The Role of Vitamin K and Its Related Compounds in Mendelian and Acquired Ectopic Mineralization Disorders


{kind=link}
{kind=link}
Abstract
1. Introduction
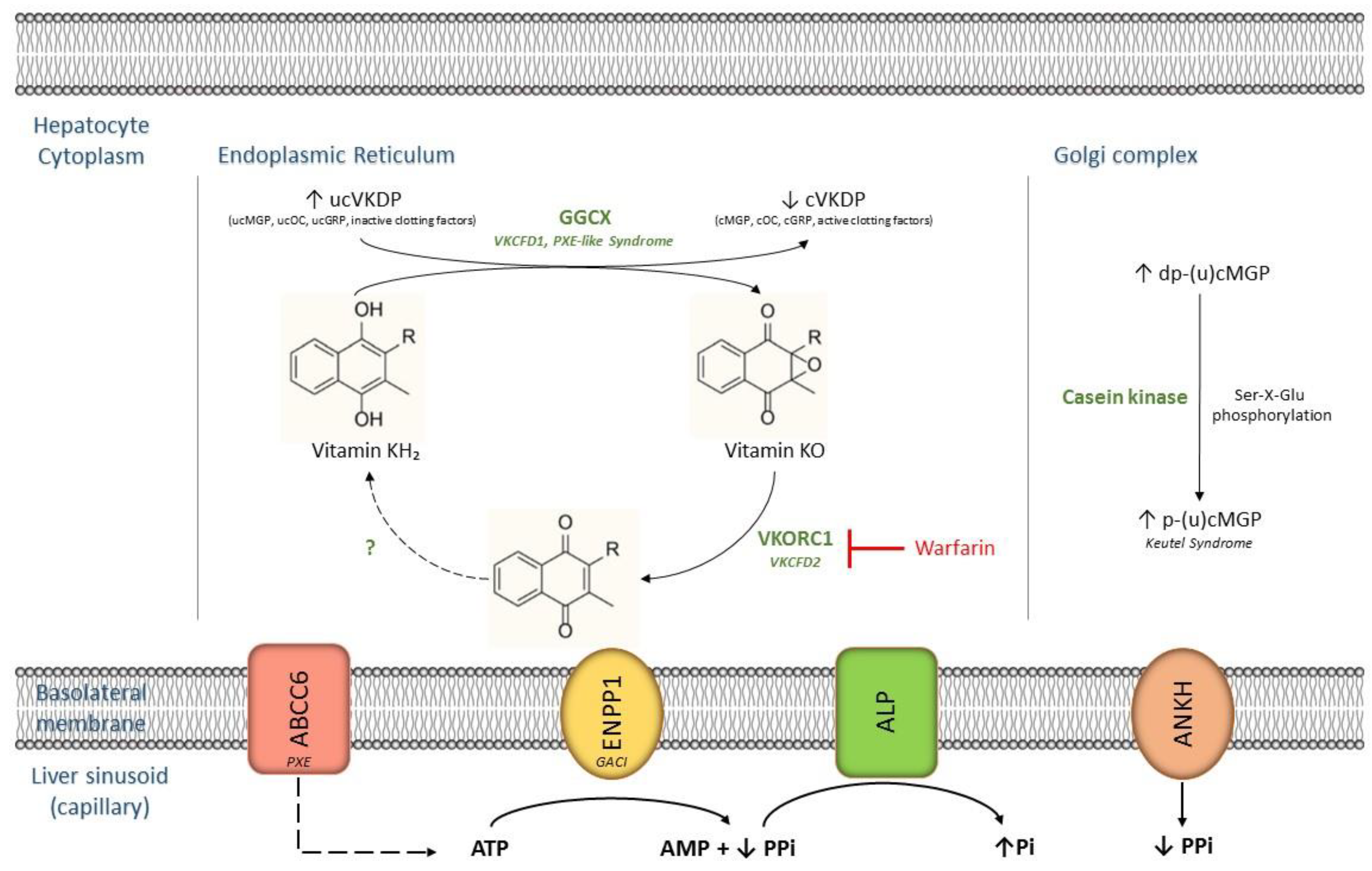
1.1. Vitamin K: Basic Chemical Structure and Function
1.2. Vitamin K is Associated with Ectopic Mineralization
2. Mendelian Ectopic Mineralization Diseases and Vitamin K
2.1. Pseudoxanthoma Elasticum
2.1.1. Phenotype
2.1.2. Molecular Etiology
2.1.3. Vitamin K: Role and Therapeutic Options in PXE
2.2. PXE-Like Syndrome with Multiple Coagulation Factor Deficiency
2.2.1. Phenotype
2.2.2. Molecular Etiology
2.2.3. Role of Vitamin K in the PXE-Like Syndrome
2.3. Keutel Syndrome
2.3.1. Phenotype
2.3.2. Molecular Etiology and Role of Vitamin K
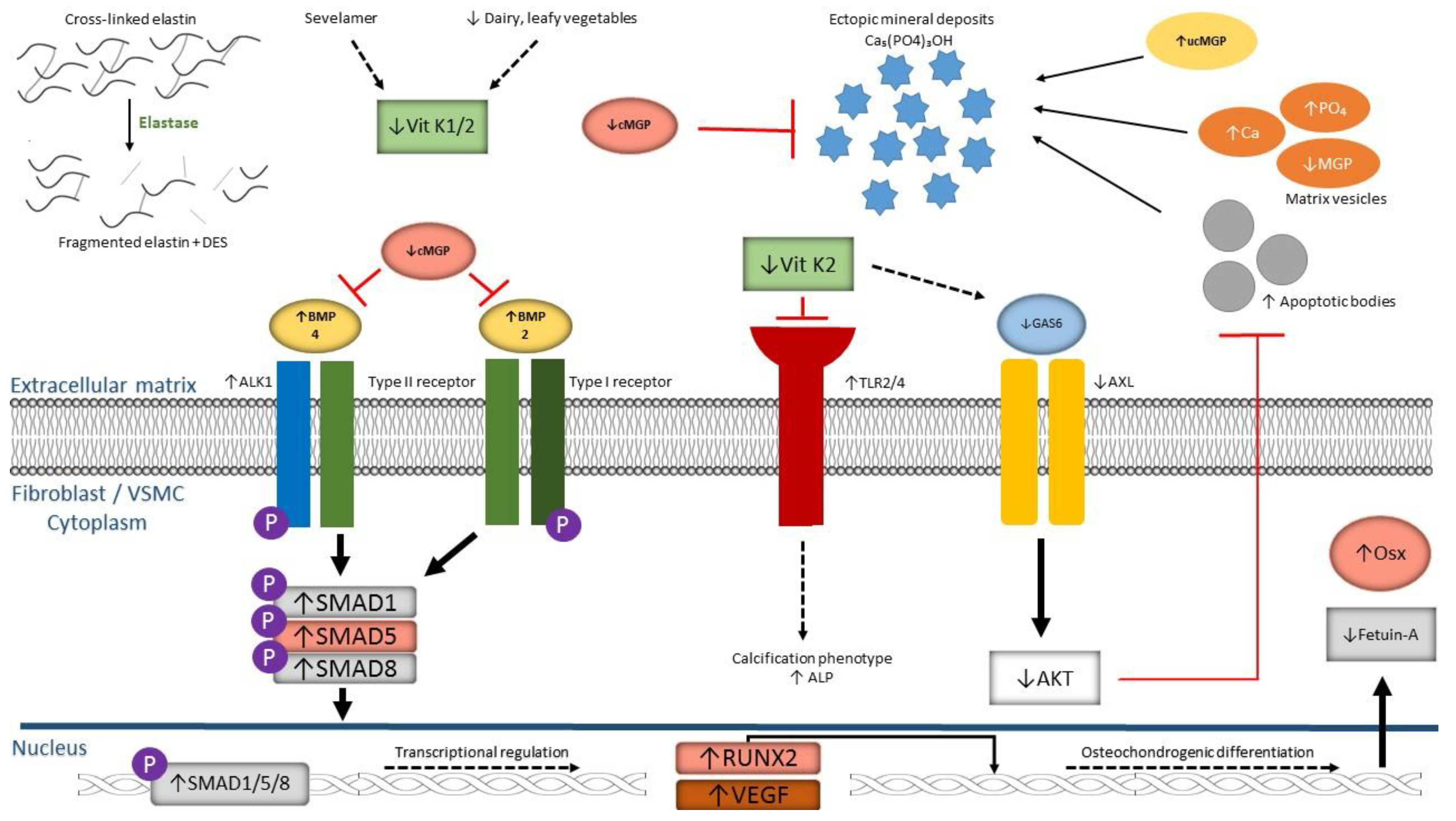
3. Acquired (Vascular) Calcification Disorders and Vitamin K
Phenotypes and Vitamin K Supplementation
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AATD | alpha-1 antitrypsin deficiency |
| AB | apoptotic body |
| ABCC6 | ATP-binding cassette transporter subfamily C member 6 |
| AKT | AK strain transforming |
| ALK1 | activin receptor-like kinase 1 |
| ALP | alkaline phosphatase |
| AMP | adenosine monophosphate |
| ANKH | progressive ankylosis homolog protein |
| ApoE | apolipoprotein E |
| ATP | adenosine triphosphate |
| AXL | AXL receptor tyrosine kinase |
| BMP | bone morphogenetic protein |
| Ca | calcium |
| Ca5(PO4)3OH | calcium hydroxyapatite |
| CAD | coronary artery disease |
| CKD | chronic kidney disease |
| CNV | choroidal neovascularization |
| COPD | chronic obstructive pulmonary disease |
| CTC | connective tissue calcifying disorder |
| DES | (iso)desmosine |
| EC | endothelial cell |
| ENPP1 | ectonucleotide pyrophosphatase-phosphodiesterase 1 |
| ER | endoplasmic reticulum |
| GACI | generalized arterial calcification of infancy |
| GAS6 | growth arrest specific-6 |
| GGCX | gamma-glutamyl carboxylase |
| Gla | γ-carboxylated glutamate |
| Glu | glutamate |
| GRP | gla-rich protein |
| HE | hematoxylin-eosin |
| IHC | immunohistochemistry |
| KS | Keutel syndrome |
| LRP1 | lipoprotein receptor-related protein 1 |
| MGP | matrix gla protein |
| MRP6 | multi-drug resistance-associated protein 6 |
| OC | osteocalcin |
| OPN | osteopontin |
| OSX | osterix |
| PAD | peripheral arterial disease |
| Pi | inorganic phosphate |
| PIVKA-II | protein induced by vitamin K absence or antagonist-II |
| PO4 | phosphate |
| PPi | inorganic pyrophosphate |
| PXE | pseudoxanthoma elasticum |
| RUNX2 | runt-related transcription factor 2 |
| SMAD | small body size mothers against decapentaplegic |
| SNP | single nucleotide polymorphism |
| TGF-β | transforming growth factor-β |
| TLR | toll-like receptor |
| VC | vascular calcification |
| VEGF | vascular endothelial growth factor |
| Vit K | vitamin K |
| VKCFD | vitamin K-dependent coagulation factor deficiency |
| VKDP | vitamin K-dependent protein |
| VKORC1 | vitamin K 2,3-epoxide reductase complex subunit 1 |
| VSMC | vascular smooth muscle cell |
| WT | wild type |
References
- Rashdan, N.A.; Rutsch, F.; Kempf, H.; Varadi, A.; Leftheriotis, G.; MacRae, V.E. New perspectives on rare connective tissue calcifying diseases. Curr. Opin. Pharmacol. 2016, 28, 14–23. [Google Scholar] [CrossRef]
