamada, H. Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell 2004, 6, 411–422. [Google Scholar] [CrossRef]
- Uehara, M.; Yashiro, K.; Mamiya, S.; Nishino, J.; Chambon, P.; Dollé, P.; Sakai, Y. CYP26A1 and CYP26C1 cooperatively regulate anterior-posterior patterning of the developing brain and the production of migratory neural crest cells in the mouse. Dev. Biol. 2007, 302, 399–411. [Google Scholar] [CrossRef]
- Sockanathan, S.; Jessell, T.M. Motor neuron-derived retinoic acid specifies the subtype identity of spinal motor neurons. Cell 1998, 94, 503–514. [Google Scholar] [CrossRef]
- Novitch, B.G.; Wichterle, H.; Jessell, T.M.; Stockanathan, S. A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 2003, 40, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, L.; Starr, A.; Nelson, A.T.; Sattler, R. Representing diversity in the dish: Using patient-derived in vitro models to recreate the heterogeneity of neurological disease. Front. Neurosci. 2018, 12, 56. [Google Scholar] [CrossRef]
- Tsukui, T.; Capdevila, J.; Tamura, K.; Ruiz-Lozano, P.; Rodriguez-Esteban, C.; Yonei-Tamura, S.; Magallón, J.; Chandraratna, R.A.; Chien, K.; Blumberg, B.; et al. Multiple left-right asymmetry defects in Shh−/− mutant mice unveil a convergence of the Shh and retinoic acid pathways in the control of Lefty-1. Proc. Natl. Acad. Sci. USA 1999, 96, 11376–11381. [Google Scholar] [CrossRef]
- Schneider, R.A.; Hu, D.; Rubenstein, J.L.R.; Maden, M.; Helms, J.A. Local retinoid signaling coordinates forebrain and facial morphogenesis by maintaining FGF8 and SHH. Development 2001, 128, 2755–2767. [Google Scholar] [PubMed]
- Tanaka, K.; Okada, Y.; Hirokawa, N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right detemination. Nature 2005, 435, 172–177. [Google Scholar] [CrossRef]
- Ribes, V.; Wang, Z.; Dollé, P.; Niederreither, K. Retinaldehyde dehydrogennase 2 (RALDH2)-mediated retinoic acid synthesis regulates early mouse embryonic forebrain development by controlling FGF and sonic hedgehog signaling. Development 2006, 133, 351–361. [Google Scholar] [CrossRef]
- Ribes, V.; Le Roux, I.; Rhinn, M.; Schuhbaur, B.; Dollé, P. Early mouse caudal development relies on crosstalk between retinoic acid, Shh and Fgf signaling pathways. Development 2009, 136, 665–676. [Google Scholar] [CrossRef]
- Mich, J.K.; Chen, J.K. Hedgehog and retinoic acid signaling cooperate to promote motoneurogenesis in zebrafish. Development 2011, 138, 5113–5119. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ribes, V.; Stutzmann, F.; Bianchetti, L.; Guillemot, F.; Dollé, P.; Le Roux, I. Combinatorial signaling controls Neurogenin2 expression at the onset of spinal neurogenesis. Dev. Biol. 2008, 321, 470–481. [Google Scholar] [CrossRef]
- Probst, S.; Kraemer, C.; Demougin, P.; Sheth, R.; Martin, G.R.; Shiratori, H.; Hamada, H.; Iber, D.; Zeller, R.; Zuniga, A. SHH propagates distal limb bud development by enhancing CYP26B1-mediated retinoic acid clearance via AER-FGF signaling. Development 2011, 138, 1913–1923. [Google Scholar] [CrossRef]
- Alonso, S.; Hernandez, D.; Chang, Y.T.; Gocke, C.B.; McCray, M.; Varadham, R.; Matsui, W.H.; Jones, R.J.; Ghiaur, G. Hedgehog and retinoic acid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. J. Clin. Investig. 2016, 126, 4460–4468. [Google Scholar] [CrossRef] [PubMed]
- El Shahawy, M.; Reibring, C.G.; Neben, C.L.; Hallberg, K.; Marangoni, P.; Harfe, B.D.; Klein, O.D.; Linde, A.; Gritli-Linde, A. Cell fate specification in the lingual epithelium is controlled by antagonistic activities of Sonic hedgehog and retinoic acid. PLoS Genet. 2017, 13, e1006914. [Google Scholar] [CrossRef]
- Dassule, H.; Lewis, P.; Bei, M.; McMahon, A.P. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 2000, 127, 4775–4785. [Google Scholar]
- Choi, K.S.; Harfe, B.D. Hedgehog signaling is required for formation of the notochord sheath and patterning of nuclei pulposi within the intervertebral discs. Proc. Natl. Acad. Sci. USA 2011, 108, 9484–9489. [Google Scholar] [CrossRef] [PubMed]
- Harfe, B.D.; Scherz, P.J.; Nissim, S.; Tian, H.; McMahon, A.P.; Tabin, C. Evidence for expansion-based temporal Shh gradient in specifying vertebrate digit identities. Cell 2004, 118, 517–528. [Google Scholar] [CrossRef]
- Chiang, C.; Litingtun, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and defective axial patterning in mice lacking Sonic Hedgehog gene function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef]
- Götz, W.; Kasper, M.; Fischer, G.; Herken, R. Intermediate filament typing of the human embryonic and fetal notochord. Cell Tissue Res. 1995, 280, 455–462. [Google Scholar] [CrossRef]
