Targeting Tyrosine kinases in Renal Cell Carcinoma: “New Bullets against Old Guys”


Abstract
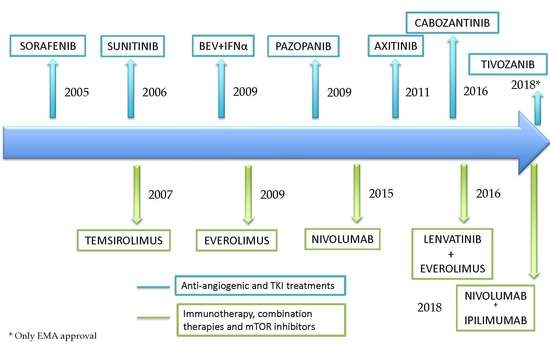
1. Introduction
2. Molecular Biology of Kidney Cancer
2.1. Von Hippel Lindau (VHL)
2.2. Other Genetic Alterations
3. Tyrosine Kinases and Coupled Intracellular Signaling Involved in RCC
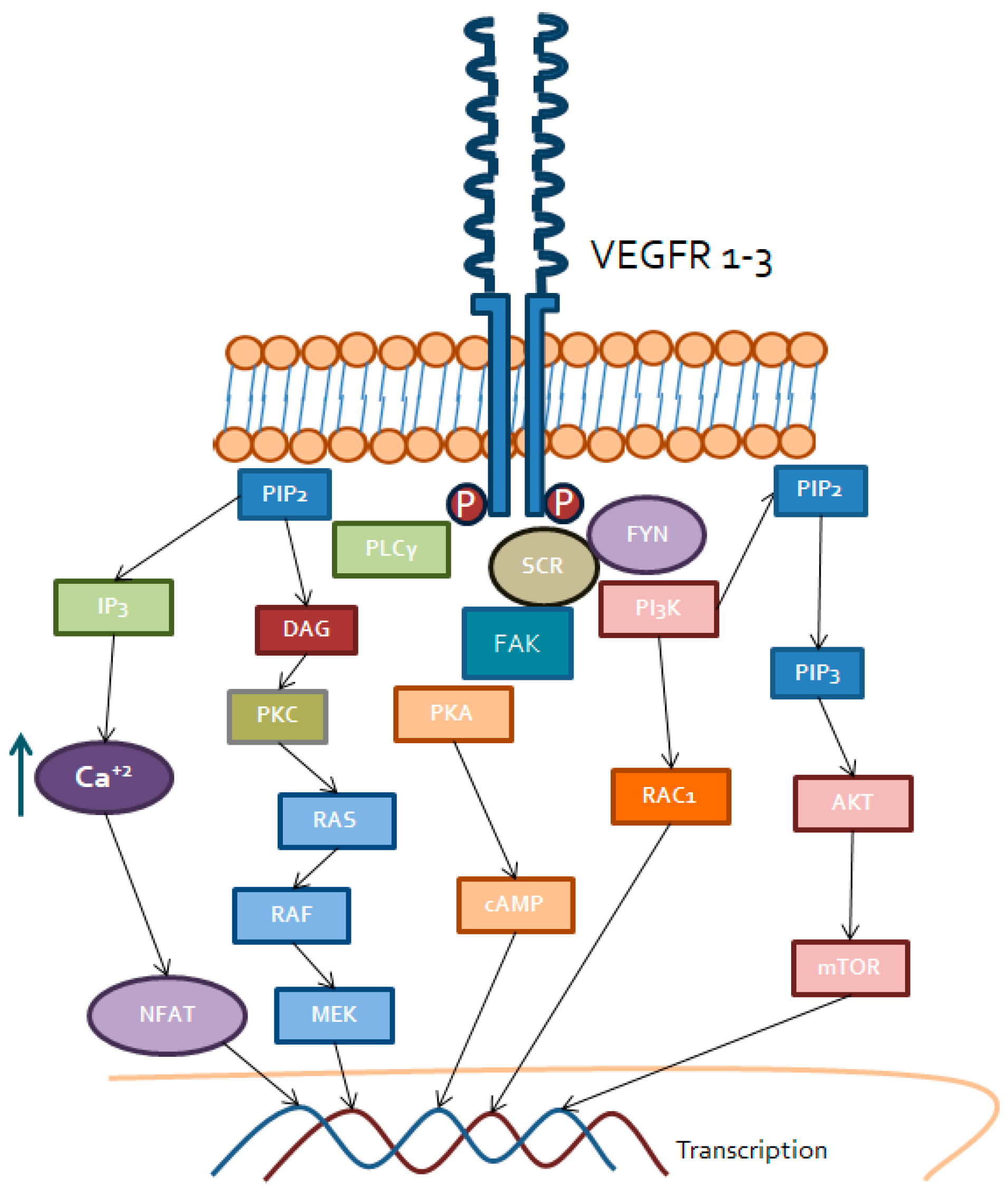
3.1. Vascular Endothelial Growth factor Receptor (VEGFR)
3.1.1. PI3K/AKT/mTOR
3.1.2. Phospholipase C (PLCγ) Pathway
3.2. Platelet Derived Growth Factor Receptor (PDGFR)
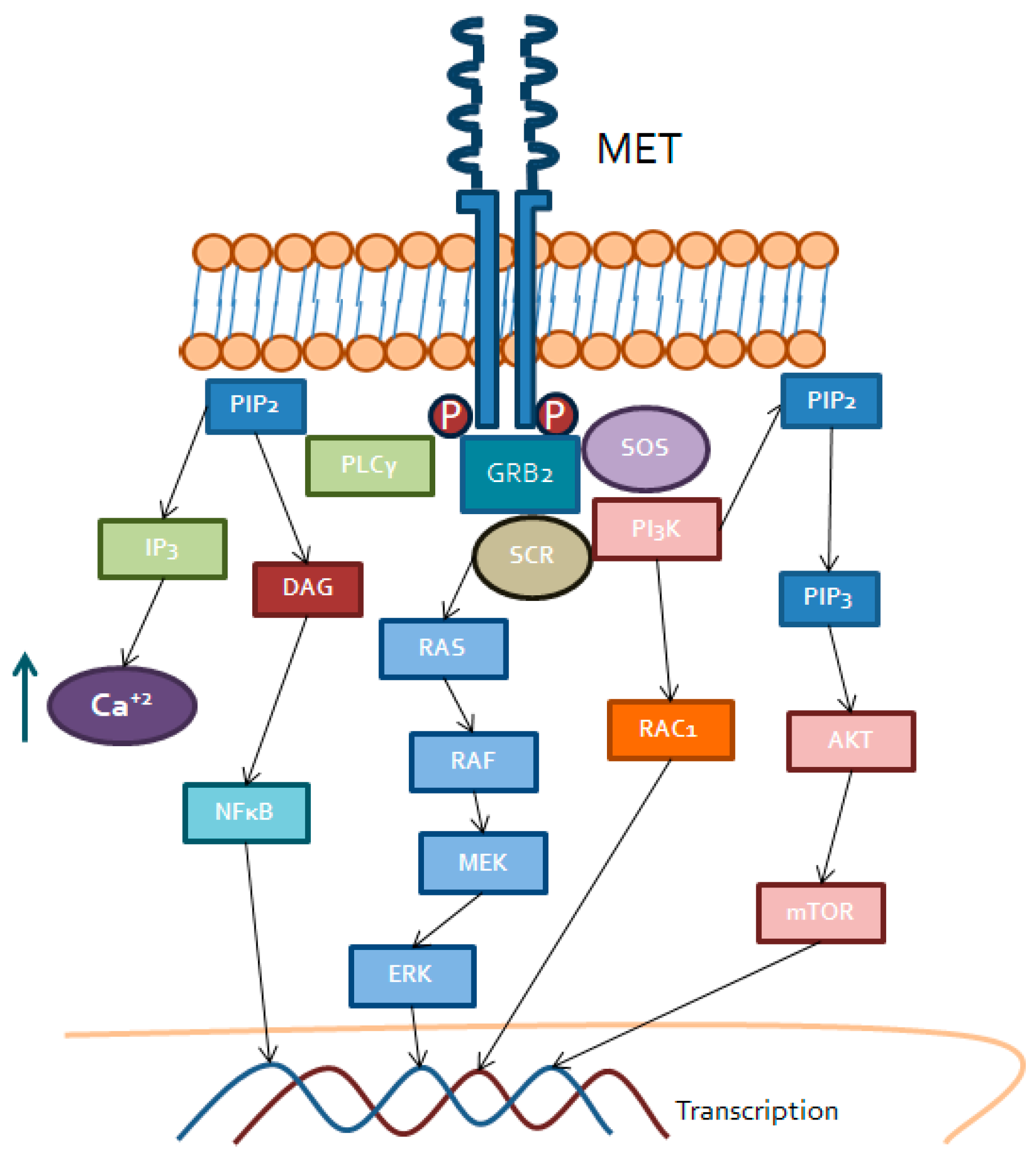
3.3. Tyrosine-Protein Kinase Met (MET)
3.4. Axl
3.5. Fibroblast Growth factor Receptor (FGFR)
- FGF1: Constituted by FGF1, is also known as aFGF or FGF acid and FGF2 or bFGF (basic FGF);
- FGF4: Constituted by FGF4, FGF5 and FGF6;
- FGF7: Constituted by FGF3, FG7, FGF10 and FGF22;
- FGF9: Constituted by FGF9, FGF16 and FGF20;
- FGF8: Constituted by FGF8, FGF17 and FGF18.
- FGF11: Constituted by FGF11, FGF12, FGF13 and FGF14.
- FGF15/19: Constituted by FGF15/19, FGF21 and FGF23.
4. Tyrosine Kinases and Coupled Intracellular Signaling Involved in RCC
4.1. Sunitinib
4.2. Pazopanib
4.3. Tivozanib
4.4. Axitinib
4.5. Cabozantinib
4.6. Lenvatinib
4.7. Sorafenib
5. The Impact of Immunotherapy in an Angiogenic Disease
6. Discussion: Considerations in RCC Treatment
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
| Yes (points) | No (points) | |
| Hemoglobin < Lower Limit of Normal | 1 | 0 |
| Time from diagnosis to systemic treatment <1 year | 1 | 0 |
| Calcium >10mg/dL (>2.5 mmol/L) | 1 | 0 |
| Performance status <80% (Karnofsky) | 1 | 0 |
| LDH > 1.5x Upper Limit of Normal | 1 | 0 |
Appendix B
| Yes (points) | No (points) | |
| Performance status <80% (Karnofsky) | 1 | 0 |
| Less than one year from time of diagnosis to systemic therapy | 1 | 0 |
| Calcium > upper limit of normal | 1 | 0 |
| Hemoglobin < lower limit of normal | 1 | 0 |
| Neutrophils > upper limit of normal | 1 | 0 |
| Platelets > upper limit of normal | 1 | 0 |
References
- Ferlay, J.; Steliarova-Foucher, E.; Lortet-Tieulent, J.; Rosso, S.; Coebergh, J.W.; Comber, H.; Forman, D.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries in 2012. Eur. J. Cancer 2013, 49, 1374–1403. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Znaor, A.; Lortet-Tieulent, J.; Laversanne, M.; Jemal, A.; Bray, F. International variations and trends in renal cell carcinoma incidence and mortality. Eur. Urol. 2015, 67, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Dabestani, S.; Thorstenson, A.; Lindblad, P.; Harmenberg, U.; Ljungberg, B.; Lundstam, S. Renal cell carcinoma recurrences and metastases in primary on-metastatic patients: A population-based study. World J. Urol. 2016, 34, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Kane, C.J.; Mallin, K.; Ritchey, J.; Cooperberg, M.R.; Carroll, P.R. Renal cell cancer stage migration: Analysis of the National Cancer Data Base. Cancer 2008, 113, 78–83. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; Oudard, S.; Negrier, S.; Szczylik, C.; Kim, S.T.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2007, 356, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Oudard, S.; Negrier, S.; Szczylik, C.; Pili, R.; Bjarnason, G.A.; et al. Overall Survival and Updated Results for Sunitinib Compared with Interferon Alfa in Patients with Metastatic Renal Cell Carcinoma. J. Clin. Oncol. 2009, 27, 3584–3590. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Oudard, S.; Hutson, T.E.; Porta, C.; Bracarda, S.; Grünwald, V.; Thompson, J.A.; Figlin, R.A.; Hollaender, N.; et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet 2008, 372, 449–456. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Nickerson, M.L.; Jaeger, E.; Shi, Y.; Durocher, J.A.; Mahurkar, S.; Zaridze, D.; Matveev, V.; Janout, V.; Kollarova, H.; Bencko, V.; et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin. Cancer Res. 2008, 14, 4726–4734. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, J.R.; Tory, K.; Weng, Y.; Schmidt, L.; Wei, M.H.; Li, H.; Latif, F.; Liu, S.; Chen, F.; Duh, F.-M.; et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat. Genet. 1994, 7, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Latif, F.; Tory, K.; Gnarra, J.R.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Kibel, A.; Iliopoulos, O.; DeCaprio, J.A.; Kaelin, W.G., Jr. Binding of the von Hippel-Lindau tumor suppressor protein to elongin B and C. Science 1995, 269, 1444–1446. [Google Scholar] [CrossRef] [PubMed]
