Straightforward Protein-Protein Interaction Interface Mapping via Random Mutagenesis and Mammalian Protein Protein Interaction Trap (MAPPIT)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
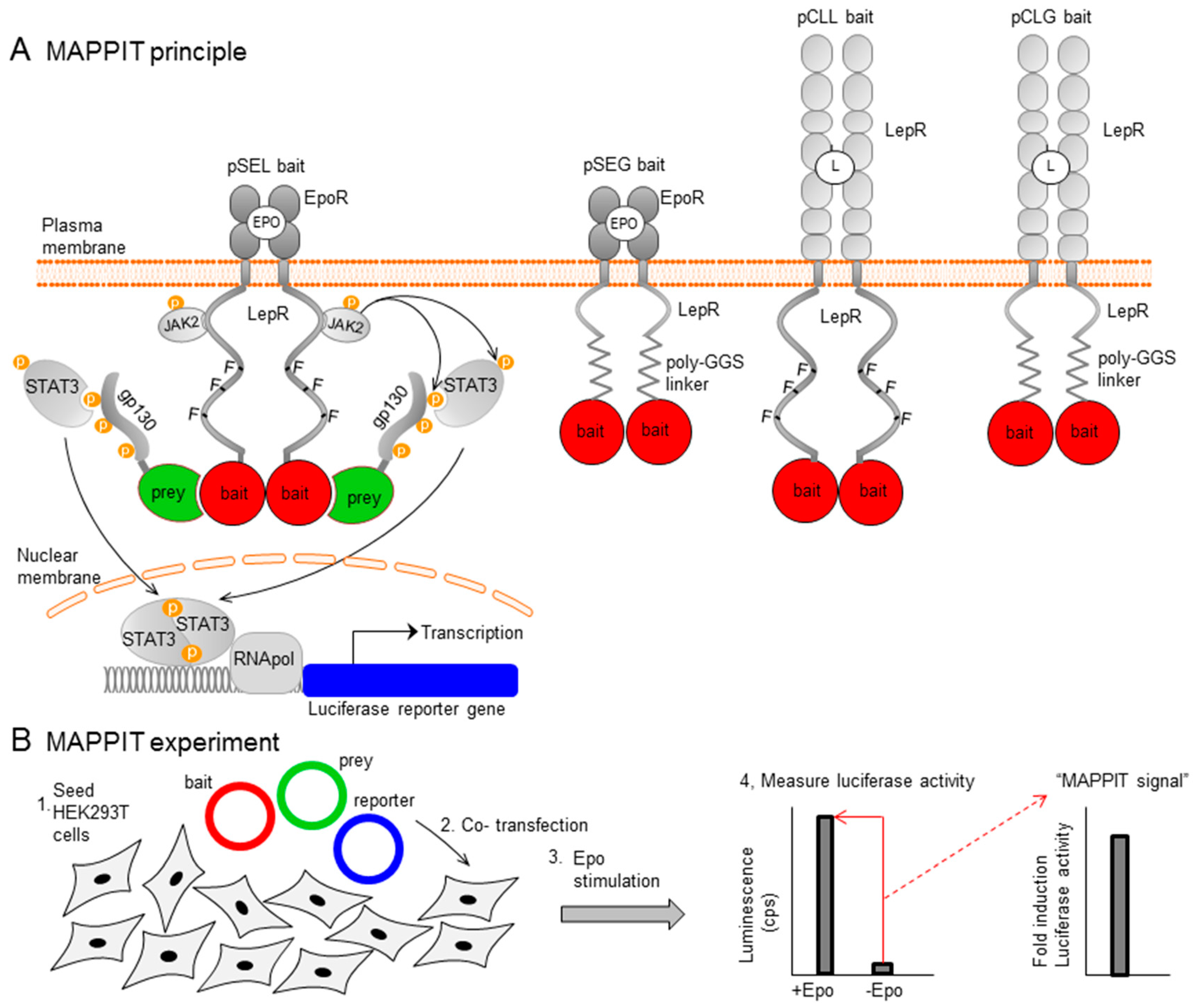
2. Materials and Methods
2.1. Step 1: Optimization of the MAPPIT Readout
2.2. Step 2: Random Mutagenesis via Error-Prone PCR
2.3. Step 3: Generation of a Mutant Plasmid Library in 96-Well Format
2.4. Step 4: MAPPIT in 384-Well Assay
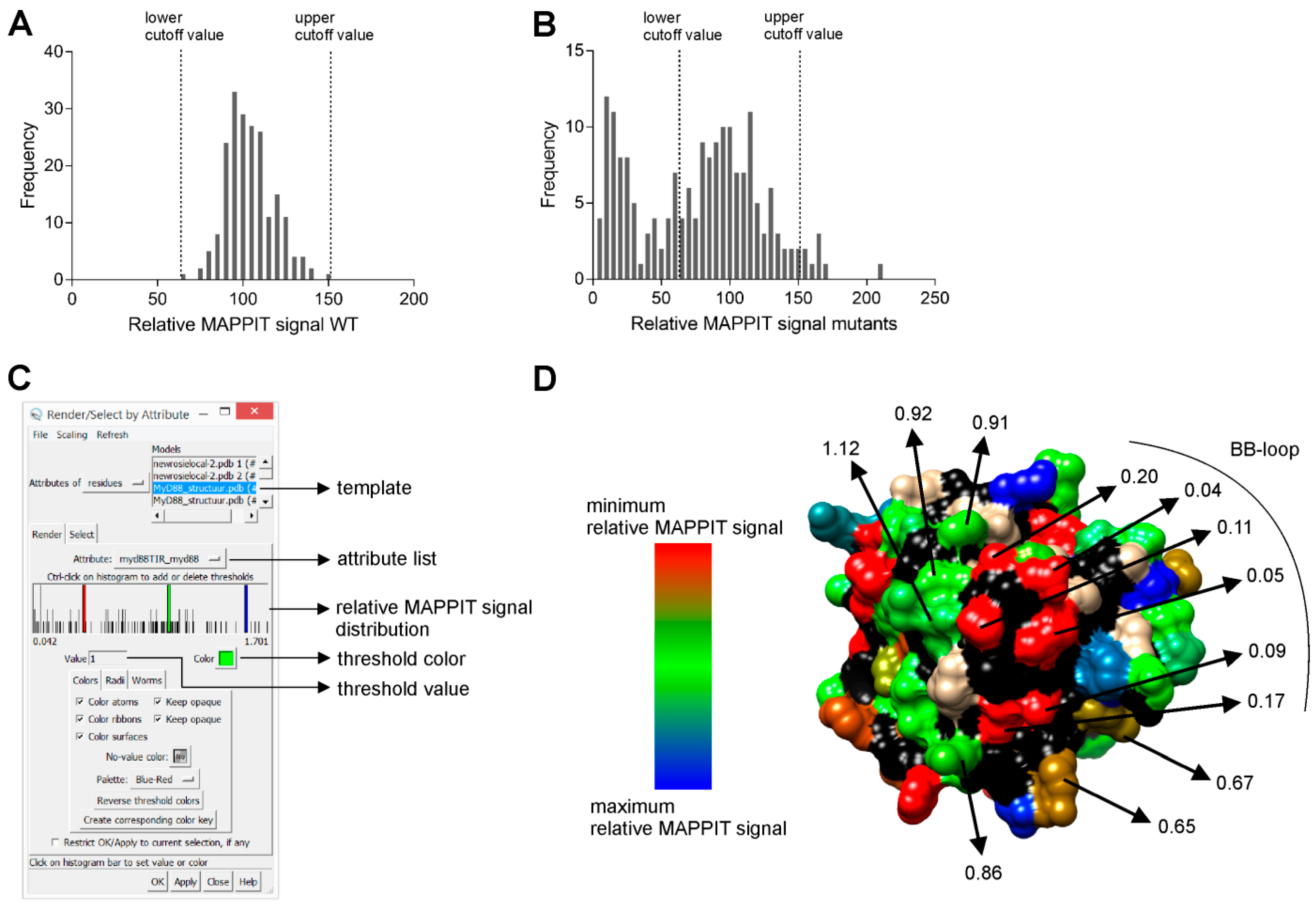
2.5. Step 5: Data Analysis
2.6. Step 6: Mapping of Relative MAPPIT Signals
3. Results and Discussion
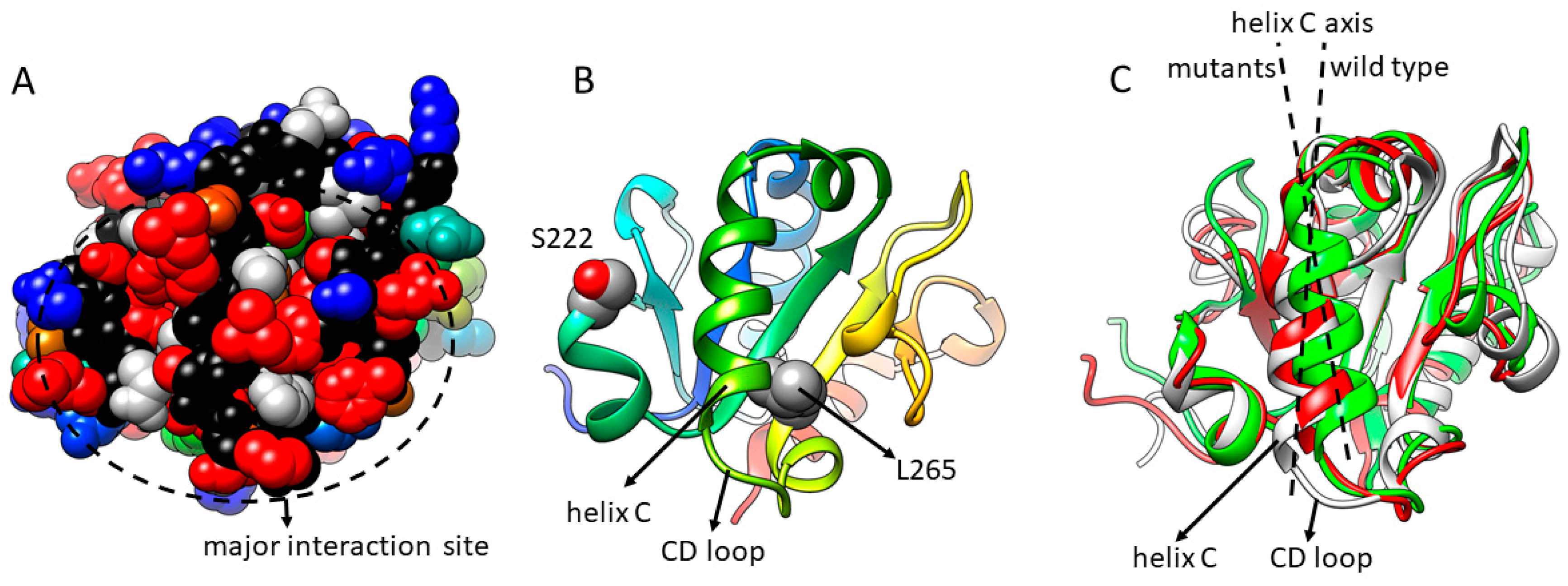
3.1. MAPPIT Detects Interfaces in Diverse Target Proteins
3.2. Interfaces Detected via MAPPIT Are Confirmed via Other Methods and Studies
3.3. MAPPIT and Mutagenesis Provide Insight in Disease Mechanisms
3.4. Interactions Requiring Additional Ligands or Phosphorylation Can Be Studied via MAPPIT
3.5. Critical Considerations in Data Interpretation
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Apobec3G | apolipoprotein B messenger RNA-editing catalytic polypeptide-like G |
| Epo | erythropoietin |
| gp130 | glycoprotein 130 |
| JAK | Janus kinase |
| LepR | leptin receptor |
| Mal | MyD88 adapter-like |
| MAPPIT | mammalian protein-protein interaction trap |
| MyD88 | myeloid differentiation primary response gene 88 |
| NMR | nuclear magnetic resonance |
| PPAR-α | peroxisome proliferator-activated receptor α |
| PPI | protein-protein interaction |
| PTM | post-translational modification |
| RNF41 | ring finger protein 41 |
| STAT | signal transducers and activators of transcription |
| TIR | Toll/interleukin-1 receptor |
| TRIF | TIR-domain-containing adapter inducing interferon-β |
References
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef]