- Richardson, S.M.; Ludwinski, F.E.; Gnanalingham, K.K.; Atkinson, R.A.; Freemont, A.J.; Hoyland, J.A. Notochordal and nucleus pulposus marker expression is maintained by subpopulations of adult human nucleosus pulposus cells through aging and regeneration. Sci. Rep. 2017, 7, 1501–1511. [Google Scholar] [CrossRef]
- Gofflot, F.; Hall, M.; Morriss-Kay, G.M. Genetic patterning of the developing mouse tail at the time of posterior neuropore closure. Dev. Dyn. 1997, 210, 431–445. [Google Scholar] [CrossRef]
- Niederreither, K.; McCaffery, P.; Dräager, U.C.; Chambon, P.; Dollé, P. Restricted expression and retinoic acid-induced downregulation of the retinaldehyde dehydrogenenase type 2 (RALDH-2) gene during mouse development. Mech. Dev. 1997, 62, 67–78. [Google Scholar] [CrossRef]
- Haselbeck, R.J.; Hoffmann, I.; Duester, G. Distinct functions for Aldh1 and Raldh2 in the control of ligand production for embryonic retinoid signaling. Dev. Genet. 1999, 25, 353–364. [Google Scholar] [CrossRef]
- Dollé, P.; Ruberte, E.; Leroy, P.; Morris-Kay, G.; Chambon, P. Retinoic acid receptors and cellular retinoid binding proteins. I. A systematic study of their differential patterns of transcription during mouse organogenesis. Development 1990, 110, 1133–1151. [Google Scholar]
- Mollard, R.; Viville, S.; Ward, S.J.; Décimo, D.; Chambon, P.; Dollé, P. Tissue-specific expression of retinoic acid receptor isoform transcripts in the mouse embryo. Mech. Dev. 2000, 94, 223–232. [Google Scholar] [CrossRef]
- Abu-Abed, S.; Dollé, P.; Metzger, D.; Wood, C.; MacLean, G.; Chambon, P.; Petkovich, M. Developing with lethal RA levels: Genetic ablation of RARg can restore the viability of mice lacking Cyp26a1. Development 2003, 130, 1449–1459. [Google Scholar] [CrossRef]
- White, R.J.; Schilling, T.F. How degrading: Cyp26s in hindbrain development. Dev. Dyn. 2008, 237, 2775–2790. [Google Scholar] [CrossRef]
- Pennimpede, T.; Cameron, D.A.; MacLean, G.A.; Li, H.; Abu-Abed, S.; Petkovitch, M. The role of CYP26 enzymes in defining appropriate retinoic acid exposure during embryogenesis. Birth Defects Res. 2010, 88, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Sato, T.; Kanedo, S.; Gotoh, O.; Fujii-Kuriyama, Y.; Osawa, K.; Kato, S.; Hamada, H. Metabolic inactivation of retinoic acid by a novel P450 differentially expressed in developing mouse embryos. EMBO J. 1997, 16, 4163–4173. [Google Scholar] [CrossRef] [PubMed]
- De Roos, K.; Sonneveld, E.; Compaan, B.; ten Berge, D.; Durston, A.J.; van der Saag, P.T. Expression of retinoic acid 4-hydroxylase (CYP26) during mouse and Xenopus laevis embryogenesis. Mech. Dev. 1999, 82, 205–211. [Google Scholar] [CrossRef]
- MacLean, G.; Abu-Abed, S.; Dollé, P.; Tahayato, A.; Chambon, P.; Petkovitch, M. Cloning of a novel retinoic acid metabolizing cytochrome P450 Cyp26B1, and comparative expression analysis with Cyp26A1 during early murine development. Mech. Dev. 2001, 107, 195–201. [Google Scholar] [CrossRef]
- Molotkova, N.; Molotkov, A.; Sirbu, I.O.; Duester, G. Requirement of mesodermal retinoic acid generated by Raldh2 for posterior neural transformation. Mech. Dev. 2005, 122, 145–155. [Google Scholar] [CrossRef]
- Sakai, Y.; Meno, C.; Fujii, H.; Nishino, J.; Shiratori, H.; Saijoh, Y.; Rossant, J.; Hamada, H. The retinoic acid-inactivating enzyme Cyp26 is essential for establishing an uneven distribution of retinoic acid along the anterio-posterior axis within the mouse embryo. Genes Dev. 2001, 15, 213–225. [Google Scholar] [CrossRef]
- Niederreither, K.; Abu-Abed, S.; Schuhbar, B.; Petkovich, M.; Chambon, P.; Dollé, P. Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet. 2002, 31, 84–88. [Google Scholar] [CrossRef]
- Shenefelt, R.E. Morphogenesis of malformations in hamsters caused by retinoic acid: Relation to dose and stage at treatment. Teratology 1972, 5, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Geelen, J.A.G.; Peters, P.W.J. Hypervitaminosis A-induced teratogenesis. CRC Crit. Rev. Toxicol. 1979, 6, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, Y.; Okamoto, M.; Konishi, H.; Matsuo, T.; Kihara, T.; Tanimura, T. Developmental anomalies induced by all-trans retinoic acid in fetal mice: Macroscopic findings. Teratology 1986, 34, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Elmazar, M.M.A.; Reichert, B.; Scroot, B.; Nau, H. Patterns of retinoid-induced teratogenenic effects: Possible relationships with relative selectivity for nuclear retinoid receptors RARa, RARb and RARg. Teratology 1996, 53, 158–167. [Google Scholar] [CrossRef]
- Padmanabhan, R. Retinoic-acid-induced caudal regression syndrome in the mouse fetus. Reprod. Toxicol. 1998, 12, 139–151. [Google Scholar] [CrossRef]
- Wiley, M.J. The pathogenesis of retinoic acid-induced vertebral anomalies in golden syrian hamster fetuses. Teratology 1983, 28, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Tahayato, A.; Dollé, P.; Petkovitch, M. Cyp26c1 encodes a novel retinoic acid-metabolizing enzyme expressed in the hindbrain, inner ear, first brachial arch and tooth buds during murine development. Gene Exp. Patterns 2003, 3, 449–454. [Google Scholar] [CrossRef]