- Aldo, P.; Elisabetta, C. Role of HIF-1 in cancer progression: Novel insights. A review. Curr. Mol. Med. 2018, 18, 343–351. [Google Scholar]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Nabi, S.; Kessler, E.R.; Bernard, B.; Flaig, T.W.; Lam, E.T. Renal cell carcinoma: A review of biology and Pathophysiology. F1000Res 2018, 7, 307. [Google Scholar] [CrossRef]
- Thomas, G.V.; Tran, C.; Mellinghoff, I.K.; Welsbie, D.S.; Chan, E.; Fueger, B.; Czernin, J.; Sawyers, C.L. Hypoxia- inducible factor determines sensitivity to inhibitors of mTOR in kidney cancer. Nat. Med. 2006, 12, 122–127. [Google Scholar] [CrossRef]
- Linehan, W.; Srinivasan, R.; Schmidt, L. The genetic basis of kidney cancer: A metabolic disease. Nat. Rev. Urol. 2010, 7, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Porta, C.; Schmidinger, M.; Rioux-Leclercq, N.; Bex, A.; Khoo, V.; Grünwald, V.; Gillessen, S.; Horwich, A. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hakimi, A.A.; Pham, C.G.; Hsieh, J.J. A clear picture of renal cell carcinoma. Nat. Genet. 2013, 45, 849–850. [Google Scholar] [CrossRef] [PubMed]
- Kapur, P.; Peña-Llopis, S.; Christie, A.; Zhrebker, L.; Pavía-Jiménez, A.; Rathmell, W.K.; Xie, X.J.; Brugarolas, J. Effects on survival of BAP1 and PBRM1 mutations in sporadic clear-cell renal-cell carcinoma: A retrospective analysis with independent validation. Lancet Oncol. 2013, 14, 159–167. [Google Scholar] [CrossRef]
- Manley, B.J.; Zabor, E.C.; Casuscelli, J.; Tennenbaum, D.M.; Redzematovic, A.; Becerra, M.F.; Benfante, N.; Sato, Y.; Morikawa, T.; Kume, H.; et al. Integration of Recurrent Somatic Mutations with Clinical Outcomes: A Pooled Analysis of 1049 Patients with Clear Cell Renal Cell Carcinoma. Eur. Urol. Focus 2017, 3, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.S.; Junker, K.; Nakaigawa, N.; Kinjerski, T.; Weirich, G.; Miller, M.; Lubensky, I.; Neumann, H.P.; Brauch, H.; Decker, J.; et al. Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 1999, 18, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Fuh, K.C.; Castellini, L.; Viswanathan, K.; Finger, E.C.; Diep, A.N.; LaGory, E.L.; Kariolis, M.S.; Chan, A.; Lindgren, D.; et al. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 2014, 111, 13373–13378. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, X.D.; Sun, M.; Zhang, X.; German, P.; Bai, S.; Ding, Z.; Tannir, N.; Wood, C.G.; Matin, S.F.; et al. Targeting MET and AXL overcomes resistance to sunitinib therapy in renal cell carcinoma. Oncogene 2016, 35, 2687–2697. [Google Scholar] [CrossRef]
- Parveen, A.; Subedi, L.; Kim, H.W.; Khan, Z.; Zahra, Z.; Farooqi, M.Q.; Kim, S.Y. Phytochemicals Targeting VEGF and VEGF-Related Multifactors as Anticancer Therapy. J. Clin. Med. 2019, 8, 350. [Google Scholar] [CrossRef]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef]
- Chung, A.S.; Ferrara, N. Developmental and pathological angiogenesis. Annu. Rev. Cell Dev. Biol. 2011, 27, 563–584. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Iring, A.; Strilic, B.; Albarrán-Juárez, J.; Kaur, H.; Troidl, K.; Tonack, S.; Burbiel, J.C.; Müller, C.E.; Fleming, I.; et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J. Clin. Investig. 2015, 125, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014, 343, 1235681. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Ballmer-Hofer, K.; Hristova, K. VEGFR-2 conformational switch in response to ligand binding. Elife 2016, 5, e13876. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, A.; Leppänen, V.M.; Tvorogov, D.; Zarkada, G.; Jeltsch, M.; Holopainen, T.; Kaijalainen, S.; Alitalo, K. The basis for the distinct biological activities of vascular endothelial growth factor eceptor-1 ligands. Sci. Signal. 2013, 6, ra52. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, X.; Simons, M. Molecular controls of lymphatic VEGFR3 signaling. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Padhan, N.; Sjöström, E.O.; Roche, F.P.; Testini, C.; Honkura, N.; Sáinz-Jaspeado, M.; Gordon, E.; Bentley, K.; Philippides, A.; et al. VEGFR2 pY949 signalling regulates adherens junction integrity and metastatic spread. Nat. Commun. 2016, 7, 11017. [Google Scholar] [CrossRef]