- Kavukcuoglu, N.B.; Li, Q.; Pleshko, N.; Uitto, J. Connective tissue mineralization in Abcc6−/− mice, a model for pseudoxanthoma elasticum. Matrix Biol. 2012, 31, 246–252. [Google Scholar] [CrossRef][Green Version]
- De Vilder, E.Y.; Vanakker, O.M. From variome to phenome: Pathogenesis, diagnosis and management of ectopic mineralization disorders. World J. Clin. Cases. 2015, 3, 556–574. [Google Scholar] [CrossRef] [PubMed]
- De Vilder, E.Y.; Hosen, M.J.; Vanakker, O.M. The ABCC6 Transporter as a Paradigm for Networking from an Orphan Disease to Complex Disorders. Biomed. Res. Int. 2015, 2015, 648569. [Google Scholar] [CrossRef]
- Schantl, A.E.; Ivarsson, M.E.; Leroux, J.C. Investigational Pharmacological Treatments for Vascular Calcification. Adv. Ther. 2018, 2, 1800094. [Google Scholar] [CrossRef]
- Turner, M.E.; Adams, M.A.; Holden, R.M. The Vitamin K Metabolome in Chronic Kidney Disease. Nutrients 2018, 10, 1076. [Google Scholar] [CrossRef] [PubMed]
- Willems, B.A.; Vermeer, C.; Reutelingsperger, C.P.; Schurgers, L.J. The realm of vitamin K dependent proteins: Shifting from coagulation toward calcification. Mol. Nutr. Food Res. 2014, 58, 1620–1635. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine Panel on Micronutrients. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc; National Academies Press: Washington, DC, USA, 2001. [Google Scholar]
- Gorgels, T.G.; Waarsing, J.H.; Herfs, M.; Versteeg, D.; Schoensiegel, F.; Sato, T.; Schlingemann, R.O.; Ivandic, B.; Vermeer, C.; Schurgers, L.J.; et al. Vitamin K supplementation increases vitamin K tissue levels but fails to counteract ectopic calcification in a mouse model for pseudoxanthoma elasticum. J. Mol. Med. 2011, 89, 1125–1135. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Martin, L.; Schurgers, L.J.; Quaglino, D.; Costrop, L.; Vermeer, C.; Pasquali-Ronchetti, I.; Coucke, P.J.; De Paepe, A. Low serum vitamin K in PXE results in defective carboxylation of mineralization inhibitors similar to the GGCX mutations in the PXE-like syndrome. Lab. Invest. 2010, 90, 895–905. [Google Scholar] [CrossRef]
- Watzka, M.; Geisen, C.; Scheer, M.; Wieland, R.; Wiegering, V.; Dorner, T.; Laws, H.J.; Gumruk, F.; Hanalioglu, S.; Unal, S.; et al. Bleeding and non-bleeding phenotypes in patients with GGCX gene mutations. Thromb. Res. 2014, 134, 856–865. [Google Scholar] [CrossRef]
- Michaux, A.; Matagrin, B.; Debaux, J.V.; Schurgers, L.J.; Benoit, E.; Lattard, V. Missense mutation of VKORC1 leads to medial arterial calcification in rats. Sci Rep. 2018, 8, 13733. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Martin, L.; Gheduzzi, D.; Leroy, B.P.; Loeys, B.L.; Guerci, V.I.; Matthys, D.; Terry, S.F.; Coucke, P.J.; Pasquali-Ronchetti, I.; et al. Pseudoxanthoma elasticum-like phenotype with cutis laxa and multiple coagulation factor deficiency represents a separate genetic entity. J. Invest. Dermatol. 2007, 127, 581–587. [Google Scholar] [CrossRef]
- Theuwissen, E.; Smit, E.; Vermeer, C. The role of vitamin K in soft-tissue calcification. Adv. Nutr. 2012, 3, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.D.; Pinero, G.J.; Loyer, E.; Behringer, R.R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386, 78–81. [Google Scholar] [CrossRef]