- MacLean, G.; Dollé, P.; Petkovitch, M. Genetic disruption of CYP26B1 severely affects development of the neural crest derived head structures but does not compromise hindbrain patterning. Dev. Dyn. 2009, 238, 732–745. [Google Scholar] [CrossRef]
- Abu-Abed, S.; MacLean, G.; Fraulob, V.; Chambon, P.; Petkovitch, M.; Dollé, P. Differential expression of the retinoic acid-metabolizing enzymes CYP26A1 and CYP26B1 during murine organogenesis. Mech. Dev. 2002, 110, 173–177. [Google Scholar] [CrossRef]
- Kaufman, M.H. The Atlas of Mouse Development, Revised ed.; Academic Press: London, UK, 2003; pp. 172–174. [Google Scholar]
- Abzhanov, A.; Rodda, S.J.; McMahon, A.P.; Tabin, C.J. Regulation of skeletogenic differentiation in cranial dermal bone. Development 2007, 134, 3133–3144. [Google Scholar] [CrossRef]
- Ferguson, C.; Alpern, E.; Miclau, T.; Helms, J.A. Does adult fracture repair recapitulate embryonic skeletal formation? Mech. Dev. 1999, 87, 57–66. [Google Scholar] [CrossRef]
- Rossant, J.; Zirngibl, R.; Cado, D.; Shago, M.; Giguère, V. Expression of a retinoic acid response element-hsplacZ transgene defines specific domains of transcriptional activity during mouse embryogenesis. Genes Dev. 1991, 5, 1333–1344. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, C.; Larkin, S.; Mark, M.; LeMeur, M.; Clifford, J.; Zelent, A.; Chambon, P. RARb isoforms: Distinct transcriptional control by retinoic acid and specific spatial patterns of promoter activity during mouse embryonic development. Mech. Dev. 1994, 45, 227–241. [Google Scholar] [CrossRef]
- Sigenthaler, J.A.; Ashique, A.M.; Kostantino, Z.; Patterson, K.P.; Hecht, J.H.; Jane, M.A.; Folias, A.E.; Choe, Y.; May, S.R.; Kume, T.; et al. Retinoic acid from meninges regulates cortical neuron generation. Cell 2009, 139, 597–609. [Google Scholar] [CrossRef]
- Dollé, P.; Fraulob, V.; Gallego-Llamas, J.; Vermot, J.; Niederreither, K. Fate of retinoic acid-activated embryonic cell lineages. Dev. Dyn. 2010, 239, 3260–3274. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Kimura, W.; Papaionnou, V.E.; Miura, N.; Yamada, G.; Shiota, K.; Sakai, Y. The regulation of endogenous retinoic acid level through CYP26B1 is required for elevation of palatal shelves. Dev. Dyn. 2012, 241, 1744–1756. [Google Scholar] [CrossRef]
- Dollé, P. Developmental expression of retinoic acid receptors (RARs). Nucl. Recept. Signal. 2009, 7, e006. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.S.; Poon, L.L.; Tang, W.W.; Koide, T.; Chan, B.W.; Leung, Y.C.; Shiroishi, T.; Copp, A.J. Retinoic acid induces down-regulation of Wnt-3a, apoptosis and diversion of tail bud cells to neural fate in the mouse embryo. Mech. Dev. 1999, 84, 17–30. [Google Scholar] [CrossRef]
- Gritli-Linde, A. Molecular control of secondary palate development. Dev. Biol. 2007, 301, 309–326. [Google Scholar] [CrossRef]
- Gritli-Linde, A. The etiopathogenesis of cleft lip and cleft palate: Usefulness and caveats of mouse models. Curr. Top. Dev. Biol. 2008, 84, 37–138. [Google Scholar]
- Dixon, M.J.; Marazita, M.L.; Beaty, T.H.; Murray, J.C. Cleft lip and palate: Understanding genetic and environmental influences. Nat. Rev. Genet. 2011, 12, 167–178. [Google Scholar] [CrossRef]
- Rahimov, F.; Jugessur, A.; Murray, J.C. Genetics of nonsyndromic orofacial clefts. Cleft Palate Craniofac. J. 2012, 49, 73–91. [Google Scholar] [CrossRef]
- Lan, Y.; Xu, J.; Jiang, R. Cellular and molecular mechanisms of palatogenesis. Curr. Top. Dev. Biol. 2015, 115, 59–84. [Google Scholar]
- Xu, J.; Liu, H.; Lan, Y.; Aronow, B.J.; Kalinichenko, V.V.; Jiang, R. A Shh-Foxf-Fgf18-Shh molecular circuit regulating palate development. PLoS Genet. 2016, 12, e1005769. [Google Scholar] [CrossRef] [PubMed]
- Economou, A.D.; Ohazama, A.; Porntaveetus, T.; Sharpe, P.T.; Kondo, S.; Basson, M.A.; Gritli-Linde, A.; Cobourne, M.T.; Green, J.B. Periodic stripe formation by a Turing mechanism operating at growth zones in the mammalian palate. Nat. Genet. 2012, 44, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Mao, J.; Tenzen, T.; Kottman, A.H.; McMahon, A.P. Hedgehog signaling in the neural crest cells regulates the patterning and growth of facial primordia. Genes Dev. 2004, 18, 937–951. [Google Scholar] [CrossRef]
- Rice, R.; Spencer-Dene, B.; Connor, E.C.; Gritli-Linde, A.; McMahon, A.P.; Dickson, C.; Thesleff, I.; Rice, D.P. Disruption of Fgf10/Fgfr2b-coordinated epithelial mesenchymal interactions causes cleft palate. J. Clin. Investig. 2004, 113, 1692–1700. [Google Scholar] [CrossRef] [PubMed]
