1. Introduction
In recent years, increasing attention has been paid to the contribution of lysosomes to autoimmune diseases [
1,
2]. Of all the cellular components, the lysosome is an obvious candidate to play a role in the inflammatory process [
3]. The relatively recent, intensively developing field of “lysosomics” provides a range of important information regarding the role of lysosomes in the inflammatory response and autoimmune conditions [
4,
5,
6,
7], despite the fact that the first signs of lysosomal involvement in inflammatory diseases were identified decades ago [
8]. Unfortunately, these investigations were only superficially concerned with inflammatory skin diseases, including the most common human skin autoimmune disease, psoriasis (PsO) [
9,
10,
11,
12]. PsO, considered to develop through the crosstalk between the epidermal keratinocytes and immunocytes, is a polygenic, chronic, inflammatory skin disease affecting approximately 2−3% of the population worldwide. It is characterized by keratinocyte hyperproliferation, abnormal epidermal differentiation, and infiltration of immune cells into lesions [
13]. Research carried out in the 1960’s highlighted the differences in the presence of lysosomes in the keratinocytes of psoriatic skin in relation to healthy skin [
14]. Several subsequent studies have shown that affected psoriatic skin differs from normal skin with respect to the level of activity of lysosomal hydrolytic enzymes and membrane proteins [
15,
16,
17,
18]. Nevertheless, the outcomes of these studies are generally contradictory, since both increases and decreases in the activity of these enzymes were observed [
18]. It should be mentioned, however, that this phenomenon was determined in various tissues and cells such as skin, scales covering psoriatic papules, lymphocytes, granulocytes, neutrophils, and blood serum, which could have led to these discrepancies. The observations have become increasingly widespread, since multiple studies have reported on the manner by which disruption of normal lysosomal function leads to abnormalities in inflammation and immunity [
1,
6,
18,
19,
20]. The link between lysosomes and skin inflammation was established by studies conducted in the 1970’s [
21,
22,
23]. Inflammation in skin diseases, such as PsO, is regulated by a complex network of keratinocytes and immune cells that infiltrate the epidermis [
24]. Understanding of the lysosome signaling in the immune response is still in its infancy, same as knowledge of the potential involvement of lysosomes and lysosomal compartments in the inflammatory signaling network in respect to inflammatory skin diseases is residual. The question is, how can we move forward to gain further information and a greater understanding of this issue. To carry out studies assessing the potential contribution of lysosomes to the disturbed processes of apoptosis and autophagy, abnormalities of sphingolipid and ceramide metabolism pathways, incorrect presentation of antigens by major histocompatibility complex (MHC) class I and II molecules or increased transcription of genes encoding selected proinflammatory cytokines and chemokines—processes being deregulated in this dermatosis, the gaps in knowledge must be filled using recently developed “lysosomics”, therein in relevance to psoriasis. Hence, in vitro and in situ study of the pattern of the lysosomal system, a potential target for therapeutic intervention in human autoimmune-mediated inflammatory skin diseases, was the focus of the present research report. In addition to the human immortalized keratinocyte HaCaT model, skin tissue from biopsies of controls, healthy individuals, and patients with psoriasis, from the involved and noninvolved psoriatic skin, were evaluated.
3. Discussion
The involvement and specific role of lysosomes and their components in the maintenance of cellular architecture is already well-documented in scientific reports on health and disease [
4,
25,
26]; however, the potential impact of lysosomal signaling on autoimmune-mediated inflammation in skin diseases such as psoriasis (PsO) requires further verification. It appears that apart from human immune system cells, the epidermis—composed mostly of keratinocytes—may also be an important area of research, if only due to the proven interaction of both these cell types within a wide-range network regulating the biological pattern of the largest and fastest-growing organ of the human body—the skin.
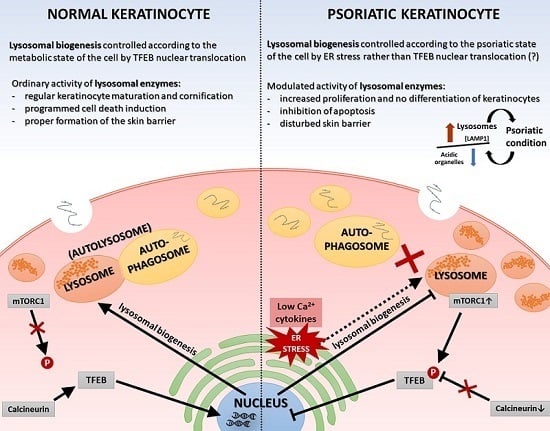
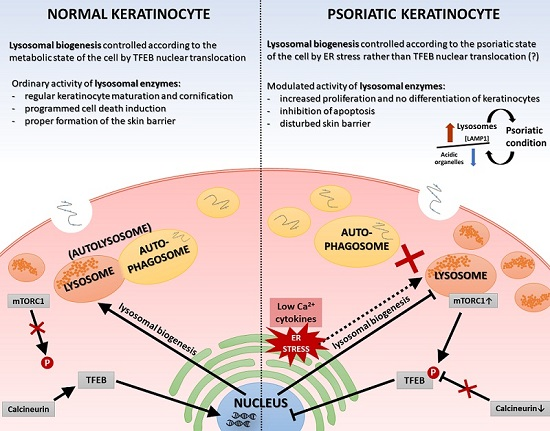
Normal epidermal maturation is associated with an increased number of lysosomes involved in intracellular degradation. It is important to determine the potential consequences of changes in the amount and morphology of lysosomes and other acidic organelles in psoriasis, which may be manifested, among others, in disturbed processes of apoptosis and autophagy, abnormalities of sphingolipid and ceramide metabolism pathways, incorrect presentation of antigens by major histocompatibility complex (MHC) class II molecules or increased transcription of genes encoding selected proinflammatory cytokines and chemokines—processes being deregulated in this dermatosis. A significant number of alterations at the level of a single cell, affecting the homeostasis of the whole organism, can be largely explained by the characteristics of the action of lysosomes. They can play a key role in the modulation of the immune response, and thus may be a potential therapeutic target for many inflammatory disorders, such as rheumatoid arthritis, atopic dermatitis, and psoriasis. For this reason, in our study we undertook research on the verification of vicissitudes in the endosomal-lysosomal system, at this stage of work in cells mimicking psoriasis (keratinocyte model of HaCaT cells with “psoriasis-like” inflammation), with simultaneous analysis of material driven from people suffering from this dermatosis.
Despite the many controversies surrounding the use of proper epidermal keratinocytes for in vitro studies relevant to PsO, the most widely used human immortalized keratinocyte cell line, HaCaT, has been employed as a cellular model to investigate hyperproliferative skin diseases [
27,
28,
29,
30]. HaCaT keratinocytes are often used instead of primary keratinocytes (NHEK) for in vitro experiments related to psoriasis, since the susceptibility of primary cells to treatment may change with increasing numbers of passages, whereas HaCaT provides an almost unlimited supply of identical cells, ensuring high reproducibility [
31]. On the other hand, it is necessary to add here that HaCaT keratinocytes are not perfect in this sense. They have a sufficient limitation as a cellular model of psoriasis due to their treatment with proinflammatory cytokines, which may not sufficiently increase proliferation rate of cells. Thus, the studies of psoriasis based on HaCaT cells as an experimental model shall take into account this important phenomenon, and if only possible, accompanied by additional tests, e.g. using tissues taken from patients. In turn, it is also worth adding that, cultured primary human keratinocytes are frequently employed for studies of psoriasis; however, interpretation of experimental data may be complicated by donor to donor variability, the relatively short culture lifetime, and variations between passages. Here, we investigated the use of HaCaT cells, a long-lived, spontaneously immortalized human keratinocyte line which is able to differentiate in vitro, as a suitable model to become undifferentiated cells under appropriate experimental conditions, mimicking the clinical psoriatic situation as closely as possible. Going forward, an important issue regarding psoriasis research is the involvement of in vivo experiments; thus, in addition to the HaCaT model with “psoriasis-like” inflammation, skin specimens of lesioned and non-lesioned tissue were examined in comparison with healthy control samples.