- Munroe, P.B.; Olgunturk, R.O.; Fryns, J.P.; Van Maldergem, L.; Ziereisen, F.; Yuksel, B.; Gardiner, R.M.; Chung, E. Mutations in the gene encoding the human matrix Gla protein cause Keutel syndrome. Nat. Genet. 1999, 21, 142–144. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Teunissen, K.J.; Knapen, M.H.; Kwaijtaal, M.; van Diest, R.; Appels, A.; Reutelingsperger, C.P.; Cleutjens, J.P.; Vermeer, C. Novel conformation-specific antibodies against matrix gamma-carboxyglutamic acid (Gla) protein: Undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscl. Throm. Vas. 2005, 25, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; D’Souza, R.; Hogue, D.; Karsenty, G. The matrix Gla protein gene is a marker of the chondrogenesis cell lineage during mouse development. J. Bone Miner. Res. 1995, 10, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.; Skepper, J.N.; Hackeng, T.M.; Shanahan, C.M.; Vermeer, C.; Weissberg, P.L.; Proudfoot, D. Post-translational modifications regulate matrix Gla protein function: Importance for inhibition of vascular smooth muscle cell calcification. J. Thromb. Haemost. 2007, 5, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis regulates human vascular calcification in vitro: Evidence for initiation of vascular calcification by apoptotic bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Stormont, R.M.; Gangahar, D.M.; Agrawal, D.K. Role of matrix Gla protein in angiotensin II-induced exacerbation of vascular calcification. Am. J. Physio. Heart Circ. Physiol. 2012, 303, H523–H532. [Google Scholar] [CrossRef] [PubMed]
- Pi, W.; Guo, X.; Su, L.; Xu, W. BMP-2 up-regulates PTEN expression and induces apoptosis of pulmonary artery smooth muscle cells under hypoxia. PloS ONE 2012, 7, e35283. [Google Scholar] [CrossRef] [PubMed]
- Finger, R.P.; Charbel Issa, P.; Ladewig, M.S.; Gotting, C.; Szliska, C.; Scholl, H.P.; Holz, F.G. Pseudoxanthoma elasticum: Genetics, clinical manifestations and therapeutic approaches. Surv. Ophthalmol. 2009, 54, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Uitto, J.; Jiang, Q.; Varadi, A.; Bercovitch, L.G.; Terry, S.F. Pseudoxanthoma elasticum: Diagnostic features, classification and treatment options. Expert. Opin. Orphan. Drugs 2014, 2, 567–577. [Google Scholar] [CrossRef]
- Marconi, B.; Bobyr, I.; Campanati, A.; Molinelli, E.; Consales, V.; Brisigotti, V.; Scarpelli, M.; Racchini, S.; Offidani, A. Pseudoxanthoma elasticum and skin: Clinical manifestations, histopathology, pathomechanism, perspectives of treatment. Intractable Rare Dis. Res. 2015, 4, 113–122. [Google Scholar] [CrossRef]
- Combrinck, M.; Gilbert, J.D.; Byard, R.W. Pseudoxanthoma elasticum and sudden death. J. Forensic. Sci. 2011, 56, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Plomp, A.S.; Toonstra, J.; Bergen, A.A.; van Dijk, M.R.; de Jong, P.T. Proposal for updating the pseudoxanthoma elasticum classification system and a review of the clinical findings. Am. J. Med. Genet. A. 2010, 152, 1049–1058. [Google Scholar] [CrossRef]