- Rice, R.; Connor, E.; Rice, D.P.C. Expression patterns of Hedgehog signaling pathway members during mouse palate development. Gene Exp. Patterns 2006, 6, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Pantalacci, S.; Prochazka, J.; Martin, A.; Rothova, M.; Lambert, A.; Bernard, L.; Charles, C.; Viriot, L.; Peterkova, R.; Laudet, V. Patterning of palatal rugae through sequential addition reveals an anterior-posterior boundary in palate development. BMC Dev. Biol. 2008, 8, 116. [Google Scholar] [CrossRef]
- Welsh, I.C.; O’Brian, T.P. Signaling integration in the rugae growth zone directs sequential SHH signaling center formation during the rostral outgrowth of the palate. Dev. Biol. 2009, 336, 53–67. [Google Scholar] [CrossRef]
- Naitoh, H.; Mori, C.; Nishimura, Y.; Shiota, K. Altered expression of retinoic acid (RA) receptor mRNAs in the fetal mouse secondary palate by all-trans and 13-cis Ras: Implications for RA-induced teratogenesis. J. Craniofac. Genet. Dev. Biol. 1998, 18, 202–210. [Google Scholar]
- Padmanabhan, R.; Ahmed, I. Retinoic acid-induced asymmetric craniofacial growth and cleft palate in the mouse fetus. Reprod. Toxicol. 1997, 11, 843–860. [Google Scholar] [CrossRef]
- Ikemi, N.; Kawata, M.; Yasuda, M. All-trans-retinoic acid-induced variant patterns of palatal rugae in CRJ:Sd rat fetuses and their potential as indicators for teratogenesis. Reprod. Toxicol. 1995, 9, 369–377. [Google Scholar] [CrossRef]
- Vaziri Sani, F.; Kaartninen, V.; El Shahawy, M.; Linde, A.; Gritli-Linde, A. Developmental changes and extracellular structural molecules in the secondary palate and in the nasal cavity of the mouse. Eur. J. Oral Sci. 2010, 118, 212–236. [Google Scholar] [CrossRef]
- Cohn, M.J. Development of the external genitalia: Conserved and divergent mechanisms of appendage patterning. Dev. Dyn. 2011, 240, 1108–1115. [Google Scholar] [CrossRef]
- Seifert, A.W.; Bouldin, C.M.; Choi, K.S.; Harfe, B.D.; Cohn, M.J. Multiphasic and tissue-specific roles for sonic hedgehog in cloacal septation and external genitalia development. Development 2009, 136, 3949–3957. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yin, Y.; Veith, G.M.; Fisher, A.V.; Long, F.; Ma, L. Temporal and spatial dissection of Shh signaling in genital tubercle development. Development 2009, 136, 3959–3967. [Google Scholar] [CrossRef] [PubMed]
- Miyagawa, S.; Moon, A.; Haraguchi, R.; Inoue, C.; Harada, M.; Nakahara, C.; Suzuki, K.; Matsumaru, D.; Kaneko, T.; Matsuo, I.; et al. Dosage-dependent hedgehog signals integrated with Wnt/beta-catenin signaling regulate external genitalia formation as an appendicular program. Development 2009, 136, 3969–3978. [Google Scholar] [CrossRef]
- Seifert, A.W.; Zheng, Z.; Ormerod, B.K.; Cohn, M.J. Sonic hedgehog controls growth of external genitalia by regulating cell cycle kinetics. Nat. Commun. 2010, 1, 23. [Google Scholar] [CrossRef]
- Liu, L.; Suzuki, K.; Nakagata, N.; Mihara, K.; Matsumaru, D.; Ogino, Y.; Yashiro, K.; Hamada, H.; Liu, Z.; Evans, S.M.; et al. Retinoic acid signaling regulates Sonic hedgehog and Bone morphogenetic protein signaling during genital tubercle development. Birth Defects Res. 2012, 95, 79–88. [Google Scholar] [CrossRef]
- Ruberte, E.; Dollé, P.; Krust, A.; Zelent, A.; Morris-Kay, G.; Chambon, P. Specific spacial and temporal distribution of retinoic acid receptor gamma transcripts during mouse embryogenesis. Development 1990, 108, 213–222. [Google Scholar] [PubMed]
- Armfield, B.A.; Seifert, A.W.; Zheng, Z.; Merton, E.M.; Rock, J.R.; Lopez, M.C.; Baker, H.V.; Cohn, M.J. Molecular characterization of the genital organizer: Gene expression profile of the mouse urethral plate epithelium. J. Urol. 2016, 196, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.A.; Kim, C.H.; Minkoff, R.; Thaller, C.; Eichele, G. Sonic hedgehog participates in craniofacial morphogenesis and is down-regulated by teratogenic doses of retinoic acid. Dev. Biol. 1997, 187, 25–35. [Google Scholar] [CrossRef]
- Gritli-Linde, A.; Bei, M.; Maas, R.; Zhang, X.M.; Linde, A.; McMahon, A.P. Shh signaling within the dental epithelium is necessary for cell proliferation, growth and polarization. Development 2002, 129, 5323–5337. [Google Scholar] [CrossRef] [PubMed]
- Nakatomi, M.; Hovorakova, M.; Gritli-Linde, A.; Blair, H.J.; MacArthur, K.; Peterka, M.; Lesot, H.; Peterkova, R.; Ruiz-Perez, V.L.; Goodship, J.A.; et al. Evc regulates a symmetrical response to Shh signaling in molar development. J. Dent. Res. 2013, 92, 222–228. [Google Scholar] [CrossRef]
- Niederreither, K.; Fraulob, V.; Garnier, J.M.; Chambon, P.; Dollé, P. Differential expression of retinoic acid-synthesizing (RALDH) enzymes during fetal development and organ differentiation in the mouse. Mech. Dev. 2002, 110, 167–171. [Google Scholar] [CrossRef]