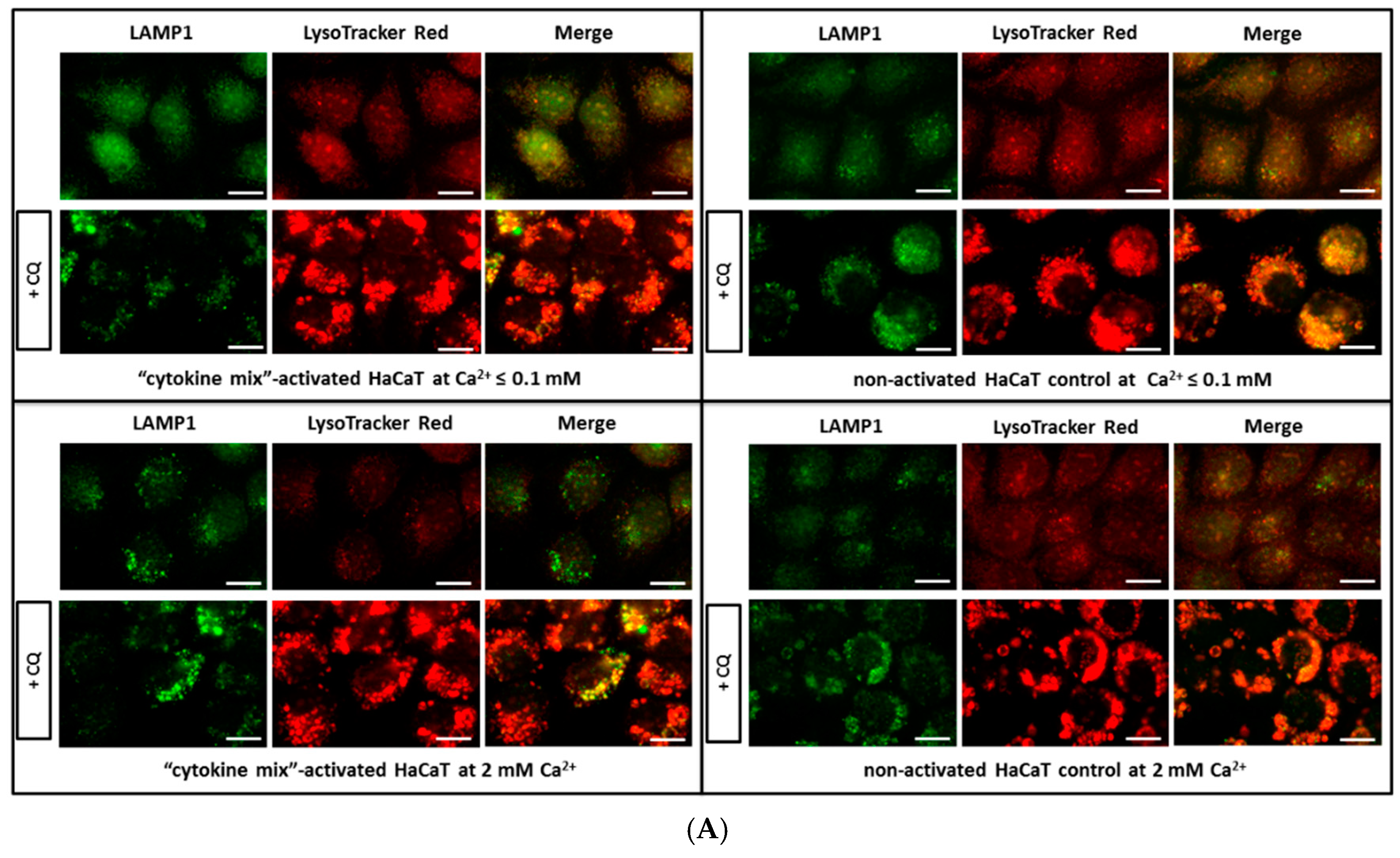
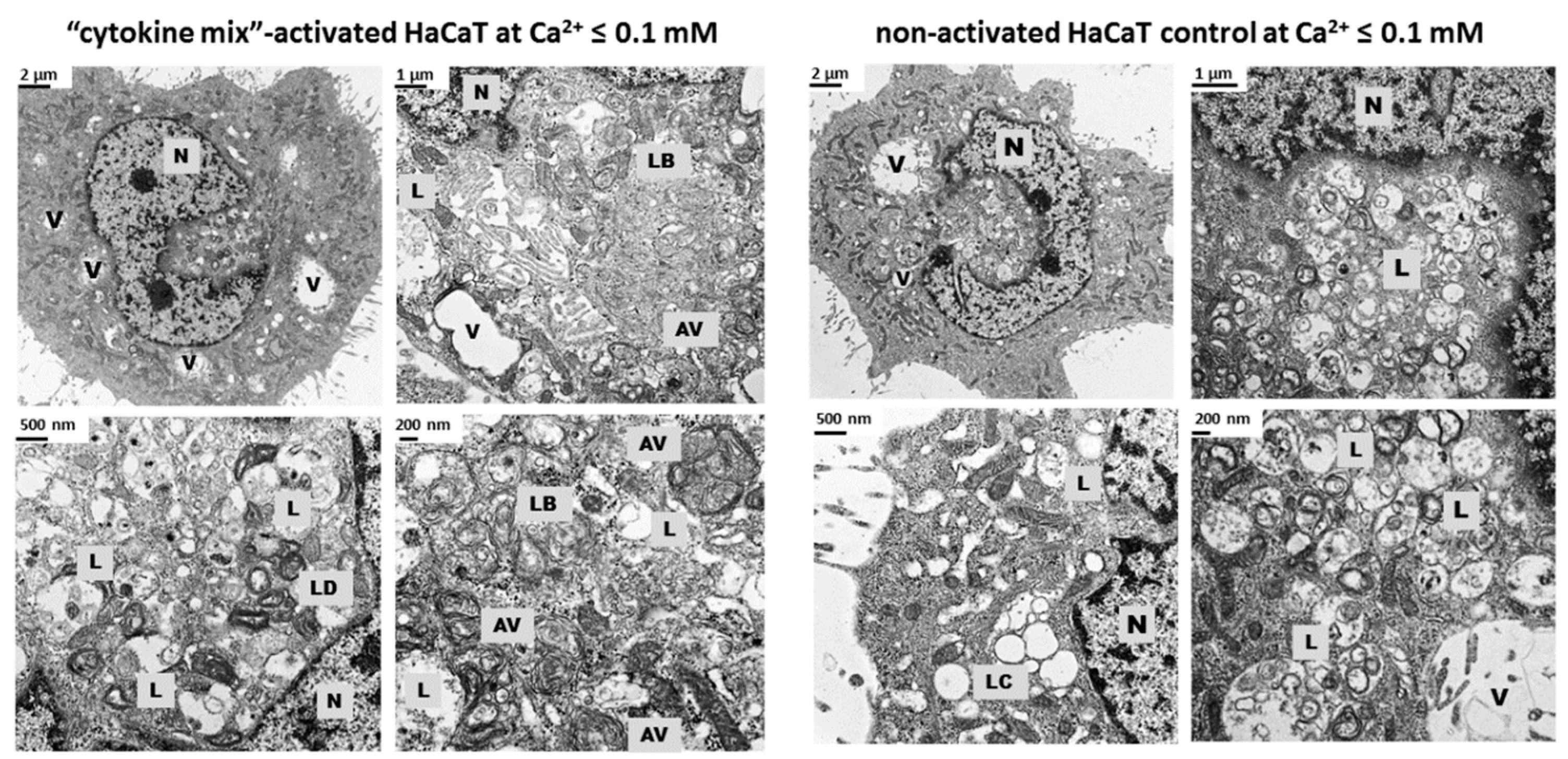
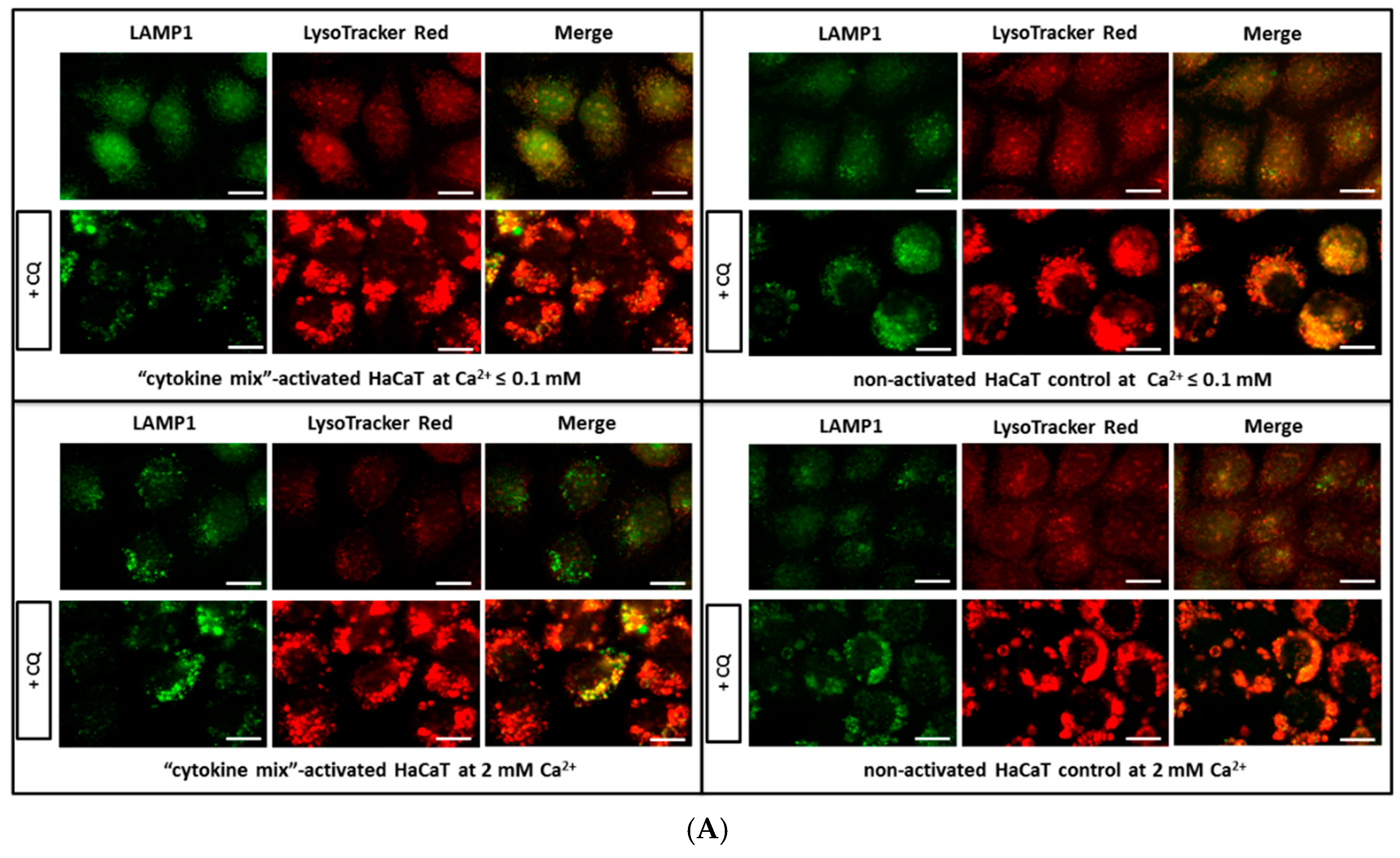
To verify the possible quantitative changes in organelles belonging to the lysosomal system among the tested keratinocytes, fluorescence microscopy was first applied. Indeed, the data demonstrates alterations in the cellular status at the level of lysosomal compartments and lysosome-related organelles (LROs) (
Figure 1). In all staining tests, i.e., in the presence of LysoTracker, anti-lysosomal-associated membrane protein 1 (LAMP1) antibodies, and acridine orange (AO), an augmented level of lysosomes in HaCaT cells with “psoriasis-like” inflammation was found relative to the non-activated HaCaT cell control cultured in medium containing either Ca
2+ ≤ 0.1 mM or 2 mM Ca
2+. A similar tendency was noticed in the immunohistochemical study of skin samples taken from psoriasis patients (
Figure 4).