- Hosen, M.J.; Lamoen, A.; De Paepe, A.; Vanakker, O.M. Histopathology of pseudoxanthoma elasticum and related disorders: Histological hallmarks and diagnostic clues. Scientifica 2012, 2012, 598262. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Leroy, B.P.; Coucke, P.; Bercovitch, L.G.; Uitto, J.; Viljoen, D.; Terry, S.F.; Van Acker, P.; Matthys, D.; Loeys, B.; et al. Novel clinico-molecular insights in pseudoxanthoma elasticum provide an efficient molecular screening method and a comprehensive diagnostic flowchart. Hum. Mutat. 2018, 29, 205. [Google Scholar] [CrossRef]
- Bertamino, M.; Severino, M.; Grossi, A.; Rusmini, M.; Tortora, D.; Gandolfo, C.; Pederzoli, S.; Malattia, C.; Picco, P.; Striano, P.; et al. ABCC6 mutations and early onset stroke: Two cases of a typical Pseudoxanthoma Elasticum. Eur. J. Paediatr. Neurol 2018, 22, 725–728. [Google Scholar] [CrossRef]
- Campens, L.; Vanakker, O.M.; Trachet, B.; Segers, P.; Leroy, B.P.; De Zaeytijd, J.; Voet, D.; De Paepe, A.; De Backer, T.; De Backer, J. Characterization of cardiovascular involvement in pseudoxanthoma elasticum families. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2646–2652. [Google Scholar] [CrossRef]
- De Vilder, E.Y.G.; Cardoen, S.; Hosen, M.J.; Le Saux, O.; De Zaeytijd, J.; Leroy, B.P.; De Reuck, J.; Coucke, P.J.; De Paepe, A.; Hemelsoet, D.; et al. PATHOGENIC VARIANTS IN THE ABCC6 GENE ARE ASSOCIATED WITH AN INCREASED RISK FOR ISCHEMIC STROKE. Brain Pathol. 2018, 28, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Varadi, A.; Plomp, A.; Bergen, A.A.; Oude Elferink, R.P.; Borst, P.; et al. ABCC6-mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation-brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef]
- Jansen, R.S.; Kucukosmanoglu, A.; de Haas, M.; Sapthu, S.; Otero, J.A.; Hegman, I.E.; Bergen, A.A.; Gorgels, T.G.; Borst, P.; van de Wetering, K. ABCC6 prevents ectopic mineralization seen in pseudoxanthoma elasticum by inducing cellular nucleotide release. Proc. Natl. Acad. Sci. USA 2013, 110, 20206–20211. [Google Scholar] [CrossRef]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.F.; et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef]
- Li, Q.; Aranyi, T.; Varadi, A.; Terry, S.F.; Uitto, J. Research Progress in Pseudoxanthoma Elasticum and Related Ectopic Mineralization Disorders. J. Invest. Dermatol. 2016, 136, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Dabisch-Ruthe, M.; Kuzaj, P.; Gotting, C.; Knabbe, C.; Hendig, D. Pyrophosphates as a major inhibitor of matrix calcification in Pseudoxanthoma elasticum. J. Dermatol. Sci. 2014, 75, 109–120. [Google Scholar] [CrossRef]
- Hosen, M.J.; Coucke, P.J.; Le Saux, O.; De Paepe, A.; Vanakker, O.M. Perturbation of specific pro-mineralizing signalling pathways in human and murine pseudoxanthoma elasticum. Orphanet. J. Rare Dis. 2014, 9, 66. [Google Scholar] [CrossRef]
- Li, Q.; Uittom, J. Mineralization/anti-mineralization networks in the skin and vascular connective tissues. Am. J. Pathol. 2013, 183, 10–18. [Google Scholar] [CrossRef]
- Zhao, J.; Kingman, J.; Sundberg, J.P.; Uitto, J.; Li, Q. Plasma PPi Deficiency is the Major, But Not the Exclusive, Cause of Ectopic Mineralization in an Abcc6−/− Mouse Model of PXE. J. Invest. Dermatol. 2017, 11, 2336–2343. [Google Scholar] [CrossRef]