- Bloch-Zupan, A.; Décimo, D.; Loriot, M.; Mark, M.P.; Ruch, J.V. Expression of nuclear retinoic acid receptors during mouse odontogenesis. Differentiation 1994, 57, 195–203. [Google Scholar] [CrossRef]
- Morkmued, S.; Laugel-Haushalter, V.; Mathieu, E.; Schuhbaur, B.; Hemmerlé, J.; Dollé, P.; Bloch-Zupan, A.; Niederreither, K. Retinoic acid excess impairs amelogenesis inducing enamel defects. Front. Physiol. 2017, 7, 673. [Google Scholar] [CrossRef]
- Mark, M.P.; Bloch-Zupan, A.; Rush, J.V. Effects of retinoids on tooth morphogenesis and cytodifferentiation in vitro. Int. J. Dev. Biol. 1992, 36, 517–526. [Google Scholar]
- Irving, J.T. The effects of avitaminosis and hypervitamisosis A upon the incisor teeth and incisal alveolar bone of rats. J. Physiol. 1949, 108, 92–101. [Google Scholar] [CrossRef]
- Schour, I.; Hoffman, M.M.; Smith, M.C. Changes in the incisor teeth of albino rats with vitamin A deficiency and the effects of replacement therapy. Am. J. Pathol. 1941, 17, 529–562. [Google Scholar] [PubMed]
- McDowell, E.M.; Shores, R.L.; Spangler, E.F.; Wenk, M.L.; De Luca, L.M. Anomalous growth of rat incisor teeth during chronic intermittent vitamin A deficiency. J. Nutr. 1987, 117, 1265–1274. [Google Scholar] [CrossRef]
- Tubbs, S.S.; Malefant, J.; Loukas, M.; Oakes, W.J.; Oskouian, R.J.; Fries, F.N. Enigmatic human tails: A review of their history, embryology, classification and clinical manifestations. Clin. Anat. 2016, 29, 430–438. [Google Scholar] [CrossRef]
- Tojima, S.; Makishima, H.; Takakuwa, T.; Shigehito, Y. Tail reduction process during human embryonic development. J. Anat. 2018, 232, 806–811. [Google Scholar] [CrossRef]
- Fallon, J.F.; Simandl, B.K. Evidence of a role for cell death in the disappearance of the embryonic human tail. Am. J. Anat. 1978, 152, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Dao, A.H.; Netsky, M.G. Human tails and pseudotails. Hum. Pathol. 1984, 15, 449–453. [Google Scholar] [CrossRef]
- Cai, C.; Shi, O.; Shen, C. Surgical treatment of a patient with human tail and multiple abnormalities of the spinal cord and column. Adv. Orthop. 2011, 2011, 153797. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Robinson, C.G.; Duke, T.C.; Allison, A.W. Incidental finding of a true human tail in an adult: A case report. J. Cutan. Pathol. 2017, 44, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Das, B.C.; Thapa, P.; Karki, R.; Das, S.; Mahapatra, S.; Liu, T.C.; Torregroza, I.; Wallace, D.P.; Kambhampati, S.; Van Veldhuizen, P.; et al. Retinoic acid signaling pathway in development and diseases. Bioorg. Med. Chem. 2014, 22, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Young, K.E.; Beachy, P.A. Small molecule modulation of Smoothened activity. Proc. Natl. Acad. Sci. USA 2002, 99, 14071–14076. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Becker, S.; Wang, Z.J.; Massey, H.; Arauz, H.; Labosky, P.; Hammerschmidt, M.; St-Jacques, B.; Bumcrot, D.; McMahon, A.; Grabel, L. A role for indian hedgehog in F9 cells and the early mouse embryo. Dev. Biol. 1997, 187, 298–310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, X.M.; Ramalho-Santos, M.; McMahon, A.P. Smoothened mutants reveal redundant roles for Shh and Ihh signaling including regulation of L/R asymmetry by the mouse node. Cell 2001, 105, 781–792. [Google Scholar] [CrossRef]
- Schachter, K.A.; Krauss, R.S. Murine models of holoprosencephaly. Curr. Top. Dev. Biol. 2008, 84, 139–170. [Google Scholar]
- Seppala, M.; Xavier, G.M.; Fan, C.M.; Cobourne, M.T. Boc modifies the spectrum of holoprosencephaly in the absence of Gas1 function. Biol. Open 2014, 3, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Lévesque, M.; Morin, S.; Laniel, D.; Wilkes, B.C.; Krauss, R.S.; McMahon, A.P.; Allen, B.L.; Charron, F. Boc and Gas each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 2011, 20, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Seppala, M.; Depew, M.J.; Martinelli, D.C.; Fan, C.M.; Sharpe, P.T.; Cobourne, M.T. Gas1 is a modifier for holoprosencephaly and genetically interacts with Sonic hedgehog. J. Clin. Investig. 2007, 117, 1575–1784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hong, M.; Bae, G.U.; Kang, J.S.; Krauss, R.S. Boc modifies the holoprosencephaly spectrum of Cdo mutant mice. Dis. Models Mech. 2011, 4, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Clagett-Dame, M.; DeLuca, H.F. The role of vitamin A in mammalian reproduction and embryonic development. Ann. Rev. Nutr. 2002, 22, 347–381. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dollé, P. Embryonic retinoic acid synthesis is required for fore limb growth and anteroposterior patterning in the mouse. Development 2002, 129, 3563–3574. [Google Scholar] [PubMed]