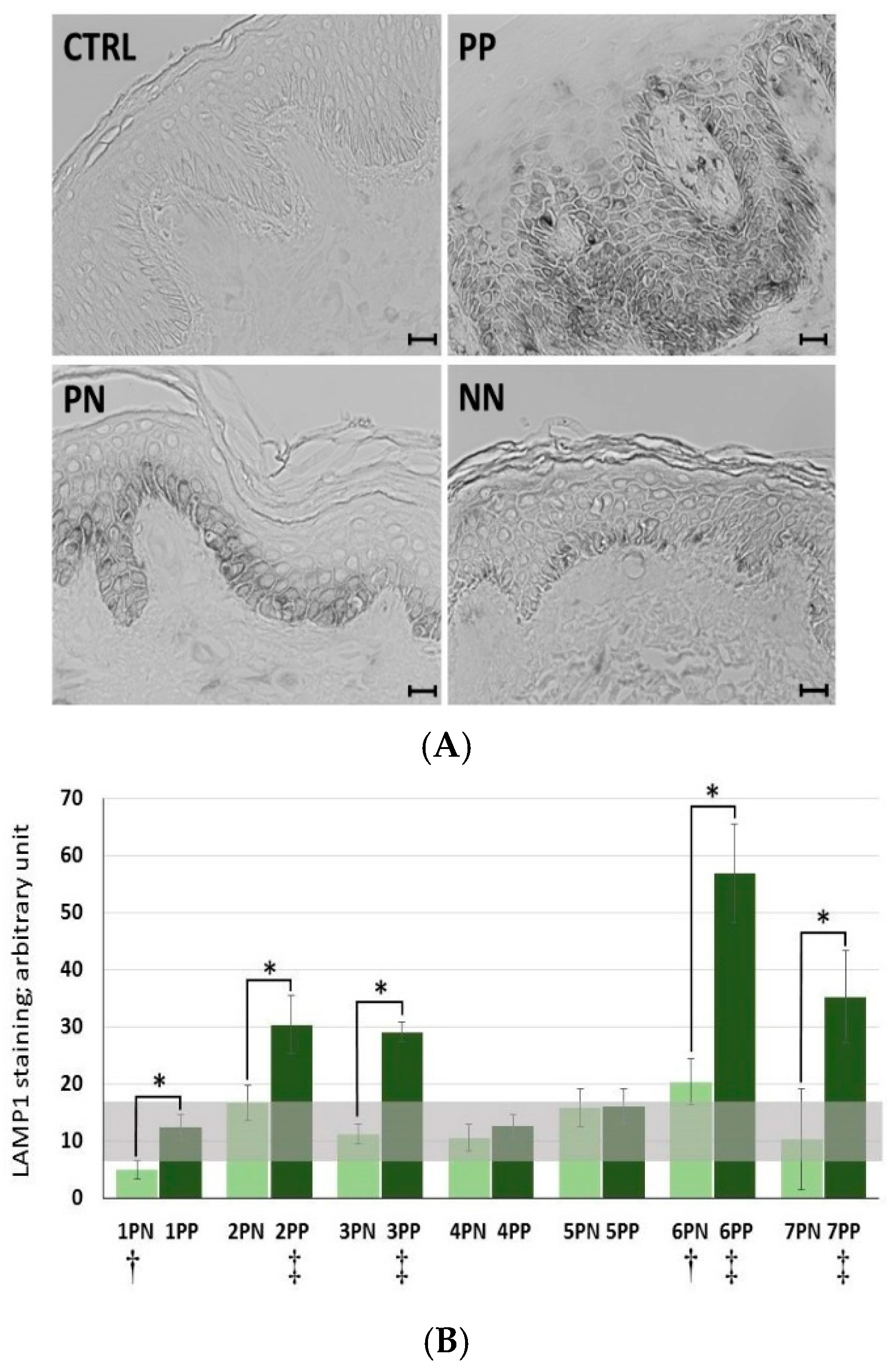
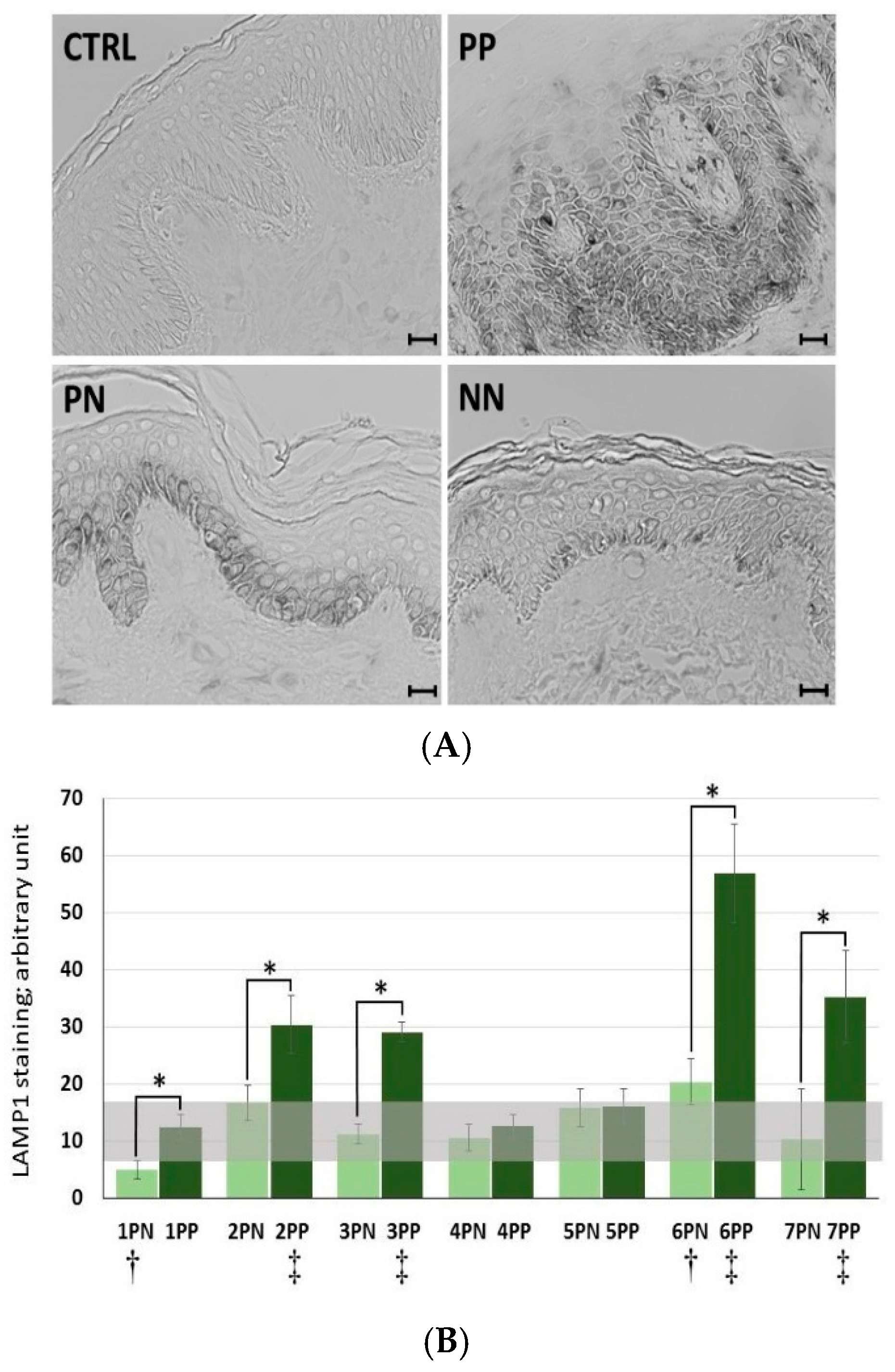
In the case of more than 70% of the tested patients (five of seven subjects), there was a statistically significant increase in LAMP1 expression in psoriatic lesioned (PP) versus non-lesioned (PN) skin, as well as in four of these five patients when PN was compared with non-psoriatic normal (NN) (
Figure 4). Interestingly, it was found that patients with the most pronounced PASI (Psoriasis Area and Severity Index), BSA (Body Surface Area), and DLQI (Dermatology Life Quality Index) scores (patients nos. 6 and 7) had a high incidence of positive LAMP1 expression, which indicates a level of correlation between particular clinical parameters and LAMP1 manifestation (
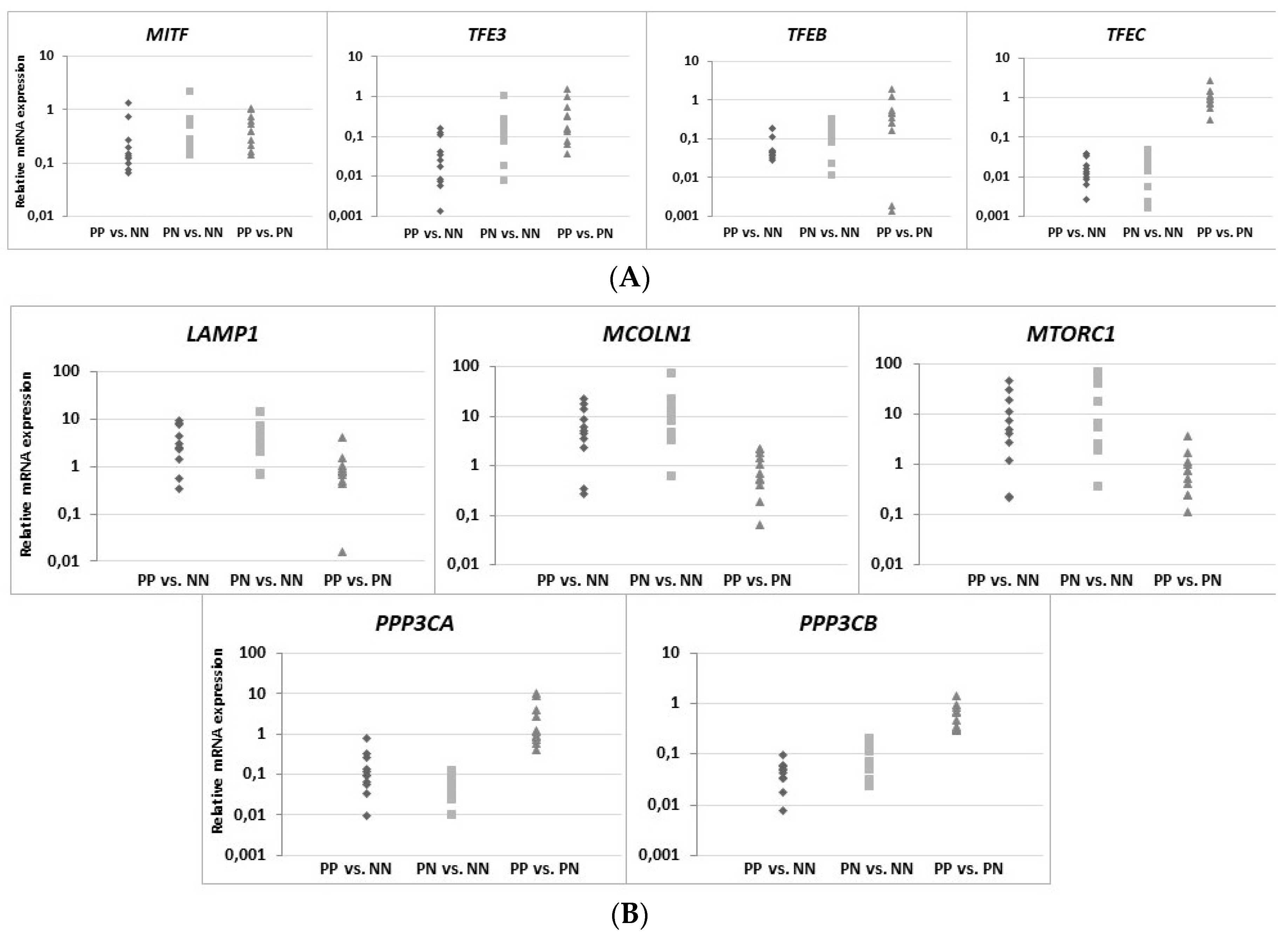
Table 2). However, no significant association was defined between LAMP1 expression and other clinical characteristics; nevertheless, proper assessment was limited due to the small number of evaluated patients. These results were confirmed by real-time qRT-PCR analysis in skin specimens (
Figure 6 and
Table S1). Up-regulation of the
LAMP1 gene (fold change, FC ≥ 1.3) was observed in 82% of psoriatic patients, including PP and PN skin specimens. However, in contrast to our results obtained in skin sections and the data of
LAMP1 expression detected in HaCaT cells (Transcripts Per Million, TPM: 298.9) [
32], there was down-regulation of
LAMP1 in our HaCaT model with “psoriasis-like” inflammation (
Table 3). Since the presence of LAMP1 is noticeable in the same cells in culture, as evidenced by immunofluorescence, the effect on
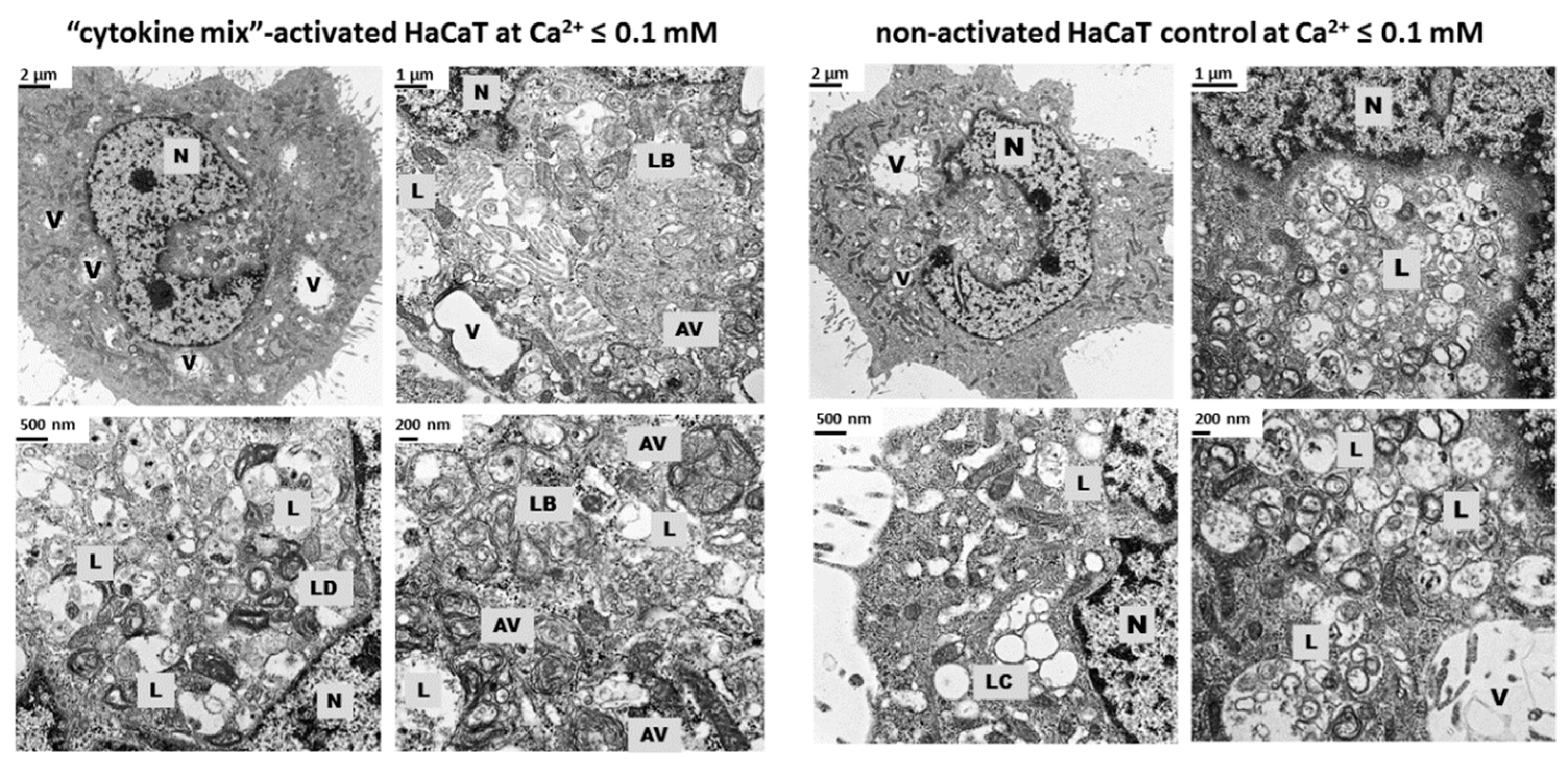
LAMP1 mRNA expression observed in the present study suggests post-transcriptional control of LAMP1 production in HaCaT cells. A possible explanation for the discrepancy in the levels of LAMP1 protein and mRNA in our cultures of keratinocytes is the presence of a feedback loop that degrades the mRNA transcript following synthesis of a large amount of protein. Moreover, electron microscopy identified a substantially increased population of autophagosomes and a reduced population of autolysosomes in keratinocytes with “psoriasis-like” inflammation as compared with the control (