- Van Gils, M.; Nollet, L.; Verly, E.; Deianova, N.; Vanakker, O.M. Cellular signaling in pseudoxanthoma elasticum: An update. Cell. Signal. 2019, 55, 119–129. [Google Scholar] [CrossRef]
- Li, Q.; Jiang, Q.; Schurgers, L.J.; Uitto, J. Pseudoxanthoma elasticum: Reduced gamma-glutamyl carboxylation of matrix gla protein in a mouse model (Abcc6−/−). Biochem. Biophys. Res. Commun. 2007, 364, 208–213. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niemeier, A.; Kassem, M.; Toedter, K.; Wendt, D.; Ruether, W.; Beisiegel, U.; Heeren, J. Expression of LRP1 by human osteoblasts: A mechanism for the delivery of lipoproteins and vitamin K1 to bone. J. Bone Miner. Res. 2005, 20, 283–293. [Google Scholar] [CrossRef]
- Bhatnagar, P.; Freund, K.B.; Spaide, R.F.; Klancnik, J., Jr.; Cooney, M.J.; Ho, I.; Fine, H.F.; Yannuzzi, L.A. Intravitreal bevacizumab for the management of choroidal neovascularization in pseudoxanthoma elasticum. Retina 2007, 27, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Finger, R.P.; Charbel Issa, P.; Schmitz-Valckenberg, S.; Holz, F.G.; Scholl, H.N. Long-term effectiveness of intravitreal bevacizumab for choroidal neovascularization secondary to angioid streaks in pseudoxanthoma elasticum. Retina 2011, 31, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Verbraak, F.D. Antivascular endothelial growth factor treatment in pseudoxanthoma elasticum patients. Dev. Ophthalmol. 2010, 46, 96–106. [Google Scholar] [PubMed]
- Li, Q.; Guo, H.; Chou, D.W.; Harrington, D.J.; Schurgers, L.J.; Terry, S.F.; Uitto, J. Warfarin accelerates ectopic mineralization in Abcc6(−/−) mice: Clinical relevance to pseudoxanthoma elasticum. Am. J. Pathol. 2013, 182, 1139–1150. [Google Scholar] [CrossRef]
- Poterucha, T.J.; Goldhaber, S.Z. Warfarin and Vascular Calcification. Am. J. Med. 2016, 129, 635.e1–635.e4. [Google Scholar] [CrossRef]
- Weijs, B.; Blaauw, Y.; Rennenberg, R.J.; Schurgers, L.J.; Timmermans, C.C.; Pison, L.; Nieuwlaat, R.; Hofstra, L.; Kroon, A.A.; Wildberger, J.; et al. Patients using vitamin K antagonists show increased levels of coronary calcification: An observational study in low-risk atrial fibrillation patients. Eur. Heart J. 2011, 32, 2555–2562. [Google Scholar] [CrossRef] [PubMed]
- Di Lullo, L.; Tripepi, G.; Ronco, C.; D’Arrigo, G.; Barbera, V.; Russo, D.; Di Iorio, B.R.; Uguccioni, M.; Paoletti, E.; Ravera, M.; et al. Cardiac valve calcification and use of anticoagulants: Preliminary observation of a potentially modifiable risk factor. Int. J. Cardiol. 2019, 278, 243–249. [Google Scholar] [CrossRef]
- Brampton, C.; Yamaguchi, Y.; Vanakker, O.; Van Laer, L.; Chen, L.H.; Thakore, M.; De Paepe, A.; Pomozi, V.; Szabo, P.T.; Martin, L.; et al. Vitamin K does not prevent soft tissue mineralization in a mouse model of pseudoxanthoma elasticum. Cell Cycle 2011, 10, 1810–1820. [Google Scholar] [CrossRef]
- Jiang, Q.; Li, Q.; Grand-Pierre, A.E.; Schurgers, L.J.; Uitto, J. Administration of vitamin K does not counteract the ectopic mineralization of connective tissues in Abcc6 (−/−) mice, a model for pseudoxanthoma elasticum. Cell Cycle 2011, 10, 701–707. [Google Scholar] [CrossRef]