- Lee, M.Y.; Luciano, A.K.; Ackah, E.; Rodriguez-Vita, J.; Bancroft, T.A.; Eichmann, A.; Simons, M.; Kyriakides, T.R.; Morales-Ruiz, M.; Sessa, W.C. Endothelial Akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc. Natl. Acad. Sci. USA 2014, 111, 12865–12870. [Google Scholar] [CrossRef]
- Chen, J.; Somanath, P.R.; Razorenova, O.; Chen, W.S.; Hay, N.; Bornstein, P.; Byzova, T.V. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005, 11, 1188–1196. [Google Scholar] [CrossRef]
- Aspenström, P. Activated Rho GTPases in Cancer-The Beginning of a New Paradigm. Int. J. Mol. Sci. 2018, 19, 3949. [Google Scholar] [CrossRef]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Yamaguchi, S.; Chida, K.; Shibuya, M. A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-γ and DNA synthesis in vascular endothelial cells. EMBO J. 2001, 20, 2768–2778. [Google Scholar] [CrossRef] [PubMed]
- Lawson, N.D.; Mugford, J.W.; Diamond, B.A.; Weinstein, B.M. Phospholipase C gamma-1 is required downstream of vascular endothelial growth factor during arterial development. Genes Dev. 2003, 17, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Rinne, A.; Banach, K.; Blatter, L.A. Regulation of nuclear factor of activated T cells (NFAT) in vascular endothelial cells. J. Mol. Cell. Cardiol. 2009, 47, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, J.D.; Kim, M.S.; Houlihan, P.R.; Shutov, L.P.; Mohapatra, D.P.; Strack, S.; Usachev, Y.M. Distinct activation properties of the nuclear factor of activated T-cells (NFAT) isoforms NFATc3 and NFATc4 in neurons. J. Biol. Chem. 2012, 287, 37594–37609. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Escobedo, J.A.; Kuang, W.J.; Yang-Feng, T.L.; Daniel, T.O.; Tremble, P.M.; Chen, E.Y.; Ando, M.E.; Harkins, R.N.; Francke, U.; et al. Structure of the receptor for platelet-derived growth factor helps define a family of closely related growth factor receptors. Nature 1986, 323, 226–232. [Google Scholar] [CrossRef]
- Heldin, C.H.; Lennartsson, J.; Westermark, B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef]
- Jitariu, A.; Raica, M.; Cîmpean, A.M.; Suciu, S.C. The role of PDGF-B/PDGFR-BETA axis in the normal development and carcinogenesis of the breast. Crit. Rev. Oncol. Hematol. 2018, 131, 46–52. [Google Scholar] [CrossRef]
- Xu, J.; Xie, L.; Guo, W. PDGF/PDGFR effects in osteosarcoma and the “add-on” strategy. Clin. Sarcoma Res. 2018, 8, 15. [Google Scholar] [CrossRef]
- Qian, H.; Appiah-Kubi, K.; Wang, Y.; Wu, M.; Tao, Y.; Wu, Y.; Chen, Y. The clinical significance of platelet-derived growth factors (PDGFs) and their receptors (PDGFRs) in gastric cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2018, 127, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. The role of small molecule platelet-derived growth factor receptor (PDGFR) inhibitors in the treatment of neoplastic disorders. Pharmacol. Res. 2018, 129, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Pal, I.; Mandal, M. PI3K and Akt as molecular targets for cancer therapy: Current clinical outcomes. Acta Pharmacol. Sin. 2012, 33, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.J.; Suh, P.G.; Lee, Y.J.; Shin, K.J.; Cocco, L.; Chae, Y.C. PLCγ1: Potential arbitrator of cancer progression. Adv. Biol. Regul. 2018, 67, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Janus kinase (JAK) inhibitors in the treatment of inflammatory and neoplastic diseases. Pharmacol. Res. 2016, 111, 784–803. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Westermark, B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef] [PubMed]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Bottaro, D.P.; Rubin, J.S.; Faletto, D.L.; Chan, A.M.; Kmiecik, T.E.; Vande-Woude, G.F.; Aaronson, S.A. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991, 251, 802–804. [Google Scholar] [CrossRef]
- Birchmeier, C.; Birchmeier, W.; Gherardi, E.; Vande-Woude, G.F. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 2003, 4, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Salgia, R. c-Met and hepatocyte growth factor: Potential as novel targets in cancer therapy. Curr. Oncol. Rep. 2007, 9, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Mo, H.N.; Liu, P. Targeting MET in cancer therapy. Chronic Dis. Transl. Med. 2017, 3, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.C.; Maulik, G.; Christensen, J.; Salgia, R. c-Met: Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003, 22, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Czyz, M. HGF/c-MET Signaling in Melanocytes and Melanoma. Int. J. Mol. Sci. 2018, 19, 3844. [Google Scholar] [CrossRef] [PubMed]