- Gingras, A.-C.; Gstaiger, M.; Raught, B.; Aebersold, R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007, 8, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Söderberg, O.; Gullberg, M.; Jarvius, M.; Ridderstråle, K.; Leuchowius, K.-J.; Jarvius, J.; Wester, K.; Hydbring, P.; Bahram, F.; Larsson, L.-G.; et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods 2006, 3, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Boute, N.; Jockers, R.; Issad, T. The use of resonance energy transfer in high-throughput screening: BRET versus FRET. Trends Pharmacol. Sci. 2002, 23, 351–354. [Google Scholar] [CrossRef]
- Barrios-Rodiles, M.; Brown, K.R.; Ozdamar, B.; Bose, R.; Liu, Z.; Donovan, R.S.; Shinjo, F.; Liu, Y.; Dembowy, J.; Taylor, I.W.; et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science 2005, 307, 1621–1625. [Google Scholar] [CrossRef]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef]
- Eyckerman, S.; Titeca, K.; Van Quickelberghe, E.; Cloots, E.; Verhee, A.; Samyn, N.; De Ceuninck, L.; Timmerman, E.; De Sutter, D.; Lievens, S.; et al. Trapping mammalian protein complexes in viral particles. Nat. Commun. 2016, 7, 11416. [Google Scholar] [CrossRef] [PubMed]
- Petschnigg, J.; Groisman, B.; Kotlyar, M.; Taipale, M.; Zheng, Y.; Kurat, C.F.; Sayad, A.; Sierra, J.R.; Mattiazzi Usaj, M.; Snider, J.; et al. The mammalian-membrane two-hybrid assay (MaMTH) for probing membrane-protein interactions in human cells. Nat. Methods 2014, 11, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Weinrich, D.; Jonkheijm, P.; Niemeyer, C.M.; Waldmann, H. Applications of protein biochips in biomedical and biotechnological research. Angew. Chem. Int. Ed. Engl. 2009, 48, 7744–7751. [Google Scholar] [CrossRef]
- Eyckerman, S.; Verhee, A.; Van der Heyden, J.; Lemmens, I.; Van Ostade, X.; Vandekerckhove, J.; Tavernier, J. Design and application of a cytokine-receptor-based interaction trap. Nat. Cell Biol. 2001, 3, 1114–1119. [Google Scholar] [CrossRef]