- Mackay, E.W.; Apschner, A.; Schulte-Merker, S. Vitamin K reduces hypermineralisation in zebrafish models of PXE and GACI. Development 2015, 142, 1095–1101. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Linares, J.L.; Garcia-Fernandez, M.I.; Morillo, M.J.; Sanchez, P.; Rioja, J.; Baron, F.J.; Ariza, M.J.; Harrington, D.J.; Card, D.; Boraldi, F.; et al. The Effects of Parenteral K1 Administration in Pseudoxanthoma Elasticum Patients Versus Controls. A Pilot Study Front. Med. 2018, 5, 86. [Google Scholar] [CrossRef]
- Li, Q.; Schurgers, L.J.; Smith, A.C.; Tsokos, M.; Uitto, J.; Cowen, E.W. Co-existent pseudoxanthoma elasticum and vitamin K-dependent coagulation factor deficiency: Compound heterozygosity for mutations in the GGCX gene. Am. J. Pathol. 2009, 174, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Gusdorf, L.; Mitcov, M.; Maradeix, S.; Cunat, S.; Martin, L.; Cribier, B. Pseudoxanthoma elasticum-like disease with deficiency of vitamin K-dependent clotting factors and cutis laxa features. Ann. Dermatol. Venereol. 2016, 143, 279–283. [Google Scholar] [CrossRef]
- Vanakker, O.M.; Leroy, B.P.; Schurgers, L.J.; Vermeer, C.; Coucke, P.J.; De Paepe, A. Atypical presentation of pseudoxanthoma elasticum with abdominal cutis laxa: Evidence for a spectrum of ectopic calcification disorders? Am. J. Med. Genet. A. 2011, 155, 2855–2859. [Google Scholar] [CrossRef] [PubMed]
- De Vilder, E.Y.; Debacker, J.; Vanakker, O.M. GGCX-Associated Phenotypes: An Overview in Search of Genotype-Phenotype Correlations. Int. J. Mol. Sci 2017, 18, 240. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Grange, D.K.; Armstrong, N.L.; Whelan, A.J.; Hurley, M.Y.; Rishavy, M.A.; Hallgren, K.W.; Berkner, K.L.; Schurgers, L.J.; Jiang, Q.; et al. Mutations in the GGCX and ABCC6 genes in a family with pseudoxanthoma elasticum-like phenotypes. J. Invest. Dermatol. 2009, 129, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Keutel, J.; Jorgensen, G.; Gabriel, P. A new autosomal-recessive hereditary syndrome. Multiple peripheral pulmonary stenosis, brachytelephalangia, inner-ear deafness, ossification or calcification of cartilages. Dtsch. Med. Wochenschr. 1971, 96, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.; VANS-Z, K.Y.; Bonafe, L.; Mittaz Crettol, L.; Rodiger, L.A.; Dikkers, F.G.; VANE, A.J.; Superti-Furga, A.; Alexandrakis, E.; Vermeer, C.; et al. Circulating matrix gamma-carboxyglutamate protein (MGP) species are refractory to vitamin K treatment in a new case of Keutel syndrome. J. Thromb. Haemost. 2011, 9, 1225–1235. [Google Scholar] [CrossRef]
- Nitschke, Y.; Rutsch, F. Inherited Arterial Calcification Syndromes: Etiologies and Treatment Concepts. Curr. Osteoporos. Rep. 2017, 15, 255–270. [Google Scholar] [CrossRef]
- Hur, D.J.; Raymond, G.V.; Kahler, S.G.; Riegert-Johnson, D.L.; Cohen, B.A.; Boyadjiev, S.A. A novel MGP mutation in a consanguineous family: Review of the clinical and molecular characteristics of Keutel syndrome. Am. J. Med. Genet. A 2005, 135, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Meier, M.; Weng, L.P.; Alexandrakis, E.; Ruschoff, J.; Goeckenjan, G. Tracheobronchial stenosis in Keutel syndrome. Eur. Respir. J. 2001, 17, 566–569. [Google Scholar] [CrossRef]