Figure 3). There may be a mechanism that moderates or even blocks efficient fusion of numerous lysosomes with autophagosomes in proinflammatory cytokine-treated HaCaT cells, resulting in a reduction in the autolysosome population in these cells. On the other hand, it is known from the literature that lysosomes and lysosome-like compartments are involved in epidermal differentiation, the stage of cell maturation that is impaired in PsO [
33,
34,
35,
36]. The role of lysosomes in cell differentiation is supported by several studies performed in various cell types including keratinocytes [
36]. In the normal human epidermis, lysosomes in keratinocytes, revealed by LAMP1 staining, were mainly observed in the granular layer, perhaps as lamellar granules involved in the keratinization and desquamation processes, whereas in cultured keratinocytes, lysosomes were abundant in subconfluent conditions. LAMP1 expression is elevated in normal proliferating keratinocytes, while it is significantly down-regulated upon entering the differentiation stage. Furthermore, maturing keratinocytes reveal deregulated lysosomal biogenesis and function, which in turn may be linked to the action of the microphthalmia family (MiT family) transcription factors. Recent studies have led to the identification of a lysosome signaling pathway and gene network that regulate cellular clearance and metabolism [
25,
26]. Using this approach, it was discovered that lysosome status is coordinated by the transcription factor EB (TFEB), a member of the MiT family, and its translocation from the cytosol to the nucleus [
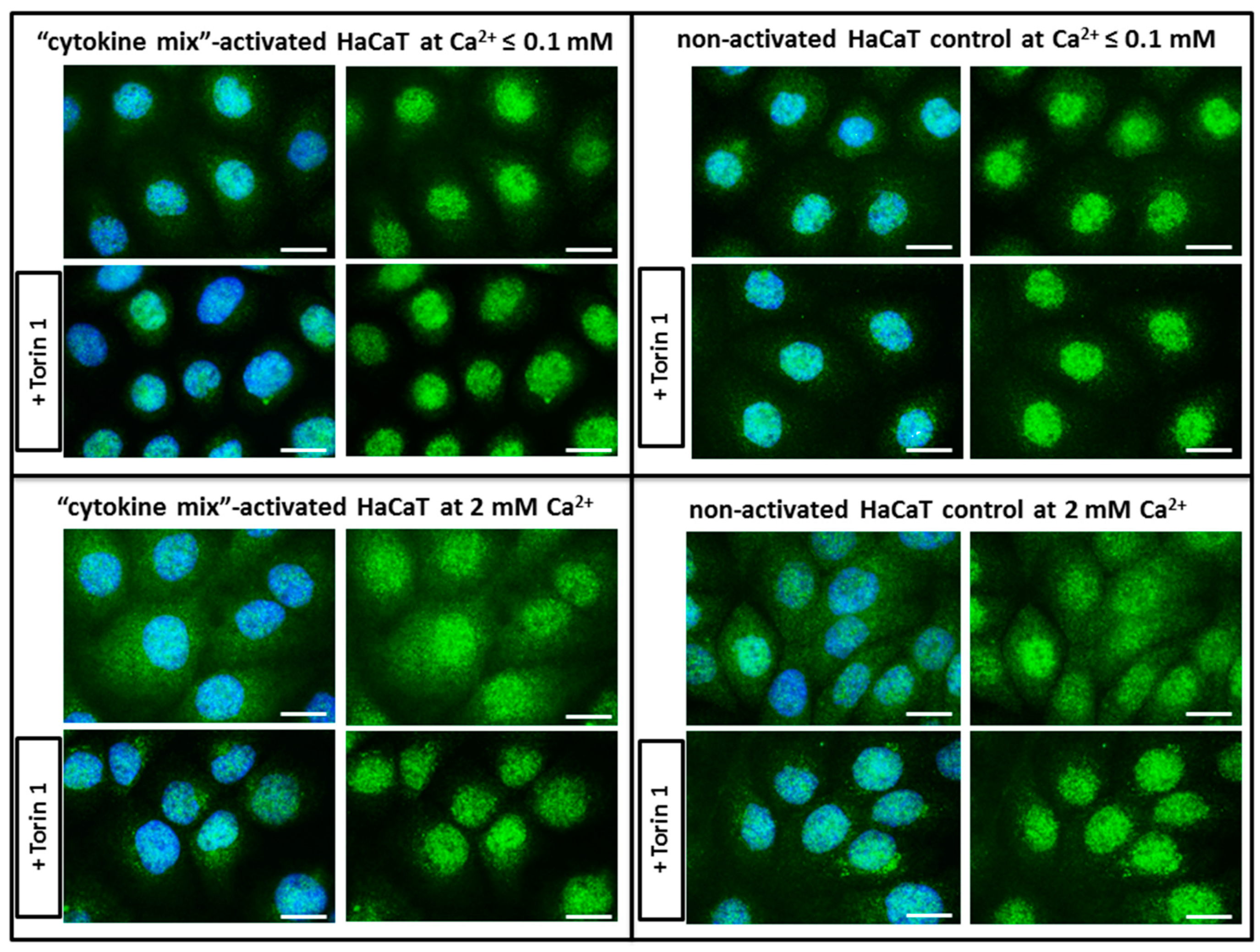
6]. Since TFEB plays a fundamental role in cell homeostasis and globally modulated lysosomal function, an investigation into the endogenous TFEB subcellular localization in our in vitro model was carried out (
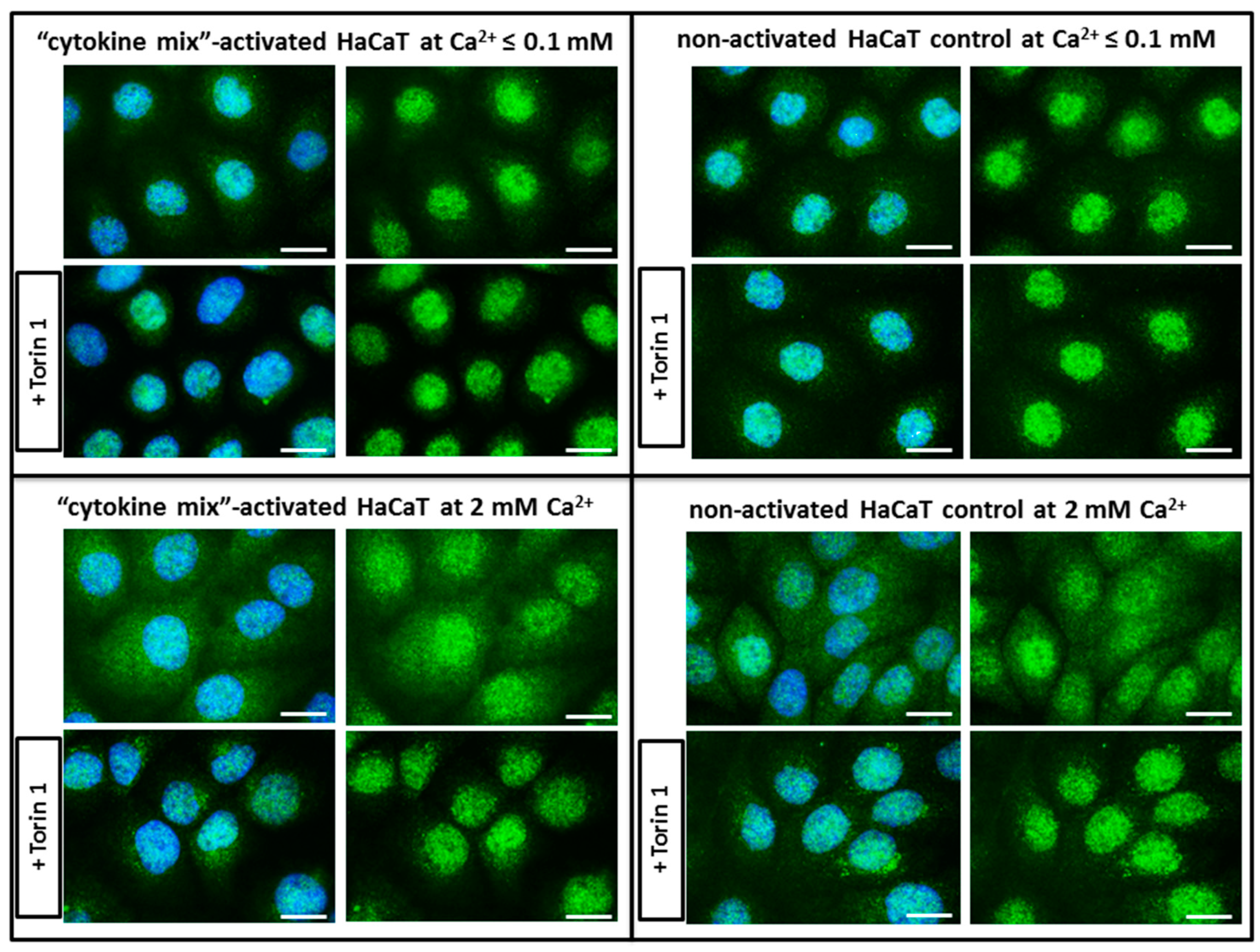
Figure 2). Surprisingly, rather than being due to proinflammatory cytokine stimulation, the cytosol-to-nucleus shuttling of TFEB was calcium-dependent.