- Yu, H.; Braun, P.; Yildirim, M.A.; Lemmens, I.; Venkatesan, K.; Sahalie, J.; Hirozane-Kishikawa, T.; Gebreab, F.; Li, N.; Simonis, N.; et al. High-quality binary protein interaction map of the yeast interactome network. Science 2008, 322, 104–110. [Google Scholar] [CrossRef]
- Braun, P.; Tasan, M.; Dreze, M.; Barrios-Rodiles, M.; Lemmens, I.; Yu, H.; Sahalie, J.M.; Murray, R.R.; Roncari, L.; de Smet, A.-S.; et al. An experimentally derived confidence score for binary protein-protein interactions. Nat. Methods 2009, 6, 91–97. [Google Scholar] [CrossRef]
- Rolland, T.; Taşan, M.; Charloteaux, B.; Pevzner, S.J.; Zhong, Q.; Sahni, N.; Yi, S.; Lemmens, I.; Fontanillo, C.; Mosca, R.; et al. A Proteome-Scale Map of the Human Interactome Network. Cell 2014, 159, 1212–1226. [Google Scholar] [CrossRef]
- Mosca, R.; Céol, A.; Aloy, P. Interactome3D: Adding structural details to protein networks. Nat. Methods 2012, 10, 47–53. [Google Scholar] [CrossRef]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.J.J.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 589, 3516–3526. [Google Scholar] [CrossRef]
- De Vries, S.J.; Melquiond, A.S.J.; Kastritis, P.L.; Karaca, E.; Bordogna, A.; van Dijk, M.; Rodrigues, J.P.G.L.M.; Bonvin, A.M.J.J. Strengths and weaknesses of data-driven docking in critical assessment of prediction of interactions. Proteins 2010, 78, 3242–3249. [Google Scholar] [CrossRef]
- Rasila, T.S.; Pajunen, M.I.; Savilahti, H. Critical evaluation of random mutagenesis by error-prone polymerase chain reaction protocols, Escherichia coli mutator strain, and hydroxylamine treatment. Anal. Biochem. 2009, 388, 71–80. [Google Scholar] [CrossRef]
- Bovijn, C.; Desmet, A.-S.; Uyttendaele, I.; Van Acker, T.; Tavernier, J.; Peelman, F. Identification of binding sites for myeloid differentiation primary response gene 88 (MyD88) and Toll-like receptor 4 in MyD88 adapter-like (Mal). J. Biol. Chem. 2013, 288, 12054–12066. [Google Scholar] [CrossRef]