- Nanda, A.; Anim, J.T.; Al-Gareeb, M.; Alsaleh, Q.A. Keutel syndrome with overlapping features of cutis laxa: A new variant. Am. J. Med. Genet. A 2006, 140, 1487–1489. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, H.E.; Sahin, S.C.; Akyuz, Y.; Ede, H. Long term follow-up of four patients with Keutel syndrome. Am. J. Med. Genet. A 2014, 164, 2849–2856. [Google Scholar] [CrossRef]
- Viegas, C.S.; Cavaco, S.; Neves, P.L.; Ferreira, A.; Joao, A.; Williamson, M.K.; Price, P.A.; Cancela, M.L.; Simes, D.C. Gla-rich protein is a novel vitamin K-dependent protein present in serum that accumulates at sites of pathological calcifications. Am. J. Pathol. 2009, 175, 2288–2298. [Google Scholar] [CrossRef]
- Chatrou, M.L.; Winckers, K.; Hackeng, T.M.; Reutelingsperger, C.P.; Schurgers, L.J. Vascular calcification: The price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev. 2012, 26, 155–166. [Google Scholar] [CrossRef]
- Bordoloi, J.; Dihingia, A.; Kalita, J.; Manna, P. Implication of a novel vitamin K dependent protein, GRP/Ucma in the pathophysiological conditions associated with vascular and soft tissue calcification, osteoarthritis, inflammation, and carcinoma. Int. J. Biol. Macromol. 2018, 113, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Villa, J.K.D.; Diaz, M.A.N.; Pizziolo, V.R.; Martino, H.S.D. Effect of vitamin K in bone metabolism and vascular calcification: A review of mechanisms of action and evidences. Crit. Rev. Food Sci. Nutr. 2017, 57, 3959–3970. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, G.; de Jong, P.A.; Mali, W.P.; Attrach, M.; Visseren, F.L.; Spiering, W. Prevalence and severity of arterial calcifications in pseudoxanthoma elasticum (PXE) compared to hospital controls. Novel insights into the vascular phenotype of PXE. Atherosclerosis 2017, 256, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Leftheriotis, G.; Omarjee, L.; Le Saux, O.; Henrion, D.; Abraham, P.; Prunier, F.; Willoteaux, S.; Martin, L. The vascular phenotype in Pseudoxanthoma elasticum and related disorders: Contribution of a genetic disease to the understanding of vascular calcification. Front. Genet. 2013, 4, 4. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Bloch, D.B.; Nazarian, R.M.; Vermeer, C.; Booth, S.L.; Xu, D.; Thadhani, R.I.; Malhotra, R. Vitamin K-Dependent Carboxylation of Matrix Gla Protein Influences the Risk of Calciphylaxis. J. Am. Soc. Nephrol. 2017, 28, 1717–1722. [Google Scholar] [CrossRef] [PubMed]
- Vissers, L.E.T.; Dalmeijer, G.W.; Boer, J.M.A.; Verschuren, W.M.M.; van der Schouw, Y.T.; Beulens, J.W.J. The relationship between vitamin K and peripheral arterial disease. Atherosclerosis 2016, 252, 15–20. [Google Scholar] [CrossRef]
- Wuyts, J.; Dhondt, A. The role of vitamin K in vascular calcification of patients with chronic kidney disease. Acta. Clin. Belg. 2016, 71, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Delanaye, P.; Krzesinski, J.M.; Warling, X.; Moonen, M.; Smelten, N.; Medart, L.; Pottel, H.; Cavalier, E. Dephosphorylated-uncarboxylated Matrix Gla protein concentration is predictive of vitamin K status and is correlated with vascular calcification in a cohort of hemodialysis patients. BMC Nephrol. 2014, 15, 145. [Google Scholar] [CrossRef]