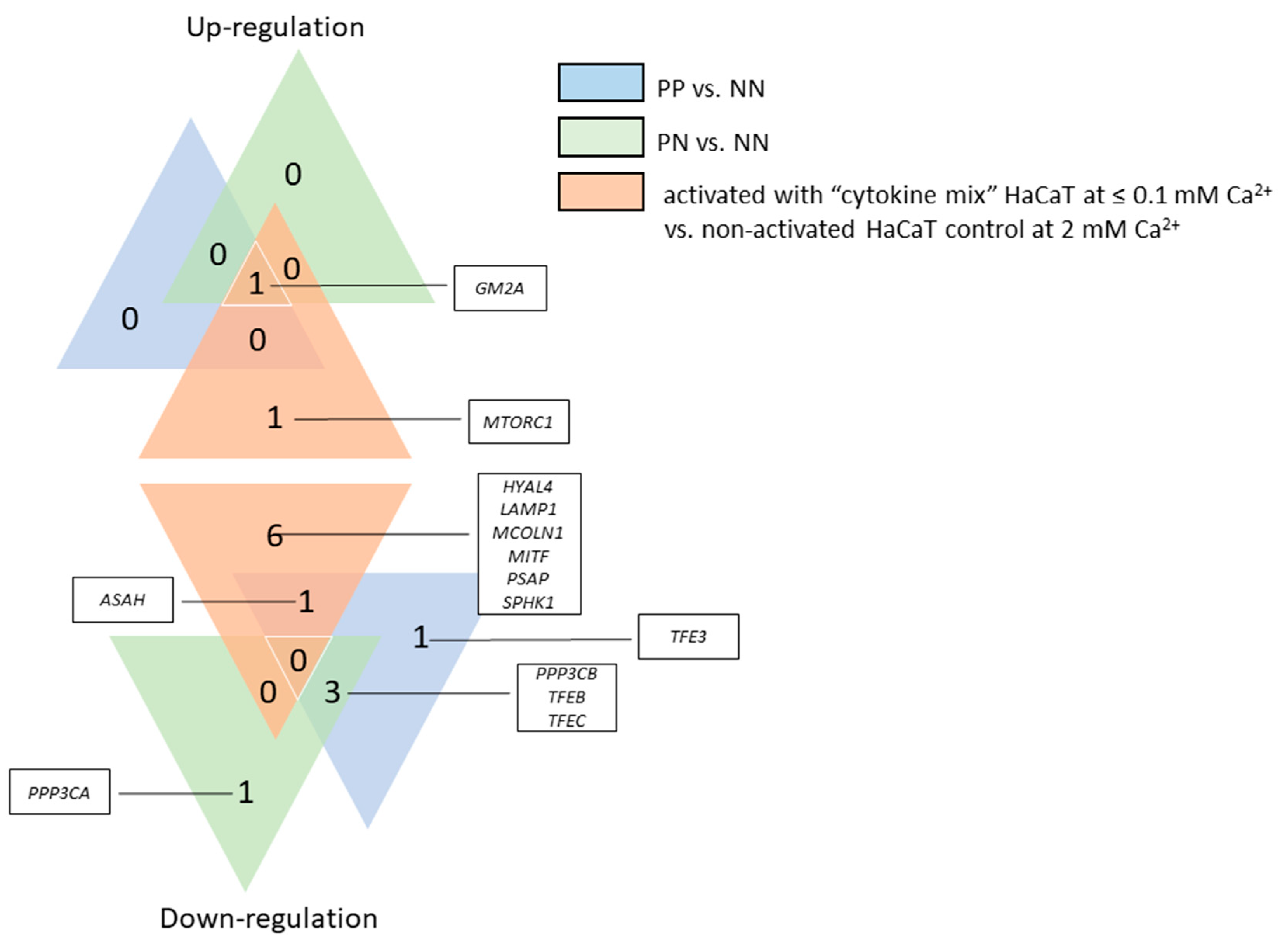
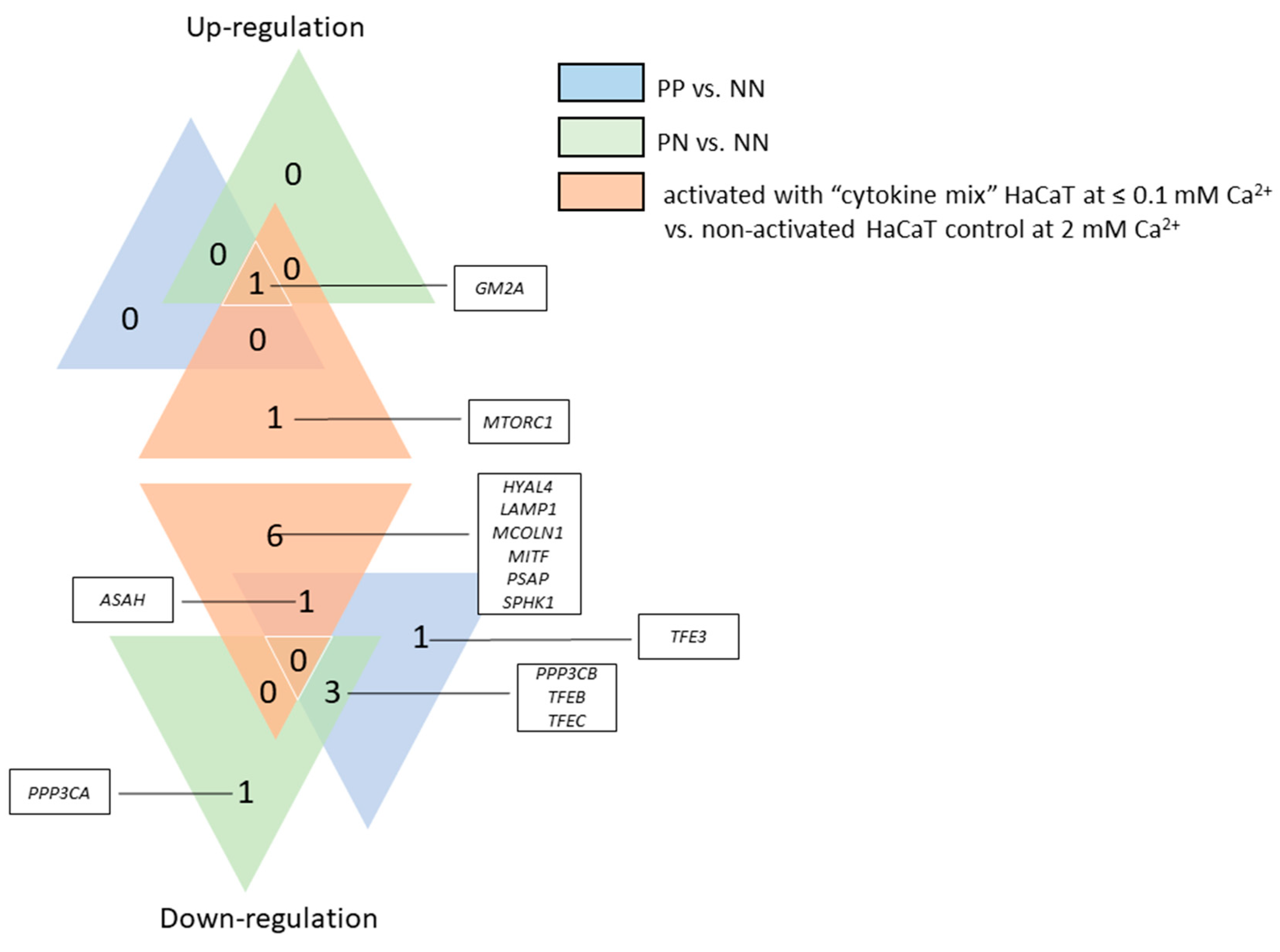
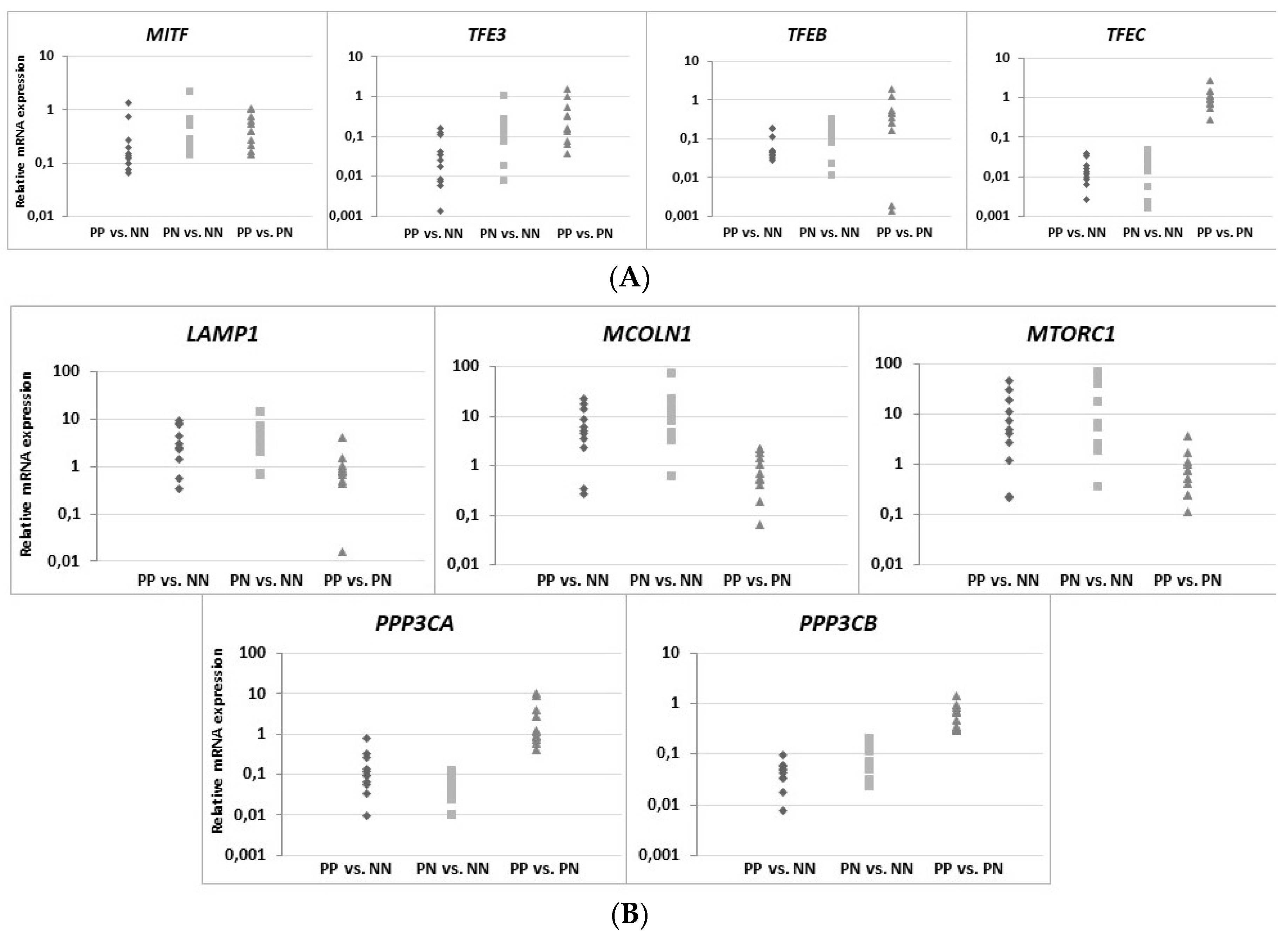
It is worth mentioning that RNA expression profiling studies, carried out to analyze differentially expressed genes (DEGs), revealed a number of changes in the expression of genes specific to lysosomal function or other signaling pathways mediating the pathogenesis of PsO [
12,
37,
38]. Numerous genes associated with lysosomes are termed coordinated lysosomal expression and regulation (CLEAR), the elements of which are recognized by the MiT family factors, promoting gene transcription. With the exception of TFEB, the MiT family of transcription factors include MITF, involved in melanogenesis; TFE3, which promotes the expression of genes downstream of transforming growth factor beta (TGFβ) signaling; and TFEC, which plays a role in cell survival, growth, and differentiation. With respect to lysosomal metabolism studies in this field, an interesting observation is the dysregulation of sphingolipid metabolism, in particular alterations in ceramides being a central hub of the sphingolipid pathway, in inflammatory skin diseases such as psoriasis and atopic dermatitis [
39]. Sphingolipids, as bioactive molecules with a putative involvement in inflammation, play an important role in epidermal signaling [
40]. A wide range of chemical structures of sphingolipids (giving various types of these compounds, i.e., ceramides, ceramide-1-phosphate, sphingomyelin, sphingosine-1-phosphate, or glycosphingolipids) is reflected in their function in the skin, specifically in proliferation signaling, survival and apoptotic processes, cell migration and adhesion, or angiogenesis. The highest percentage (almost 50%) of all sphingolipids in the epidermis are ceramides; however, this quantity is much lower in psoriatic plaque skin, which through its anti-apoptotic effect, contributes to the excessive proliferation of keratinocytes [
40,
41]. Furthermore, quantitative changes of sphingolipids can cause increased transepidermal water loss (TEWL) and epidermal barrier dysfunction. Moreover, there is altered sphingosine conversion in psoriatic patients’ skin, in which the level of the proinflammatory and prosurvival sphingosine-1-phosphate form is more common. In the present study, up-regulation of the
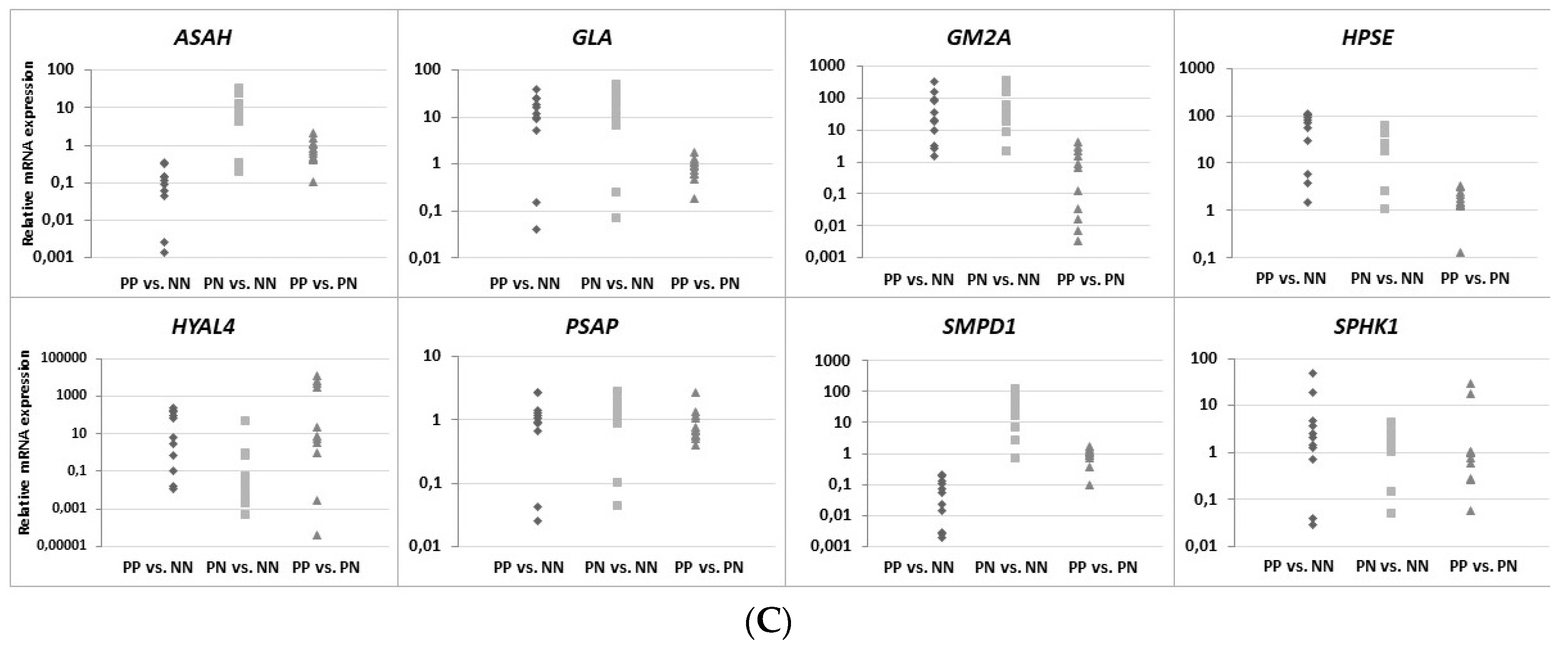
ASAH and
SMPD1 genes (encoding enzymes necessary for the production of ceramide) was observed in PN skin specimens, while down-regulation was observed in PP specimens (
Figure 6 and
Figure S1), which may indicate quantitative disturbances (decreased amounts) in ceramides, resulting in a lack of differentiation and excessive proliferation of keratinocytes, suppression of the apoptotic process, and finally impairment of the skin barrier. Increased
SPHK1 activity in PP tissue sections may additionally result in excessive synthesis of sphingosine-1-phosphate, contributing to the production of tumor necrosis factor alpha (TNF-α), activation of the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) pathway, or secretion of interleukin 8 (IL-8).
On the other hand, the function of the sphingosine form is calcium release from acidic stores, essential for TFEB translocation, which in turn is mediated by mucolipin-1 (MCOLN1) and calcineurin. This mechanism results in the biogenesis of lysosomes and the activation of autophagy processes, which are not observed in psoriatic epidermis [