- Mic, F.A.; Sirbu, I.O.; Duester, G. Retinoic acid synthesis controlled by Raldh2 is required early for limb bud initiation and then later as a proximodistal signal during apical ectodermal ridge formation. J. Biol Chem. 2004, 279, 26698–26706. [Google Scholar] [CrossRef] [PubMed]
- Mic, F.A.; Molotkov, A.; Fan, X.; Cuenca, A.E.; Duester, G. RALDH3, a retinaldehyde dehydrogenase that generates retinoic acid, is expressed in the ventral retina, otic vesicle and olfactory pit during mouse development. Mech. Dev. 2000, 97, 227–230. [Google Scholar] [CrossRef]
- Cunningham, T.J.; Duester, G. Mechanisms of retinoic acid signalling and its roles in organ and limb development. Nat. Rev. Mol. Cell. Biol. 2015, 16, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Ribes, V.; Fraulob, V.; Petkovich, M.; Dollé, P. The oxidizing enzyme CYP26A1 tightly regulates the availability of retinoic acid in the gastrulating mouse embryo to ensure proper head development and vasculogenesis. Dev. Dyn. 2007, 236, 644–653. [Google Scholar] [CrossRef]
- Uehara, M.; Yashirom, K.; Takaokam, K.; Yamamoto, M.; Hamada, H. Removal of maternal retinoic acid by embryonic CYP26 is required for correct Nodal expression during early embryonic patterning. Genes Dev. 2009, 23, 1689–1698. [Google Scholar] [CrossRef]
- Sulik, K.K.; Dehart, D.B.; Rogers, J.M.; Chernoff, N. Teratogenicity of low doses of all-trans retinoic acid in presomite mouse embryo. Teratology 1995, 51, 398–403. [Google Scholar] [CrossRef]
- Nolen, G.A. Effect of high systemic background level of vitamin A on the teratogenicity of all-trans-retinoic acid given either acutely or subacutely. Teratology 1989, 39, 333–339. [Google Scholar] [CrossRef]
- Lohnes, D.; Kastner, P.; Dierich, A.; Mark, M.; LeMeur, M.; Chambon, P. Function of retinoic acid receptor gamma in the mouse. Cell 1993, 73, 643–658. [Google Scholar] [CrossRef]
- Knudsen, P.A. Congenital malformations of upper incisors in exencephalic mouse embryos, induced by hypervitaminosis A. II. Morphology of fused upper incisors. Acta Odontol. Scand. 1965, 23, 391–409. [Google Scholar] [CrossRef]
- Kalter, H.; Warkany, J. Experimental production of congenital malformations in strains of inbred mice by maternal treatment with hypervitaminosis A. Am. J. Pathol. 1961, 38, 1–21. [Google Scholar]
- Rutledge, J.C.; Shoubaji, A.G.; Hughes, L.A.; Polifka, J.E.; Cruz, Y.P.; Bishop, J.B.; Generoso, W.M. Limb and lower-body duplications induced by retinoic acid in mice. Proc. Natl. Acad. Sci. USA 1994, 91, 5436–5440. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Shimotake, T.; Yanagihara, I.; Iwai, N. Development of anorectal malformations using etretinate. J. Pediatr. Surg. 1998, 33, 127–129. [Google Scholar] [CrossRef]
- Kubota, Y.; Shimotake, T.; Iwai, N. Congenital anomalies in mice induced by etretinate. Eur. J. Pediatr. Surg. 2000, 10, 248–251. [Google Scholar] [CrossRef]
- Mo, R.; Freer, A.M.; Zinyk, D.L.; Crackower, M.A.; Michaud, J.; Heng, H.Q.; Chik, K.W.; Shi, X.M.; Tsui, L.C.; Cheng, S.H.; et al. Specific and redundant functions of Gli2 and Gli3 zinc finger genes in skeletal patterning and development. Development 1997, 124, 113–123. [Google Scholar]
- Lan, Y.; Jiang, R. Sonic Hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal growth. Development 2009, 136, 1387–1396. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, R.J.; Song, C.; Sulik, K.K.; Everson, J.L.; Gipp, J.J.; Yan, D.; Bushman, W.; Rowland, I.J. Cleft lip and palate from hedgehog signaling antagonism in the mouse: Phenotypic characterization and clinical implications. Birth Defects Res. Clin. Mol. Teratol. 2010, 88, 232–240. [Google Scholar] [CrossRef]
- Heyne, G.W.; Melberg, C.G.; Doroodchi, P.; Parins, K.F.; Kietzman, H.W.; Everson, J.L.; Ansen-Wilson, L.J.; Lipinski, R.J. Definition of critical periods for Hedgehog signaling pathway antagonist-induced holoprosencephaly, cleft lip and cleft palate. PLoS Genet. 2015, 10, e0120517. [Google Scholar]
- Dupé, V.; Matt, N.; Garnier, J.M.; Chambon, P.; Mark, M.; Ghyselinck, N.B. A newborn lethal defect due to inactivation of retinaldehyde dehydrogenase type 3 is prevented by maternal retinoic acid treatment. Proc. Natl. Acad. Sci. USA 2003, 100, 14036–14041. [Google Scholar] [CrossRef] [PubMed]
- Molotkova, N.; Molotkov, A.; Duester, G. Role of retinoic acid during forebrain development begins late when Raldh3 generates retinoic acid in the ventral subventricular zone. Dev. Biol. 2007, 303, 601–610. [Google Scholar] [CrossRef]
- Laue, K.; Pogoda, H.M.; Daniel, P.B.; van Haeringen, A.; Alany, Y.; von Ameln, S.; Rachwalski, M.; Morgan, T.; Gray, M.J.; Breuning, M.H.; et al. Craniosynostosis and multiple skeletal anomalies in humans and zebrafish result from a defect in the localized degradation of retinoic acid. Am. J. Hum. Genet. 2011, 89, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, R.; Valencia, C.; Chandraratna, R.A.; Covarrubias, L. Programmed cell death is required for palate shelf fusion and is regulated by retinoic acid. Dev. Biol. 2002, 245, 145–156. [Google Scholar] [CrossRef]