- Silaghi, C.N.; Fodor, D.; Gheorghe, S.R.; Craciun, A.M. Serum total matrix Gla protein: Reference interval in healthy adults and variations in patients with vascular and osteoarticular diseases. Clin. Chim. Acta 2019, 490, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Pasch, A. Novel assessments of systemic calcification propensity. Curr. Opin. Nephrol. Hypertens. 2016, 25, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zheng, H.; Tao, H.; Yu, W.; Jiang, X.; Li, A.; Jin, H.; Lv, A.; Li, H. Vitamin K2 inhibits rat vascular smooth muscle cell calcification by restoring the Gas6/Axl/Akt anti-apoptotic pathway. Mol. Cell. Biochem. 2017, 433, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Z.; Zhu, J.; Long, X.; Yan, J. Vitamin K2 can suppress the expression of Toll-like receptor 2 (TLR2) and TLR4, and inhibit calcification of aortic intima in ApoE(−/−) mice as well as smooth muscle cells. Vascular 2018, 26, 18–26. [Google Scholar] [CrossRef]
- Viegas, C.S.; Rafael, M.S.; Enriquez, J.L.; Teixeira, A.; Vitorino, R.; Luis, I.M.; et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef]
- Willems, B.A.; Furmanik, M.; Caron, M.M.J.; Chatrou, M.L.L.; Kusters, D.H.M.; Welting, T.J.M.; et al. Ucma/GRP inhibits phosphate-induced vascular smooth muscle cell calcification via SMAD-dependent BMP signalling. Sci Rep. 2018, 8, 4961. [Google Scholar] [CrossRef]
- Janssen, R.; Vermeer, C. Vitamin K deficit and elastolysis theory in pulmonary elasto-degenerative diseases. Med. Hypotheses. 2017, 108, 38–41. [Google Scholar] [CrossRef]
- Shea, M.K.; Holden, R.M. Vitamin K status and vascular calcification: Evidence from observational and clinical studies. Adv Nutr. 2012, 3, 158–165. [Google Scholar] [CrossRef]
- Van Ballegooijen, A.J.; Beulens, J.W. The Role of Vitamin K Status in Cardiovascular Health: Evidence from Observational and Clinical Studies. Curr. Nutr. Rep. 2017, 6, 197–205. [Google Scholar] [CrossRef]
- Lees, J.S.; Chapman, F.A.; Witham, M.D.; Jardine, A.G.; Mark, P.B. Vitamin K status, supplementation and vascular disease: A systematic review and meta-analysis. Heart 2018. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nollet, L.; Van Gils, M.; Verschuere, S.; Vanakker, O. The Role of Vitamin K and Its Related Compounds in Mendelian and Acquired Ectopic Mineralization Disorders. Int. J. Mol. Sci. 2019, 20, 2142. https://doi.org/10.3390/ijms20092142
Nollet L, Van Gils M, Verschuere S, Vanakker O. The Role of Vitamin K and Its Related Compounds in Mendelian and Acquired Ectopic Mineralization Disorders. International Journal of Molecular Sciences. 2019; 20(9):2142. https://doi.org/10.3390/ijms20092142
Chicago/Turabian StyleNollet, Lukas, Matthias Van Gils, Shana Verschuere, and Olivier Vanakker. 2019. "The Role of Vitamin K and Its Related Compounds in Mendelian and Acquired Ectopic Mineralization Disorders" International Journal of Molecular Sciences 20, no. 9: 2142. https://doi.org/10.3390/ijms20092142
APA StyleNollet, L., Van Gils, M., Verschuere, S., & Vanakker, O. (2019). The Role of Vitamin K and Its Related Compounds in Mendelian and Acquired Ectopic Mineralization Disorders. International Journal of Molecular Sciences, 20(9), 2142. https://doi.org/10.3390/ijms20092142