- Noriega-Guerra, H.; Freitas, V.M. Extracellular Matrix Influencing HGF/c-MET Signaling Pathway: Impact on Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiang, X.; Jiang, Y.; Guo, M.; Zhang, S.; Li, J.; He, J.; Liu, J.; Wang, J.; Ouyang, L. Recent advances in the development of dual VEGFR and c-Met small molecule inhibitors as anticancer drugs. Eur. J. Med. Chem. 2016, 108, 495–504. [Google Scholar] [CrossRef]
- Miranda, O.; Farooqui, M.; Siegfried, J. Status of Agents Targeting the HGF/c-Met Axis in Lung Cancer. Cancers (Basel) 2018, 10, 280. [Google Scholar] [CrossRef] [PubMed]
- Abounader, R.; Reznik, T.; Colantuoni, C.; Martinez-Murillo, F.; Rosen, E.M.; Laterra, J. Regulation of c-Met-dependent gene expression by PTEN. Oncogene 2004, 23, 9173–9182. [Google Scholar] [CrossRef] [PubMed]
- Vouri, M.; Hafizi, S. TAM receptor tyrosine kinases in cancer drug resistance. Cancer Res. 2017, 77, 2775–2778. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; DeRyckere, D.; Davies, K.; Earp, H. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Caberoy, N.; Alvarado, G.; Bigcas, J.; Li, W. Galectin-3 is a new MerTK-specific eat-me signal. J. Cell. Physiol. 2012, 227, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Cristina, C.; Laura, G.; Cinzia, L.; Nadia, Z.; Diego, C.; Paola, P. Role of the receptor tyrosine kinase Axl and its targeting in cancer cells. Curr. Med. Chem. 2016, 23, 1496–1512. [Google Scholar]
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910. [Google Scholar] [CrossRef]
- Shen, Y.; Chen, X.; He, J.; Liao, D.; Zu, X. Axl inhibitors as novel cancer therapeutic agents. Life Sci. 2018, 198, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Antony, J.; Huang, R.Y. AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 2017, 77, 3725–3732. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar]
- Schoumacher, M.; Burbridge, M. Key Roles of AXL and MER Receptor Tyrosine Kinases in Resistance to Multiple Anticancer Therapies. Curr. Oncol. Rep. 2017, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Scaltriti, M.; Elkabets, M.; Baselga, J. Molecular Pathways: AXL, a Membrane Receptor Mediator of Resistance to Therapy. Clin. Cancer Res. 2016, 22, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.; Zhang, J.; Heckrodt, T.; Yu, J.; Ding, P.; Singh, R.; Holland, S.; Li, W.; Irving, M. Discovery of dual Axl/VEGF-R2 inhibitors as potential anti-angiogenic and anti-metastatic drugs for cancer chemotherapy. Bioorg. Med. Chem. Lett. 2017, 27, 3766–3771. [Google Scholar] [CrossRef]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef]
- Gong, S.G. Isoforms of receptors of fibroblast growth factors. J. Cell. Physiol. 2014, 229, 1887–1895. [Google Scholar] [CrossRef] [PubMed]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [PubMed]
- Ghedini, G.C.; Ronca, R.; Presta, M.; Giacomini, A. Future applications of FGF/FGFR inhibitors in cancer. Expert Rev. Anticancer Ther. 2018, 18, 861–872. [Google Scholar] [CrossRef] [PubMed]
- Porta, R.; Borea, R.; Coelho, A.; Khan, S.; Araújo, A.; Reclusa, P.; Franchina, T.; Van Der Steen, N.; Van Dam, P.; Ferri, J.; et al. FGFR a promising druggable target in cancer: Molecular biology and new drugs. Crit. Rev. Oncol. Hematol. 2017, 113, 256–267. [Google Scholar] [CrossRef]
- Du, X.; Xie, Y.; Xian, C.J.; Chen, L. Role of FGFs/FGFRs in skeletal development and bone regeneration. J. Cell. Physiol. 2012, 227, 3731–3743. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Grose, R. Fibroblast Growth Factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer. Res. 2016, 22, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef] [PubMed]
- Helsten, T.; Schwaederle, M.; Kurzrock, R. Fibroblast growth factor receptor signaling in hereditary and neoplastic disease: Biologic and clinical implications. Cancer Metastasis Rev. 2015, 34, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.C.; Lin, X.; Torres, J. The strong dimerization of the transmembrane domain of Williams SV the fibroblast growth factor receptor (FGFR) is modulated by C-terminal juxtamembrane residues. Protein Sci. 2009, 18, 450–459. [Google Scholar] [CrossRef]