- Abe, M.; Maeda, T.; Wakisaka, S. Retinoic acid affects craniofacial patterning by changing Fgf8 expression in the pharyngeal ectoderm. Dev. Growth Differ. 2008, 50, 717–729. [Google Scholar] [CrossRef]
- Litingtung, Y.; Lei, L.; Westphal, H.; Chiang, C. Sonic Hedgehog is essential to foregut development. Nat. Genet. 1998, 20, 58–61. [Google Scholar] [CrossRef]
- Pepicelli, C.V.; Lewis, P.M.; McMahon, A.P. Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr. Biol. 1998, 8, 1083–1096. [Google Scholar] [CrossRef]
- Motoyama, J.; Liu, J.; Mo, R.; Ding, Q.; Post, M.; Hui, C.C. Essential function of Gli2 and Gli3 in the formation of lung, trachea and oesophagus. Nat. Genet. 1998, 20, 54–57. [Google Scholar] [CrossRef]
- Malpel, S.; Mendelsohn, C.; Cardoso, W.V. Regulation of retinoic acid signaling during lung morphogenesis. Development 2000, 127, 3057–3067. [Google Scholar]
- Mendelsohn, C.; Lohnes, D.; Décimo, D.; Lufkin, T.; Chambon, P.; Mark, M. Function of the retinoic acid receptors (RARs) during development. II. Multiple abnormalities at various stages of organogenesis in RAR double mutants. Development 1994, 120, 2749–2771. [Google Scholar]
- Ramalho-Santos, M.; Melton, D.A.; McMahon, A.P. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 2000, 127, 2763–2772. [Google Scholar]
- Mo, R.; Kim, J.H.; Zhang, J.; Chiang, C.; Hui, C.H.; Kim, P.C.W. Anorectal malformations caused by defects in Sonic Hedgehog signaling. Am. J. Pathol. 2001, 159, 765–773. [Google Scholar] [CrossRef]
- Yu, J.; Carroll, T.J.; McMahon, A.P. Sonic Hedgehog regulates proliferation and differentiation of mesenchymal cells in the mouse metanephric kidney. Development 2002, 129, 5301–5312. [Google Scholar] [PubMed]
- Rosselot, C.; Spraggon, L.; Chia, I.; Batourina, E.; Riccio, P.; Lu, B.; Niederreither, K.; Dolle, P.; Duester, G.; Chambon, P.; et al. Non-cell autonomous retinoid signaling is crucial for renal development. Development 2010, 137, 283–292. [Google Scholar] [CrossRef]
- Bitgood, M.J.; Shen, L.; McMahon, A.P. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr. Biol. 1996, 6, 298–304. [Google Scholar] [CrossRef]
- Vernet, N.; Dennefeld, C.; Rochette-Egly, M.; Ouled Abdelghani, M.; Chambon, P.; Ghyselinck, N.B.; Mark, M. Retinoic acid metabolism and signaling pathways in adult and developing mouse testis. Endocrinology 2006, 147, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Teletin, M.; Vernet, N.; Yu, J.; Klopfenstein, M.; Jones, J.W.; Féret, B.; Kane, M.A.; Ghyselinck, N.B.; Mark, M. Two functionally redundant sources of retinoic acid secure spermatogonia differentiation in the seminiferous epithelium. Development 2019, 146. [Google Scholar] [CrossRef] [PubMed]
- Bowles, J.; Feng, C.W.; Ineson, J.; Miles, K.; Spiler, C.M.; Harley, V.R.; Sinclair, A.H.; Koopman, P. Retinoic acid antagonizes testis development in mice. Cell Rep. 2018, 24, 1330–1341. [Google Scholar] [CrossRef]
- Li, H.; MacLean, G.; Cameron, D.; Clagett-Dame, M.; Petkovitch, M. Cyp26b1 expression in murine Sertoli cells is required to maintain germ cells in an undifferentiated state during embryogenesis. PLoS ONE 2009, 4, e7501. [Google Scholar] [CrossRef]
- Biswas, N.M.; Deb, C. Testicular degeneration in rats during hypervitamisosis A. Endokrinologie 1965, 49, 64–69. [Google Scholar]
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef]
- Long, F.; Zhang, X.M.; Karp, S.; Yang, Y.; McMahon, A.P. Genetic manipulation of hedgehog signaling in the endochondral skeleton reveals a direct role in the regulation of chondrocyte proliferation. Development 2001, 128, 5099–5108. [Google Scholar]
- Minegishi, Y.; Sakai, Y.; Yahara, Y.; Akiyama, H.; Yoshikawa, H.; Hosokawa, K.; Tsumaki, N. Cyp26b1 within the growth plate regulates bone growth in juvenile mice. Biochem. Biophys. Res. Commun. 2014, 454, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Shimono, K.; Tung, W.E.; Macolino, C.; Chi, A.H.T.; Didizian, J.J.; Mundy, C.; Chandraratna, R.A.; Mishina, Y.; Enomoto-Iwamoto, M.; Pacifici, M.; et al. Potent inhibition of heterotopic ossification by nuclear retinoic acid receptor-γ agonists. Nat. Med. 2011, 17, 454–460. [Google Scholar] [CrossRef]
- Gritli-Linde, A.; Hallberg, K.; Harfe, B.D.; Reyahi, A.; Kannius-Janson, M.; Nilsson, J.; Cobourne, M.T.; Sharpe, P.T.; McMahon, A.P.; Linde, A. Abnormal hair development and apparent follicular transformation into mammary glands in the absence of hedgehog signaling. Dev. Cell 2007, 12, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Everts, H.B.; Sundberg, J.P.; King, L.E., Jr.; Ong, D.E. Immunolocalization of enzymes, binding proteins and receptors sufficient for retinoic acid synthesis and signaling during the hair cycle. J. Investig. Dermatol. 2007, 127, 1593–1604. [Google Scholar] [CrossRef] [PubMed]