42,
43,
44]. The
MTORC1 gene was up-regulated in the majority of psoriasis patients in the present study, while the opposite genes,
PPP3CA and
PPP3CB, encoding calcineurin subunits, were down-regulated, which consequently led to reduced mRNA belonging to the MiT family. Interestingly, there was both an up-regulation of
LAMP1 gene activity and an elevated level of signaling protein in psoriatic skin tissue, which may indicate that the overall number of lysosomes is increased in PP skin, but their biogenesis process dependent on the MiT family transcription factors is inhibited. Therefore, endoplasmic reticulum (ER) stress may be a potential cause of the formation of lysosomes [
34].
It must be mentioned that there are certain limitations faced by the present study. For instance, the use of a restricted number of samples may introduce bias to this study; hence, future investigations including larger sample sizes are necessary to confirm the results of the present exploration. A series of further in vitro and in vivo experiments are in progress. We anticipate that our research group will report related studies to illustrate the molecular mechanisms involved in the regulation of lysosome signaling, even considering it as a potential target for therapeutic intervention in serious inflammatory skin diseases such as psoriasis.
4. Materials and Methods
4.1. Cell Line, Culture Media, and Reagents
Spontaneously transformed keratinocytes from histologically normal skin (HaCaT cell line) were purchased from the CLS Cell Lines Service GmbH (Eppelheim, Germany) [
45]. HaCaT cells from passage 38−42 were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific) and 1% antibiotic/antimycotic solution (Gibco, Thermo Fisher Scientific) at 37 °C in a humidified atmosphere containing 5% carbon dioxide (CO
2).
4.2. Two-Dimensional (2D) Engineered Skin Psoriatic Model
HaCaT cells were cultured to 80% confluence in 25-cm2 flasks containing DMEM supplemented with 10% FBS and 1% antibiotic/antimycotic solution. Subsequently, cells were enzymatically digested using 0.025% trypsin−0.01% EDTA and seeded on 6-well plates at a density of 4 × 105 cells/well in defined serum-free keratinocyte medium (Keratinocyte-SFM, Gibco, Thermo Fisher Scientific) supplemented with bovine pituitary extract (BPE) and epidermal growth factor (EGF) (Gibco, Thermo Fisher Scientific). Following a 24-h incubation, the medium was changed to Keratinocyte-SFM without growth supplements for a further 16 h. Cells were then stimulated with a combination of a proinflammatory cytokines (“cytokine mix”): interleukin 1 alpha (IL-1α), interleukin 17A (IL-17A), interleukin 22 (IL-22), oncostatin M (OSM), and tumor necrosis factor-α (TNF-α) (Gibco, Thermo Fisher Scientific) at a concentration of 2 ng/mL each constituent. For better evaluation of the results, two control conditions were carried out: (1) Untreated cells cultured in medium with a low concentration of calcium ions (≤0.1 mM) (Keratinocyte-SFM, Gibco, Thermo Fisher Scientific); and, (2) untreated cells cultured in medium with a high concentration of calcium ions (addition of 2 mM calcium chloride for 24 h) (Keratinocyte-SFM, Gibco, Thermo Fisher Scientific).