- Klaeger, S.; Heinzlmeir, S.; Wilhelm1, M.; Polzer, H.; Vick, B.; Koenig, P.A.; Reinecke, M.; Ruprecht, B.; Petzoldt, S.; Meng, C.; et al. The target landscape of clinical kinase drugs. Science 2017, 358, 6367. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Mazumdar, M.; Bacik, J.; Berg, W.; Amsterdam, A.; Ferrara, J. Survival and prognostic stratification of 670 patients with advanced renal cell carcinoma. J. Clin. Oncol. 1999, 17, 2530–2540. [Google Scholar] [CrossRef]
- Heng, D.Y.; Xie, W.; Regan, M.M.; Warren, M.A.; Golshayan, A.R.; Sahi, C.; Eigl, B.J.; Ruether, J.D.; Cheng, T.; North, S.; et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: Results from a large, multicenter study. J. Clin. Oncol. 2009, 27, 5794–5799. [Google Scholar] [CrossRef] [PubMed]
- Faivre, S.; Delbaldo, C.; Vera, K.; Robert, C.; Lozahic, S.; Lassau, N.; Bello, C.; Deprimo, S.; Brega, N.; Massimini, G.; et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 2006, 24, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.E.; Szczylik, C.; Porta, C.; Bracarda, S.; Bjarnason, G.A.; Oudard, S.; Hariharan, S.; Lee, S.H.; Haanen, J.; Castellano, D.; et al. Safety and efficacy of sunitinib for metastatic renal-cell carcinoma: An expanded-access trial. Lancet Oncol. 2009, 10, 757–763. [Google Scholar] [CrossRef]
- Britten, C.D.; Kabbinavar, F.; Hecht, J.R.; Bello, C.L.; Li, J.; Baum, C.; Slamon, D. A phase I and pharmacokinetic study of sunitinib administered daily for 2 weeks, followed by a 1-week off period. Cancer Chemother. Pharmacol. 2008, 61, 515–524. [Google Scholar] [CrossRef]
- Lee, J.L.; Kim, M.K.; Park, I.; Ahn, J.H.; Lee, D.H.; Ryoo, H.M.; Song, C.; Hong, B.; Hong, J.H.; Ahn, H. RandomizEd phase II trial of Sunitinib four weeks on and two weeks off versus Two weeks on and One week off in metastatic clear-cell type REnal cell carcinoma: RESTORE trial. Ann. Oncol. 2015, 26, 2300–2305. [Google Scholar] [CrossRef]
- Sternberg, C.; Hawkins, R.E.; Wagstaff, J.; Salman, P.; Mardiak, J.; Barrios, C.H.; Zarba, J.J.; Gladkov, O.A.; Lee, E.; Szczylik, C.; et al. A randomised, double-blind phase III study of pazopanib in patients with advanced and/or metastatic renal cell carcinoma: Final overall survival results and safety update. Eur. J. Cancer 2013, 49, 1287–1296. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Hutson, T.E.; Cella, D.; Reeves, J.; Hawkins, R.; Guo, J.; Nathan, P.; Staehler, M.; de Souza, P.; Merchan, J.R.; et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N. Engl. J. Med. 2013, 369, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Nosov, D.; Eisen, T.; Bondarenko, I.; Lesovoy, V.; Lipatov, O.; Tomczak, P.; Lyulko, O.; Alyasova, A.; Harza, M.; et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: Results from a phase III trial. J. Clin. Oncol. 2013, 31, 3791–3799. [Google Scholar] [CrossRef] [PubMed]
- Molina, A.M.; Hutson, T.E.; Nosov, D.; Tomczak, P.; Lipatov, O.; Sternberg, C.N.; Motzer, R.; Eisen, T. Efficacy of tivozanib treatment after sorafenib in patients with advanced renal cell carcinoma: Crossover of a phase 3 study. Eur. J. Cancer 2018, 94, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Halabi, S.; Sanford, B.L.; Hahn, O.; Michaelson, M.D.; Walsh, M.K.; Feldman, D.R.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib Versus Sunitinib As Initial Targeted Therapy for Patients with Metastatic Renal Cell Carcinoma of Poor or Intermediate Risk: The Alliance A031203 CABOSUN Trial. J. Clin. Oncol. 2017, 35, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Hessel, C.; Halabi, S.; Sanford, B.; Michaelson, M.D.; Hahn, O.; Walsh, M.; Olencki, T.; Picus, J.; Small, E.J.; et al. Cabozantinib versus sunitinib as initial therapy for metastatic renal cell carcinoma of intermediate or poor risk (Alliance A031203 CABOSUN randomised trial): Progression-free survival by independent review and overall survival update. Eur. J. Cancer 2018, 94, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Pal, S.K.; Escudier, B.; Atkins, M.B.; Hutson, T.E.; Porta, C.; Verzoni, E.; Needle, M.N.; McDermott, D.F. TIVO-3: A phase III, randomized, controlled, multicenter, open-label study to compare tivozanib to sorafenib in subjects with refractory advanced renal cell carcinoma (RCC). J. Clin. Oncol. 2019, 37 (Suppl. 7), 541. [Google Scholar] [CrossRef]