- Vyncke, L.; Bovijn, C.; Pauwels, E.; Van Acker, T.; Ruyssinck, E.; Burg, E.; Tavernier, J.; Peelman, F. Reconstructing the TIR Side of the Myddosome: A Paradigm for TIR-TIR Interactions. Structure 2016, 24, 437–447. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Snyder, G.A.; Cirl, C.; Jiang, J.; Chen, K.; Waldhuber, A.; Smith, P.; Römmler, F.; Snyder, N.; Fresquez, T.; Dürr, S.; et al. Molecular mechanisms for the subversion of MyD88 signaling by TcpC from virulent uropathogenic Escherichia coli. Proc. Natl. Acad. Sci. USA 2013, 110, 6985–6990. [Google Scholar] [CrossRef]
- Uyttendaele, I.; Lavens, D.; Catteeuw, D.; Lemmens, I.; Bovijn, C.; Tavernier, J.; Peelman, F. Random mutagenesis MAPPIT analysis identifies binding sites for Vif and Gag in both cytidine deaminase domains of Apobec3G. PLoS ONE 2012, 7, e44143. [Google Scholar] [CrossRef]
- Masschaele, D.; De Ceuninck, L.; Wauman, J.; Defever, D.; Stenner, F.; Lievens, S.; Peelman, F.; Tavernier, J. RNF41 interacts with the VPS52 subunit of the GARP & EARP complexes. PLoS ONE 2017, 12, e0178132. [Google Scholar]
- Kouno, T.; Luengas, E.M.; Shigematsu, M.; Shandilya, S.M.D.; Zhang, J.; Chen, L.; Hara, M.; Schiffer, C.A.; Harris, R.S.; Matsuo, H. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 2015, 22, 485–491. [Google Scholar] [CrossRef]
- Lavens, D.; Peelman, F.; van der Heyden, J.; Uyttendaele, I.; Catteeuw, D.; Verhee, A.; van Schoubroeck, B.; Kurth, J.; Hallenberger, S.; Clayton, R.; et al. Definition of the interacting interfaces of Apobec3G and HIV-1 Vif using MAPPIT mutagenesis analysis. Nucleic Acids Res. 2009, 38, 1902–1912. [Google Scholar] [CrossRef]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: Mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [CrossRef]
- Varettoni, M.; Arcaini, L.; Zibellini, S.; Boveri, E.; Rattotti, S.; Riboni, R.; Corso, A.; Orlandi, E.; Bonfichi, M.; Gotti, M.; et al. Prevalence and clinical significance of the MYD88 (L265P) somatic mutation in Waldenstrom’s macroglobulinemia and related lymphoid neoplasms. Blood 2013, 121, 2522–2528. [Google Scholar] [CrossRef]