4.3. Ethical Consent and Study Design Regarding Skin Biopsies
The in vivo material, i.e., the skin tissue, was collected from 18 psoriasis patients (numbered from 1 to 18), i.e.,: 7 subjects (numbered from 1 to 7) for the immunohistochemistry studies and 11 subjects (numbered from 8 to 18) for the gene expression analysis; and 15 healthy individuals as a control (numbered from 1 to 15), i.e., 4 subjects (numbered from 1 to 4) for the immunohistochemistry studies and 11 subjects (numbered from 5 to 15) for the gene expression analysis, and the study was assembled within the framework of our collaboration with the Department of Dermatology, Venerology, and Allergology at the Medical University of Gdańsk. Signed informed consent was obtained from all subjects under protocols approved by the Independent BioEthics Committee of the Medical University of Gdańsk (NKBBN/161/2017 and NKBBN/161-634/2018). Patient data were classified with respect to gender, age, family history, BMI, and disease indicators such as PASI (Psoriasis Area and Severity Index), BSA (Body Surface Area), and DLQI (Dermatology Life Quality Index).
4.4. Determination of the Localization and Quantitative Changes in the Lysosomal Organelles via Fluorescence Microscopy
To visualize the acidophilic cell compartments, LAMP1 and acridine orange staining were used. Lysosomal membrane integrity and maintenance of the lysosomal−cytosolic pH gradient was assessed using acridine orange (AO) relocation techniques.
An equal number of HaCaT cells (1 × 105 cells/well) was seeded on glass slides (Millicell EZ SLIDE 8-well glass, Millipore, Burlington, MA, USA) in supplemented DMEM (Gibco, Thermo Fisher Scientific) and cultured for 24 h. The culture medium was replaced with Keratinocyte-SFM (Gibco, Thermo Fisher Scientific) supplemented with BPE and EGF (Gibco, Thermo Fisher Scientific). Following a 6-h incubation, cells were cultured in Keratinocyte-SFM without growth supplements for a further 16 h. For subsequent experiments, cells were stimulated for 24 h with a combination of proinflammatory “cytokine mix”: IL-1α, IL-17A, IL-22, OSM, and TNF-α (Gibco, Thermo Fisher Scientific) at a concentration of 2 ng/mL each constituent. The control cells were left unstimulated in Keratinocyte-SFM containing a low concentration of calcium ions (≤0.1 mM) (Gibco, Thermo Fisher Scientific). An additional control without the cytokine mixture but with 2 mM Ca2+ was applied to simulate conditions prone to keratinocyte differentiation.
Images were captured immediately using a Leica DMI4000 B microscope (Leica, Wetzlar, Germany) and the LAS X software (ver 3.0.16120.2, Leica, Wetzlar, Germany) at 100× magnification, from randomly selected ten microscopic fields containing 50–100 cells, each from three independent experiments (n = 3).
4.4.1. LAMP1 Detection Using a Specific Fluorescently Labelled Antibody
To assess the lysosomal number and distribution via LAMP1 staining, proinflammatory cytokine-stimulated HaCaT cells were fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature (RT) and subsequently permeabilized with 0.1% Triton X-100 for 15 min at RT. Cells were then blocked with 3% BSA in PBS + 0.1% Tween for 1 h at 4 °C. Immunostaining was performed by overnight incubation with a LAMP1 primary antibody (#9091, Cell Signaling, Leiden, Netherlands; 1:200) in blocking solution at 4 °C. The following day, cells were washed three times with PBS and incubated with a secondary antibody (anti-rabbit IgG (H + L), F(ab′)2 fragment, Alexa Fluor® 594 Conjugate; #8889, Cell Signaling, Leiden, Netherlands; 1:1000) in blocking solution for 1 h at 4 °C. After three washes with PBS, coverslips were mounted in SlowFade™ Diamond Antifade Mountant with DAPI (Invitrogen, Thermo Fisher Scientific). Each treatment was analyzed in triplicate in three independent experiments.
4.4.2. Visualization of Acidic Organelles Using Acridine Orange (AO) Staining
Since AO is a metachromatic fluorophore that emits red fluorescence at high acidic pH (intact lysosome) and green fluorescence at low acidic pH (cytosol and nucleus), lysosomal membrane destabilization was monitored as a decrease in granular red fluorescence or an increase in diffuse cytoplasmic green fluorescence. Chloroquine (CQ), which raises lysosomal pH, was used as a positive control. Granular red puncta were also indicators of lysosome number.
HaCaT cells cultured on glass slides were treated with 50 µM chloroquine sulphate (Sigma-Aldrich, St. Louis, MO, USA) for 24 h in conjunction with proinflammatory cytokine stimulation. After three washes with PBS, cells were incubated with 50 nM AO in Keratinocyte-SFM (Gibco, Thermo Fisher Scientific) without growth supplements, for 15 min at 37 °C, followed by three further PBS washes. Each treatment was performed in triplicate, with and without CQ, in three independent experiments.
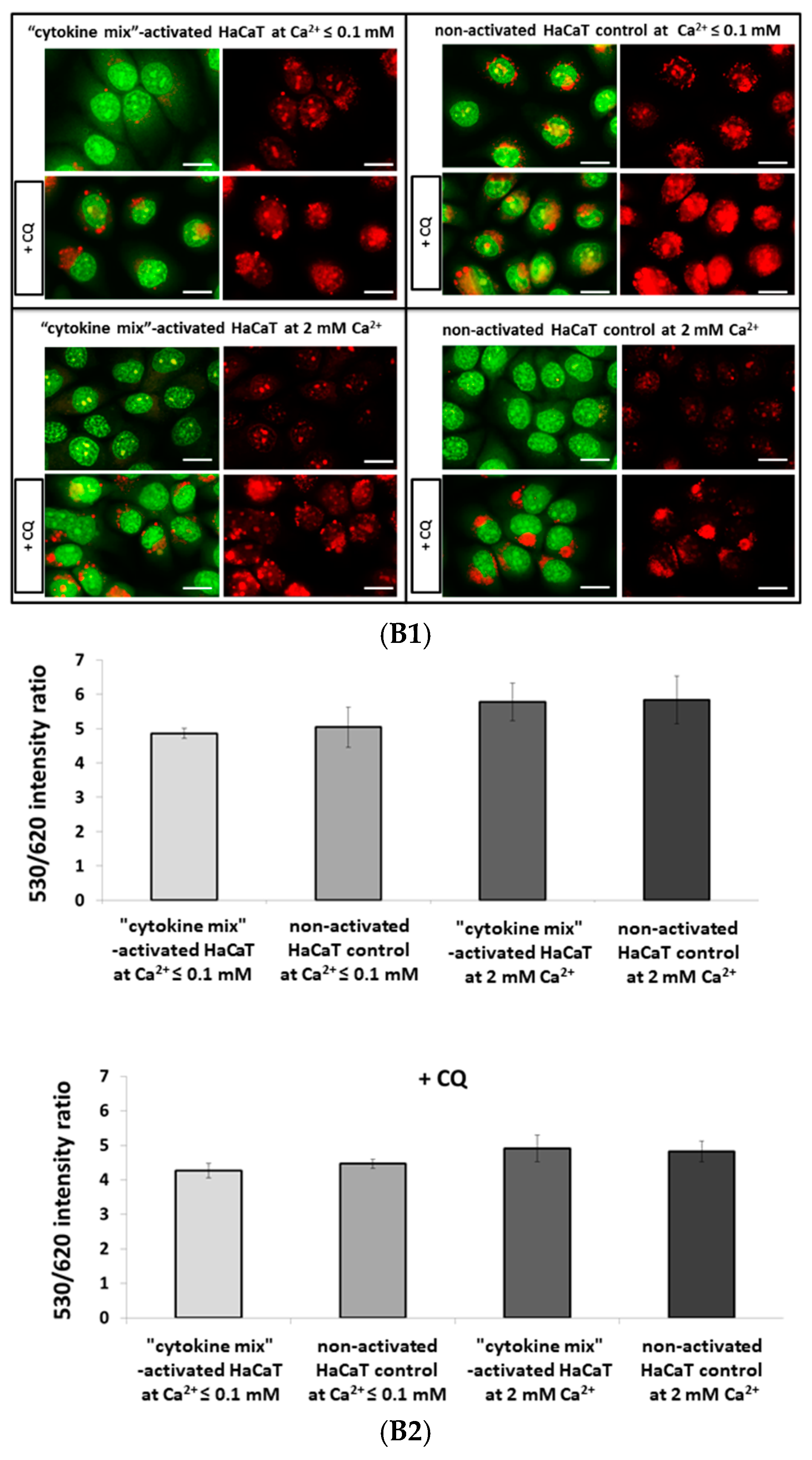
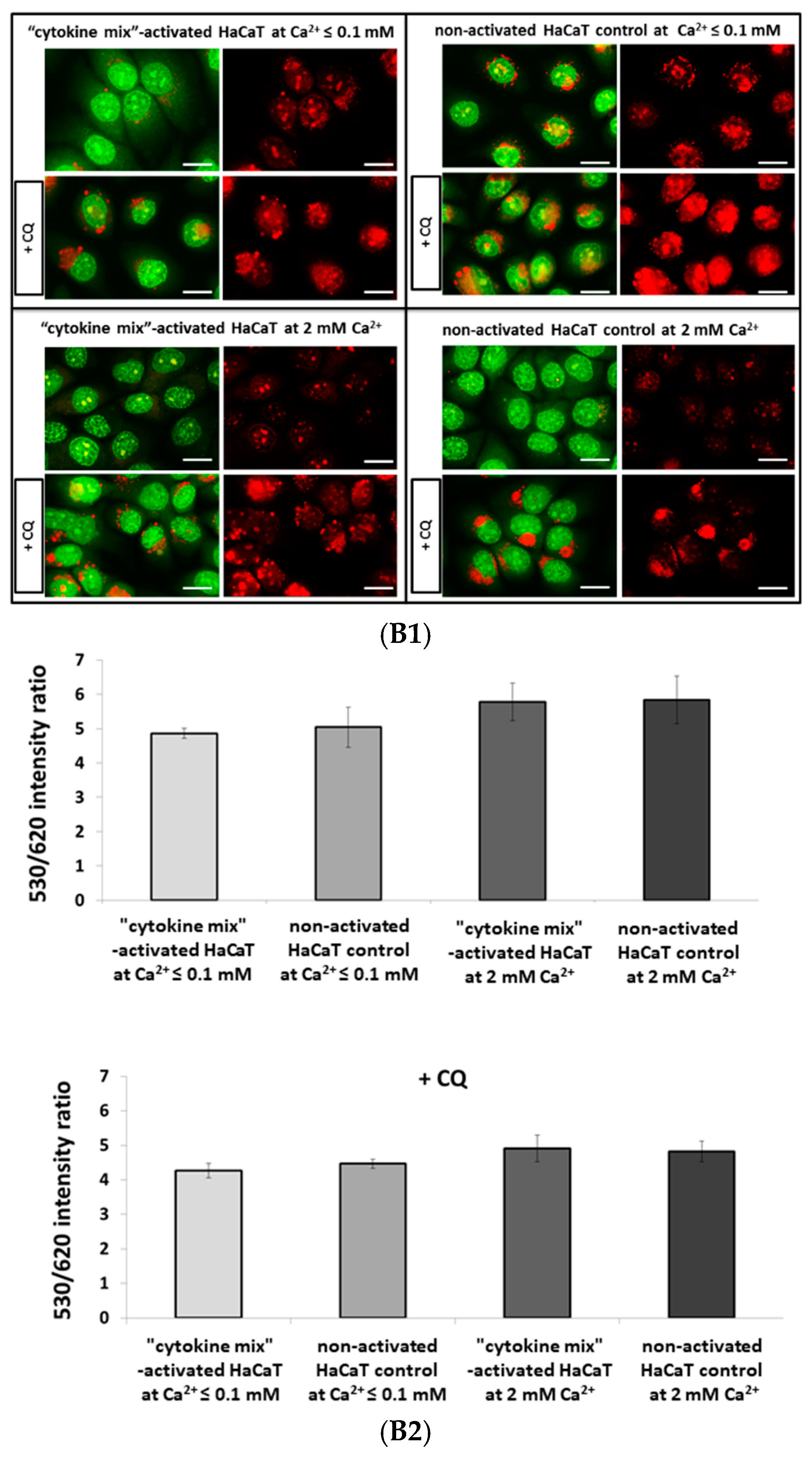
4.4.3. Quantitation of Acidic Organelles Using AO Staining
AO can be used to stain and count acidic organelles such as endosomes, lysosomes, and autolysosomes. Upon blue light excitation (485 nm) at low pH (inside organelles), it emits orange fluorescence (peak at 620 nm) and green fluorescence (peak at 530 nm) in the cytoplasm. A ratio of 530/620 was adopted to estimate the cytoplasmic to lysosomal area in proinflammatory cytokine-stimulated HaCaT cells.