- Rini, B.I.; Escudier, B.; Tomczak, P.; Kaprin, A.; Szczylik, C.; Hutson, T.E.; Michaelson, M.D.; Gorbunova, V.A.; Gore, M.E.; Rusakov, I.G.; et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): A randomised phase 3 trial. Lancet 2011, 378, 1931–1939. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; Tomczak, P.; Hutson, T.E.; Michaelson, M.D.; Negrier, S.; Oudard, S.; Gore, M.E.; Tarazi, J.; Hariharan, S.; et al. Axitinib versus sorafenib as second-line treatment for advanced renal cell carcinoma: Overall survival analysis and updated results from a randomised phase 3 trial. Lancet Oncol. 2013, 14, 552–562. [Google Scholar] [CrossRef]
- Rini, B.I.; Melichar, B.; Ueda, T.; Grünwald, V.; Fishman, M.N.; Arranz, J.A.; Bair, A.H.; Pithavala, Y.K.; Andrews, G.I.; Pavlov, D.; et al. Axitinib with or without dose titration for first-line metastatic renal-cell carcinoma: A randomised double-blind phase 2 trial. Lancet Oncol. 2013, 14, 1233–1242. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Pal, S.K.; McDermott, D.F.; Morrissey, S.; Ferguson, S.K.; Holland, J.; Kaelin, W.G.; Dutcher, J.P.; et al. A phase I study of cabozantinib (XL184) in patients with renal cell cancer. Ann. Oncol. 2014, 25, 1603–1608. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; Hammers, H.; Hutson, T.E.; Lee, J.L.; Peltola, K.; et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1814–1823. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Tannir, N.M.; Mainwaring, P.N.; Rini, B.I.; Hammers, H.J.; Donskov, F.; Roth, B.J.; Peltola, K.; et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol. 2016, 17, 917–927. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; Jassem, J.; Zolnierek, J.; Maroto, J.P.; Mellado, B.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–1482. [Google Scholar] [CrossRef]
- Escudier, B.; Eisen, T.; Stadler, W.M.; Szczylik, C.; Oudard, S.; Siebels, M.; Negrier, S.; Chevreau, C.; Solska, E.; Desai, A.A.; et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N. Engl. J. Med. 2007, 356, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Hutson, T.E.; Al-Shukri, S.; Stus, V.P.; Lipatov, O.N.; Shparyk, Y.; Bair, A.H.; Rosbrook, B.; Andrews, G.I.; Vogelzang, N.J.; et al. Axitinib Versus Sorafenib in First-Line Metastatic Renal Cell Carcinoma: Overall Survival from a Randomized Phase III Trial. Clin. Genitourin. Cancer 2017, 15, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; Tykodi, S.S.; Sosman, J.A.; Procopio, G.; Plimack, E.R.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2015, 373, 1803–1813. [Google Scholar] [CrossRef] [PubMed]
- Tannir, N.M.; Arén-Frontera, O.; Hammers, H.J.; Carducci, M.A.; McDermott, D.F.; Salman, P.; Escudier, B.; Beuselinck, B.; Amin, A.; Porta, C. Thirty-month follow-up of the phase III CheckMate 214 trial of first-line nivolumab + ipilimumab (N+I) or sunitinib (S) in patients (pts) with advanced renal cell carcinoma (aRCC). J. Clin. Oncol. 2019, 37 (Suppl. 7), 547. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1103–1115. [Google Scholar] [CrossRef]
- Motzer, R.J.; Powles, T.; Atkins, M.B.; Escudier, B.; McDermott, D.F.; Suarez, C.; Bracarda, S.; Stadler, M.W.; Donskov, F.; Lee, J.L.; et al. IMmotion151: A Randomized Phase III Study of Atezolizumab Plus Bevacizumab vs. Sunitinib in Untreated Metastatic Renal Cell Carcinoma (mRCC). J. Clin. Oncol. 2018, 36 (Suppl. 6), 578. [Google Scholar] [CrossRef]
- Schlumberger, M.; Tahara, M.; Wirth, L.J.; Robinson, B.; Brose, M.S.; Elisei, R.; Habra, M.A.; Newbold, K.; Shah, M.H.; Hoff, A.O.; et al. Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med. 2015, 372, 621–630. [Google Scholar] [CrossRef]
- Brose, M.S.; Nutting, C.M.; Jarzab, B.; Elisei, R.; Siena, S.; Bastholt, L.; de la Fouchardiere, C.; Pacini, F.; Paschke, R.; Shong, Y.K.; et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: A randomised, double-blind, phase 3 trial. Lancet 2014, 384, 319–328. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Capdevila, J.; Fazio, N.; López, C.; Teulé, A.; Valle, J.W.; Tafuto, S.; Custodio, A.; Reed, N.; Raderer, M.; Grande, E.; et al. Efficacy of lenvatinib in patients with advanced pancreatic (panNETs) and gastrointestinal (giNETs) WHO grade 1/2 (G1/G2) neuroendocrine tumors: Results of the international phase II TALENT trial (GETNE 1509). Ann. Oncol. 2018, 29 (Suppl. 8), viii467–viii478. [Google Scholar] [CrossRef]