- Sikora, K.A.; Bennett, J.R.; Vyncke, L.; Deng, Z.; Tsai, W.L.; Pauwels, E.; Layh-Schmitt, G.; Brundidge, A.; Navid, F.; Zaal, K.J.M.; et al. Germline gain-of-function myeloid differentiation primary response gene–88 (MYD88) mutation in a child with severe arthritis. J. Allergy Clin. Immunol. 2018, 141, 1943–1947. [Google Scholar] [CrossRef]
- Van Durme, J.; Delgado, J.; Stricher, F.; Serrano, L.; Schymkowitz, J.; Rousseau, F. A graphical interface for the FoldX forcefield. Bioinformatics 2011, 27, 1711–1712. [Google Scholar] [CrossRef]
- Lemmens, I.; et al. Heteromeric MAPPIT: A novel strategy to study modification-dependent protein-protein interactions in mammalian cells. Nucleic Acids Res. 2003, 15, e75. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyncke, L.; Masschaele, D.; Tavernier, J.; Peelman, F. Straightforward Protein-Protein Interaction Interface Mapping via Random Mutagenesis and Mammalian Protein Protein Interaction Trap (MAPPIT). Int. J. Mol. Sci. 2019, 20, 2058. https://doi.org/10.3390/ijms20092058
Vyncke L, Masschaele D, Tavernier J, Peelman F. Straightforward Protein-Protein Interaction Interface Mapping via Random Mutagenesis and Mammalian Protein Protein Interaction Trap (MAPPIT). International Journal of Molecular Sciences. 2019; 20(9):2058. https://doi.org/10.3390/ijms20092058
Chicago/Turabian StyleVyncke, Laurens, Delphine Masschaele, Jan Tavernier, and Frank Peelman. 2019. "Straightforward Protein-Protein Interaction Interface Mapping via Random Mutagenesis and Mammalian Protein Protein Interaction Trap (MAPPIT)" International Journal of Molecular Sciences 20, no. 9: 2058. https://doi.org/10.3390/ijms20092058
APA StyleVyncke, L., Masschaele, D., Tavernier, J., & Peelman, F. (2019). Straightforward Protein-Protein Interaction Interface Mapping via Random Mutagenesis and Mammalian Protein Protein Interaction Trap (MAPPIT). International Journal of Molecular Sciences, 20(9), 2058. https://doi.org/10.3390/ijms20092058