A total of 5 × 105/well keratinocytes were seeded on 96-well plates containing supplemented DMEM (Gibco, Thermo Fisher Scientific) and cultured for 24 h. Subsequently, the culture medium was replaced with Keratinocyte-SFM (Gibco, Thermo Fisher Scientific) supplemented with BPE and EGF (Gibco, Thermo Fisher Scientific). Following a 6-h incubation, cells were cultured in Keratinocyte-SFM without growth supplements for a further 16 h. For subsequent experiments, cells were stimulated for 24 h with a combination of proinflammatory “cytokine mix”: IL-1α, IL-17A, IL-22, OSM, and TNF-α (Gibco, Thermo Fisher Scientific) at a concentration of 2 ng/mL each constituent. The control cells were left unstimulated in Keratinocyte-SFM containing a low concentration of calcium ions (≤0.1 mM) (Gibco, Thermo Fisher Scientific). An additional control without the cytokine mix but with 2 mM Ca2+ was applied to simulate conditions prone to keratinocyte differentiation.
After treatment, cells were washed three times with PBS and incubated with 50 nM AO in Keratinocyte-SFM (Gibco, Thermo Fisher Scientific) without growth supplements for 15 min at 37 °C, followed by three further washes with PBS. Fluorescence quantitation was performed using an EnSpire Multimode Fluorescence Plate Reader (Perkin Elmer, Waltham, MA, USA). Each treatment was performed in 20 wells in three independent experiments.
4.4.4. Determination of the Subcellular Localization of Transcription Factor EB (TFEB)
To analyze the proinflammatory effect on TFEB nuclear translocation, cytokine-stimulated HaCaT cells were fixed with 4% PFA for 10 min at RT. For endogenous TFEB staining, cells were permeabilized with 0.1% Triton X-100 for 15 min at RT, followed by blocking with 3% BSA in PBS + 0.1% Tween for 1 h at 4 °C. Immunostaining was performed by overnight incubation with a TFEB primary antibody (#4240, Cell Signaling, Leiden, Netherlands; 1:1000) in blocking solution at 4 °C. The following day, cells were washed three times with PBS, and incubated for 1 h with a goat anti-rabbit IgG Alexa Fluor 488 secondary antibody (#A-11034, Invitrogen, Thermo Fisher Scientific,; 1:500) in blocking solution at 4 °C. Following an additional three washes with PBS, coverslips were mounted in SlowFade™ Diamond Antifade Mountant with DAPI (Invitrogen, Thermo Fisher Scientific). Each treatment was analyzed in triplicate in three independent experiments. An additional control with 0.3 µM Torin 1 (MTORC1 inhibitor) was applied for the last 3 h of treatment.
4.5. Lysosomal Ultrastructure Determination Using Transmission Electron Microscopy (TEM)
To determine the effect of proinflammatory stimulation on HaCaT cellular structures, 1.5 x 105 cells/well were seeded on 12-well plates and cultured overnight. The culture medium was then replaced with Keratinocyte-SFM (Gibco, Thermo Fisher Scientific) supplemented with BPE and EGF (Gibco, Thermo Fisher Scientific). Following a 6-h incubation, cells were cultured in Keratinocyte-SFM without growth supplements for a further 16 h. For subsequent experiments, cells were stimulated for 24 h with a combination of proinflammatory “cytokine mix”: IL-1α, IL-17A, IL-22, OSM, and TNF-α (Gibco, Thermo Fisher Scientific) at a concentration of 2 ng/mL each constituent. The control cells were left unstimulated. Following treatment, cells were washed three times with PBS, fixed with 2.5% glutaraldehyde and subsequently with 1% osmium tetroxide and 1% potassium hexacyanoferrate (III), and dehydrated in ethanol. Ultrasections of Epon 812 resin (Sigma-Aldrich Produktions GmbH, Fluka, Steinheim, Germany)-embedded cells were stained with lead citrate and uranyl acetate and examined using a transmission electron microscope (CM100, Philips, Amsterdam, Netherlands). All photographs were taken using the iTEM program (Olympus Soft Imaging Solution, Tokyo, Japan). The keratinocytes had an intact membrane and visible cells structures (nucleus, mitochondria, and lysosomes). At least 30 cells obtained from three independent cell cultures (n = 3) were examined, and representative electron micrographs are shown (1650× magnification).
4.6. LAMP1 Evaluation Using Immunohistochemistry
Skin biopsies with a minimum diameter of 3 mm (two clippings in the study group of 7 subjects (numbered from 1 to 7): From the psoriatic plaque and the non-lesioned skin; and one clipping in the control group of 4 healthy individuals (numbered from 1 to 4)) were taken from the buttocks, fixed with 4% PFA in PBS, and embedded in paraffin. Subsequently, 5 µm-thick sections were mounted on Superfrost adhesive slides (Fisher Scientific, Hampton, NH, USA), and the epitopes were exposed by boiling in a 10-mM citric acid solution (pH 6.0). Endogenous peroxidase activity was inhibited using 3% hydrogen peroxidase in methanol for 5 min. Sections were blocked with 10% goat serum/1% BSA in PBS for 1 h and stained with mouse anti-LAMP1 (eBioH4A3, Invitrogen, Thermo Fisher Scientific; 5 µg/mL) in 1% BSA for 1 h at RT. Sections were subsequently washed in PBS and incubated with a biotinylated goat anti-mouse IgG secondary antibody (Vector Laboratories Inc, Burlingame, CA, USA; 4 µg/mL) in 10% normal goat serum (Vector Laboratories Inc)/1% BSA in PBS for 45 min at RT. Finally, an anti-biotin tertiary antibody (VECTASTAIN Elite ABC-Peroxidase Kit, Vector Laboratories Inc) was added for 30 min according to the manufacturer’s instructions. The reaction was developed using DAB substrate (Vector Laboratories Inc) according to the manufacturer’s instructions. For quantification analysis, nickel was added to the DAB substrate to obtain black staining. A non-specific mouse IgG was used as a control in place of the primary antibody. All comparative sections were performed at the same time under identical conditions. Images were taken using an IX51 Olympus inverted light microscope (Olympus, Tokyo, Japan) with a color CCD camera and the cellSens imaging software (ver 1.12, Olympus). Comparative images were taken under the same light conditions and processed in the same way.
Four to ten non-overlapping low power (20× magnification) fields of view were digitally photographed from each section using an IX51 Olympus inverted light microscope (Olympus) with a color CCD camera and the cellSens imaging software (ver 1.12, Olympus). Identical exposure settings were used for each stain, and all photographs were taken during one session. Images were transformed to eight-bit grey resolution and stored in a TIFF format. The ImageJ software (ver.1.8.0_112, National Institutes of Health, Bethesda, MD, USA) was used for quantitation of LAMP1 staining. The entire non-manipulated field of view was blinded and quantitated for each stain, whilst an unstained area was used to determine (and subtract) background staining for each section. An average optical density for each section and each patient was calculated, which were subsequently averaged to give a group mean for the sample for each patient. Statistical analyses of LAMP1 expression in skin biopsies was made by comparisons among particular paired PP vs. PN, PP vs. mean NN, and PN vs. mean NN, and statistical differences are observed following application of a nonparametric Mann–Whitney U test, with continuity correction (p ≤ 0.05).
4.7. RNA Processing
4.7.1. Cell Cultures
Total RNA was extracted from cells using the High Pure RNA Isolation Kit (Roche Applied Science, Penzberg, Germany) following the manufacturer’s instructions. The quantity of each RNA sample was evaluated using Quant-iT™ RiboGreen™ RNA Assay Kit (Invitrogen, Thermo Fisher Scientific). In addition, the quality of each RNA sample was assessed using the RNA 6000 Nano Assay on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). The synthesis of cDNA from an RNA template was conducted using Transcriptor First-Strand cDNA Synthesis Kit (Roche Applied Science).
4.7.2. Skin Specimens
Total RNA was extracted from skin biopsies of 11 psoriasis patients (numbered from 8 to 18) and 11 control group subjects (numbered from 5 to 15) with the RNeasy Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions. The quality and quantity of each RNA sample was evaluated using the RNA 6000 Nano Assay on the Agilent 2100 Bioanalyzer (Agilent Technologies) and Quant-iT™ RiboGreen™ RNA Assay Kit (Invitrogen, Thermo Fisher Scientific). The synthesis of cDNA from an RNA template was conducted by Transcriptor First-Strand cDNA Synthesis Kit (Roche Applied Science).
4.8. Real-Time Quantitative Reverse Transcription PCR Assays for mRNA Analysis (Real-Time qRT-PCR).
Real-time quantitative reverse transcription polymerase chain reaction (real-time qRT-PCR) was carried out with the Single Tube Custom TaqMan Gene Expression Assays (Thermo Fisher Scientific) and the LightCycler1 480 Probes Master (Roche Applied Science) using the Light Cycler 480 II detection system (Roche Applied Science). Expression values were normalized against control genes: GAPDH and TBP. The TaqMan probes used were as follows: ASAH (Hs00602774_m1), GAPDH (Hs02786624_g1), GLA (Hs00609238_m1), GM2A (Hs00166197_m1), HPSE (Hs00935036_m1), HYAL4 (Hs00202177_m1), IVL (Hs00846307_s1), LAMP1 (Hs00931461_m1), MCOLN1 (Hs01100653_m1), MITF (Hs01117294_m1), MTORC1 (Hs00234508_m1), PPP3CA (Hs00174223_m1), PPP3CB (Hs00236113_m1), PSAP (Hs01551096_m1), S100A7 (Hs01923188_u1), S100A9 (Hs00610058_m1), SMPD1 (Hs03679346_g1), SPHK1 (Hs00184211_m1), TBP (Hs00427620_m1), TFE3 (Hs00232406_m1), TFEB (Hs00292981_m1), and TFEC (Hs00992838_m1). Each experiment of real-time qRT-PCR analysis was repeated at least three times (n). In case of HaCaT cells they were biological replications and data are reported as the mean SD with p < 0.05 considered statistically significant. For skin specimens technical repetitions were conducted.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}