- Chan, J.A.; Faris, J.E.; Murphy, J.E.; Blaszkowsky, L.S.; Kwak, E.L.; McCleary, N.J.; Fuchs, C.S.; Meyerhardt, J.A.; Ng, K.; Zhu, A.; et al. Phase II trial of cabozantinib in patient with carcinoid and pancreatic neuroendocrine tumors (Pnet). J. Clin. Oncol. 2017, 35 (Suppl. 4), 228. [Google Scholar] [CrossRef]
- Fallick, M.L.; McDermott, D.F.; LaRock, D.; Long, J.P.; Atkins, M.B. Nephrectomy before interleukin-2 therapy for patients with metastatic renal cell carcinoma. J. Urol. 1997, 158, 1691–1695. [Google Scholar] [CrossRef]
- Méjean, A.; Ravaud, A.; Thezenas, S.; Colas, S.; Beauval, J.B.; Bensalah, K.; Geoffrois, L.; Thiery-Vuillemin, A.; Cormier, L.; Lang, H.; et al. Sunitinib Alone or after Nephrectomy in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Bex, A.; Mulders, P.; Jewett, M.; Wagstaff, J.; van Thienen, J.V.; Blank, C.U.; van Velthoven, R.; Del Pilar Laguna, M.; Wood, L.; van Melick, H.H.E.; et al. Cytoreductive Nephrectomy in Patients with Synchronous Metastatic Renal Cell Carcinoma Receiving Sunitinib. The SURTIME Randomized Clinical Trial. JAMA Oncol. 2019, 5, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B. Combination Therapy as First-Line Treatment in Metastatic Renal-Cell Carcinoma. N. Engl. J. Med. 2019, 380, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Rini, B. New strategies in kidney cancer: Therapeutic advances through understanding the molecular basis of response and resistance. Clin. Cancer Res. 2010, 16, 1348–1354. [Google Scholar] [CrossRef]
- Atkins, M.B.; Gravis, G.; Drosik, K.; Demkow, T.; Tomczak, P.; Wong, S.S.; Michaelson, M.D.; Choueiri, T.K.; Wu, B.; Navale, L.; et al. Trebananib (AMG 386) in Combination with Sunitinib in Patients with Metastatic Renal Cell Cancer: An Open-Label, Multicenter, Phase II Study. J. Clin. Oncol. 2015, 33, 3431–3438. [Google Scholar] [CrossRef]
- Rini, B.; Appleman, L.J.; Figlin, R.A.; Plimack, E.R.; Merchan, J.R.; Wang, K.; Thamake, T.; Zojwalla, J.N.; Choueiri, T.K.; McDermott, D.F.; et al. Results from a phase I expansion cohort of the first-in-class oral HIF-2α inhibitor PT2385 in combination with nivolumab in patients with previously treated advanced RCC. J. Clin. Oncol. 2019, 37 (Suppl. 7), 558. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Lee, R.J.; Carthon, B.C.; Iliopoulos, O.; Mier, J.W.; Patelet, M.R.; Tannir, N.; Taofeek, O.; Haas, B.; Henner, N.; Voss, M. CB-839, a glutaminase inhibitor, in combination with cabozantinib in patients with clear cell and papillary metastatic renal cell cancer (mRCC): Results of a phase I study. J. Clin. Oncol. 2019, 37 (Suppl. 7), 549. [Google Scholar] [CrossRef]
- Molina-Cerrillo, J.; Alonso-Gordoa, T.; Gajate, P.; Grande, E. Bruton’s tyrosine kinase (BTK) as a promising target in solid tumors. Cancer Treat. Rev. 2017, 58, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Thomas, S.; Pawlowska, N.; Bartelink, I.; Grabowsky, J.; Jahan, T.; Cripps, A.; Harb, A.; Leng, J.; Reinert, A.; et al. Inhibiting Histone Deacetylase as a Means to Reverse Resistance to Angiogenesis Inhibitors: Phase I Study of Abexinostat Plus Pazopanib in Advanced Solid Tumor Malignancies. J. Clin. Oncol. 2017, 35, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Fragiadaki, M.; Zeidler, M.P. Ankyrin repeat and single KH domain 1 (ANKHD1) drives renal cancer cell proliferation via binding to and altering a subset of miRNAs. J. Biol. Chem. 2018, 293, 9570–9579. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Giovannetti, E.; Poel, D.; Nowak-Sliwinska, P.; Griffioen, A.W. miRNAs: Micro-managers of anticancer combination therapies. Angiogenesis 2017, 20, 269–285. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| TKIs | Sunitinib [97] | Pazopanib COMPARZ [98] | Tivozanib TIVO-1 [99] | Cabozantinib CABOSUN [101,102] |
|---|---|---|---|---|
| Treatment line | 1st | 1st | 1st | 1st |
| Study design | Phase III | Phase III Non inferiority | Phase III | Phase II |
| N | 750 | 1110 | 517 | 157 |