- Okano, J.; Lichti, U.; Mamiya, S.; Aronova, M.; Zhang, G.; Yuspa, S.H.; Hamada, H.; Sakai, Y.; Morasso, M.I. Increased retinoic acid levels through ablation of Cyp26b1 determine the processs of embryonic skin barrier formation and peridermal development. J. Cell Sci. 2012, 125, 1827–1836. [Google Scholar] [CrossRef]
- Hardy, M.H. Glandular metaplasia of hair follicles and other responses to vitamin A excess in cultures of rodent skin. J. Embryol. Exp. Morph. 1968, 19, 157–180. [Google Scholar]
- Viallet, J.P.; Dhouailly, D. Retinoic acid and mouse skin morphogenesis. II. Role of epidermal competence in hair glandular metaplasia. Dev. Biol. 1994, 166, 277–288. [Google Scholar] [CrossRef]
- Blanchet, S.; Favier, B.; Chevalier, G.; Kastner, P.; Michaille, J.J.; Chambon, P.; Dhouailly, D. Both retinoic acid receptors alpha (RARalpha) and gamma (RAR gamma) are able to initiate mouse upper-lip skin glandular metaplasia. J. Investig. Dermatol. 1998, 111, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Fell, H.B.; Mellanby, E. Metaplasia produced in cultures of chick ectoderm by high vitamin A. J. Physiol. 1953, 119, 470–488. [Google Scholar] [CrossRef]
- Lawrence, D.J.; Bern, H.A. Mucous gland formation in keratinized adult epithelium in situ treated with vitamin A. Exp. Cell Res. 1960, 21, 443–446. [Google Scholar] [CrossRef]
- Lawrence, D.J.; Bern, H.A.; Steadman, M.G. Vitamin A and keratinization. Studies on the hamster cheek pouch. Ann. Otol. Rhinol. Laryngol. 1960, 69, 645–661. [Google Scholar] [CrossRef]
- Covant, H.A.; Hardy, M.H. Stability of the glandular morphogenesis produced by retinoids in the newborn hamster cheek pouch in vitro. J. Exp. Zool. 1988, 246, 139–149. [Google Scholar] [CrossRef]
- So, P.L.; Lee, K.; Hebert, J.; Walker, P.; Lu, Y.; Hwang, J.; Kopelovich, L.; Athar, M.; Bickers, D.; Aszterbaum, M.; et al. Topical tazarotene chemoprevention reduces basal cell carcinoma number and size in Ptch+/− mice exposed to ultraviolet or ionizing radiation. Cancer Res. 2004, 64, 4385–4390. [Google Scholar] [CrossRef]
- Riddle, R.D.; Johnson, R.L.; Laufer, E.; Tabin, C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell 1993, 75, 1401–1416. [Google Scholar] [CrossRef]
- Tickle, C.; Towers, M. Sonic hedgehog signaling in limb development. Front. Cell Dev. Biol. 2017, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Bouldin, C.M.; Gritli-Linde, A.; Ahn, S.; Harfe, B.D. Shh pathway activation is present and required within the vertebrate limb bud apical ectodermal ridge for normal auropod patterning. Proc. Natl. Acad. Sci. USA 2010, 107, 5489–5494. [Google Scholar] [CrossRef] [PubMed]
- Scherz, P.J.; McGlinn, E.; Nissim, S.; Tabin, C.J. Extended exposure to Sonic hedgehog is required for patterning the posterior digits of the vertebral limb. Dev. Biol. 2007, 308, 343–354. [Google Scholar] [CrossRef]
- Zhang, R.; Lee, C.; Lawson, L.Y.; Svete, L.J.; McIntyre, L.M.; Harfe, B.D. SHH protein variance in the limb bud is constrained by feedback regulation and correlates with altered digit patterning. Genes Genomes Genet. 2017, 7, 851–858. [Google Scholar] [CrossRef]
- Zhou, J.; Kochlar, D.M. Cellular anomalies underlying retinoic-acid-induced phocomelia. Reprod. Toxicol. 2004, 19, 103–110. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El Shahawy, M.; Reibring, C.-G.; Hallberg, K.; Neben, C.L.; Marangoni, P.; Harfe, B.D.; Klein, O.D.; Linde, A.; Gritli-Linde, A. Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development. Int. J. Mol. Sci. 2019, 20, 2275. https://doi.org/10.3390/ijms20092275
El Shahawy M, Reibring C-G, Hallberg K, Neben CL, Marangoni P, Harfe BD, Klein OD, Linde A, Gritli-Linde A. Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development. International Journal of Molecular Sciences. 2019; 20(9):2275. https://doi.org/10.3390/ijms20092275
Chicago/Turabian StyleEl Shahawy, Maha, Claes-Göran Reibring, Kristina Hallberg, Cynthia L. Neben, Pauline Marangoni, Brian D. Harfe, Ophir D. Klein, Anders Linde, and Amel Gritli-Linde. 2019. "Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development" International Journal of Molecular Sciences 20, no. 9: 2275. https://doi.org/10.3390/ijms20092275
APA StyleEl Shahawy, M., Reibring, C.-G., Hallberg, K., Neben, C. L., Marangoni, P., Harfe, B. D., Klein, O. D., Linde, A., & Gritli-Linde, A. (2019). Sonic Hedgehog Signaling Is Required for Cyp26 Expression during Embryonic Development. International Journal of Molecular Sciences, 20(9), 2275. https://doi.org/10.3390/ijms20092275