| Comparator arm | IFN α | Sunitinib | Sorafenib | Sunitinib |
| ORR (%) | 31 vs. 6 | 31 vs. 25 | 33.1 vs. 23.4 | 20 vs. 9 |
| PFS (months) | 11 vs. 5 (HR 0.42; p < 0.001) | 8.4 vs. 9.5 (HR 1.05) | 11.9 vs. 9.1 (HR 0.79; p = 0.042) | 8.6 vs. 5.3 (HR 0.66; p = 0.012) |
| OS (months) | 26.4 vs. 21.8 (HR 0.82) | 28.4 vs. 29.3 (HR 0.91) | 29.3 vs. 28.8 (HR 1.24; p = 0.105) | 26.6 vs. 21.2 (HR 0.80) † |
| Adverse events | Hypertension, Diarrhea, Fatigue, HFS, Leukopenia, Thrombocytopenia | Fatigue, PPE AST, ALT, Br increase | Hypertension, Dysphonia | Hypertension, Diarrhea, Anorexia, PPE, Weight loss |
| Approval regulatory authorities | 2006 (FDA) 2006 (EMA) | 2009 (FDA) 2010 (EMA) | 2017 (EMA) | 2017 (FDA) 2018 (EMA) |
| TKIs | Axitinib AXIS [102] | Cabozantinib METEOR [106,107] | Lenvatinib + Everolimus HOPE 205 [110] | Tivozanib TIVO-3 [101] | Sorafenib [111] |
|---|---|---|---|---|---|
| Treatment line | 2nd | ≥2nd | 2nd | 3rd and 4th | ≥2nd cytokine-based therapy |
| N | 723 | 375 | 153 | 350 | 903 |
| Comparator arm | Sorafenib | Everolimus | Lenvatinib vs. Everolimus | Sorafenib | Placebo |
| ORR (%) | 19 vs. 9 | 17 vs. 3 | 30 vs. 19 vs. 20 | 18 vs. 8 | 44 vs. 2 |
| PFS (months) | 6.7 vs. 4.7 (HR 0.665; p = 0.0001) | 7.4 vs. 3.8 (HR 0.58; p < 0.001) | 14.6 vs. 7.4 vs. 5.5 (HR 0.4; p = 0.0005 and HR 0.6; p = 0.048) | 5.6 vs. 3.9 (HR 0.73; p = 0.0165) | 5.5 vs. 2.8 (HR 0.44; p < 0.001) |
| OS (months) | 20.1 vs. 19.2 (HR 0.97; p = 0.37) | 21.4 vs. 16.5 (HR 0.66; p = 0.0003) | 25.5 vs. 19.1 vs. 15.4 (HR 0.51; p = 0.024 and HR 0.68; p = 0.12) † | 16.4 vs. 19.7 (HR 1.12; p = 0.4) * | NR vs. 14.7 (HR 0.72; p = 0.02) ‡ 19.3 vs. 15.9 (HR 0.77; p = 0.02) ‡‡ |
| Adverse events | Hypertension, Diarrhea, Fatigue, Anorexia, Asthenia, PPE# | Hypertension, Diarrhea, Fatigue, PPE#, anaemia, ionic disorders | Diarrhea, Hypertension, Fatigue, Anorexia, Proteinuria, Hypertrygliceridaemia, Nausea/Vomiting, Decreased weight, Hyperglycaemia, Dyspnoea | Hypertension, Fatigue, Diarrhea, Anorexia, Dysphonia | PPE, Fatigue, Hypertension, Anaemia, Dyspnea, Diarrhea |
| Approval regulatory authorities | 2012 (FDA) 2012 (EMA) | FDA 2016 EMA 2016 | FDA 2016 EMA 2016 | - | 2005 (FDA) |
| Treatment and Study | Checkmate 214 Nivolumab + Ipilimumab [10,114] | MK426 Pembrolizumab + Axitinib [115] | IMmotion 151 Atezolizumab +bevacizumab [117] | JAVELIN RENAL 101 Avelumab + Axitinib [116] | CLEAR Pembrolizumab + Lenvatinib | CheckMate 9ER Nivolumab + Cabozantinib | Tivozanib + Nivolumab |
|---|---|---|---|---|---|---|---|
| Comparator arm | Sunitinib | Sunitinib | Sunitinb | Sunitinib | Sunitinib | Sunitinib | - |
| Primary endpoint | ORR PFS OS | PFS OS | PFS (Investigator) PDL1+ OS ITT | PFS PDL1ve OS PDL1ve | PFS | PFS | Safety/security |
| Results initially presented | ESMO 2017 | ASCO GU 2019 | ASCO GU 2018 | ESMO 2018 | Recruiting | Recruiting | Recruiting |
| Median Follow Up (months) | 32.4 | 12.8 | 15 | 9.9 | NRe | NRe | NRe |
| ORR (%) | 42 | 59.3 | 43 | 55.2 | NRe | NRe | NRe |
| PFS (months) | 11.6 | 15.2 | 11.2 | 13.8 | NRe | NRe | NRe |
| mOS (months) | NR | NR | NRe | NR | NRe | NRe | NRe |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alonso-Gordoa, T.; García-Bermejo, M.L.; Grande, E.; Garrido, P.; Carrato, A.; Molina-Cerrillo, J. Targeting Tyrosine kinases in Renal Cell Carcinoma: “New Bullets against Old Guys”. Int. J. Mol. Sci. 2019, 20, 1901. https://doi.org/10.3390/ijms20081901
Alonso-Gordoa T, García-Bermejo ML, Grande E, Garrido P, Carrato A, Molina-Cerrillo J. Targeting Tyrosine kinases in Renal Cell Carcinoma: “New Bullets against Old Guys”. International Journal of Molecular Sciences. 2019; 20(8):1901. https://doi.org/10.3390/ijms20081901
Chicago/Turabian StyleAlonso-Gordoa, Teresa, María Laura García-Bermejo, Enrique Grande, Pilar Garrido, Alfredo Carrato, and Javier Molina-Cerrillo. 2019. "Targeting Tyrosine kinases in Renal Cell Carcinoma: “New Bullets against Old Guys”" International Journal of Molecular Sciences 20, no. 8: 1901. https://doi.org/10.3390/ijms20081901
APA StyleAlonso-Gordoa, T., García-Bermejo, M. L., Grande, E., Garrido, P., Carrato, A., & Molina-Cerrillo, J. (2019). Targeting Tyrosine kinases in Renal Cell Carcinoma: “New Bullets against Old Guys”. International Journal of Molecular Sciences, 20(8), 1901. https://doi.org/10.3390/ijms20081901