Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives

Abstract
1. Introduction
2. Development, Activation and Regulation of ILC2s
2.1. Development
2.2. Activation
2.3. Regulation of ILC2s
3. Immune Regulation in Gut and Liver
4. ILC2s in Intestinal Inflammation
4.1. Intestinal ILC2s Regulate Colitis
4.2. A Potential Role of ILC2-Derived AREG for Intestinal Maintenance
4.3. Intestinal ILC2s Are Heterogeneous
5. The Role of ILC2s in Liver Inflammation
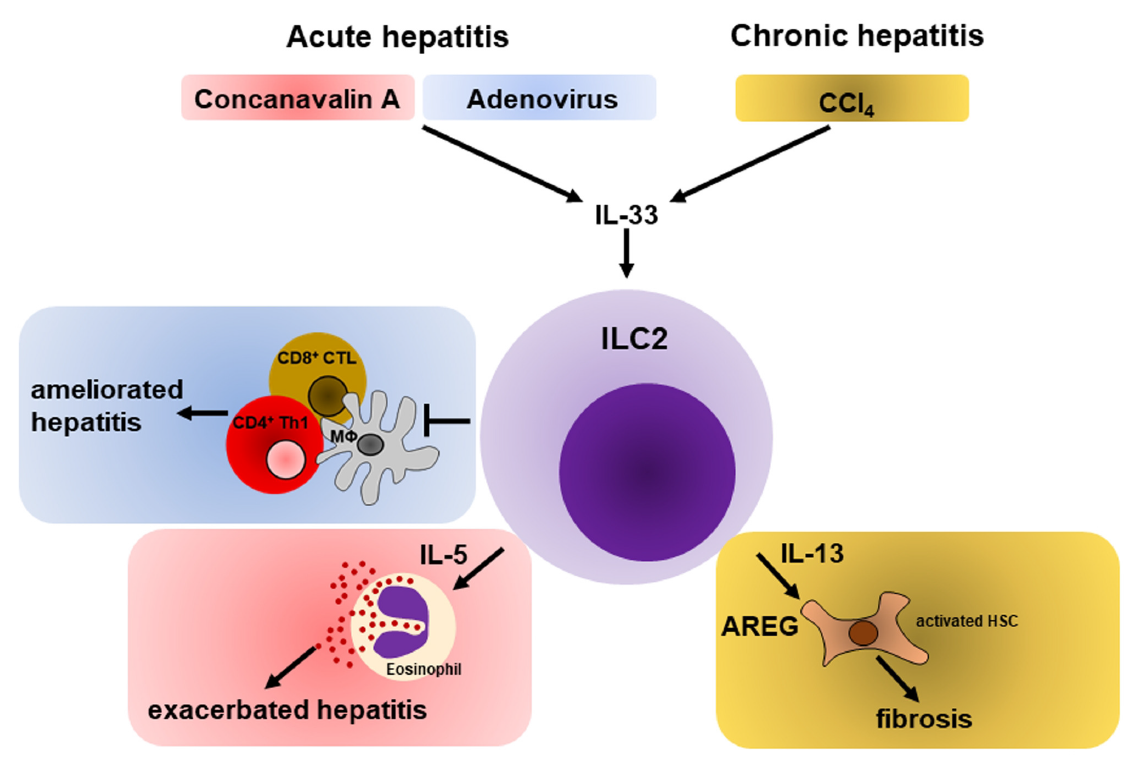
5.1. Hepatic ILC2s during Acute Liver Inflammation
5.1.1. Role of ILC2s during Immune-Mediated Hepatitis
5.1.2. ILC2s Promote Hepatic Protection during Viral Hepatitis
5.1.3. Plasticity of ILC2s
5.2. Hepatic ILC2s in Chronic Liver Inflammation
5.3. Liver Cancer
6. Potential Migration of ILC2s
7. Summary
8. Perspectives
Acknowledgments
Conflicts of Interest
References
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Artis, D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015, 21, 698–708. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef]
- Weizman, O.-E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. ILC1 confer early host protection at initial sites of viral infection. Cell 2017, 171, 795–808.e12. [Google Scholar] [CrossRef]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; Velde, A.A.T.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, E.S.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Arae, K.; Unno, H.; Miyauchi, K.; Toyama, S.; Nambu, A.; Oboki, K.; Ohno, T.; Motomura, K.; Matsuda, A.; et al. An Interleukin-33-Mast Cell-Interleukin-2 Axis Suppresses Papain-Induced Allergic Inflammation by Promoting Regulatory T Cell Numbers. Immunity 2015, 43, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.A.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef]
- Van Maele, L.; Carnoy, C.; Cayet, D.; Ivanov, S.; Porte, R.; Deruy, E.; Chabalgoity, J.A.; Renauld, J.-C.; Eberl, G.; Benecke, A.G.; et al. Activation of Type 3 Innate Lymphoid Cells and Interleukin 22 Secretion in the Lungs During Streptococcus pneumoniae Infection. J. Infect. Dis. 2014, 210, 493–503. [Google Scholar] [CrossRef]
- Satoh-Takayama, N.; Vosshenrich, C.A.J.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial Flora Drives Interleukin 22 Production in Intestinal NKp46+ Cells that Provide Innate Mucosal Immune Defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Gladiator, A.; Wangler, N.; Trautwein-Weidner, K.; LeibundGut-Landmann, S. Cutting Edge: IL-17-Secreting Innate Lymphoid Cells Are Essential for Host Defense against Fungal Infection. J. Immunol. 2013, 190, 521–525. [Google Scholar] [CrossRef]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+CD3-LTβ+cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef]
- Aparicio-Domingo, P.; Romera-Hernandez, M.; Karrich, J.J.; Cornelissen, F.; Papazian, N.; Lindenbergh-Kortleve, D.J.; Butler, J.A.; Boon, L.; Coles, M.C.; Samsom, J.N.; et al. Type 3 innate lymphoid cells maintain intestinal epithelial stem cells after tissue damage. J. Exp. Med. 2015, 212, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Razumilava, N.; Gores, G.J.; Walters, S.; Mizuochi, T.; Mourya, R.; Bessho, K.; Wang, Y.-H.; Glaser, S.S.; Shivakumar, P.; et al. Biliary repair and carcinogenesis are mediated by IL-33–dependent cholangiocyte proliferation. J. Clin. Investig. 2014, 124, 3241–3251. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Artis, D. Innate lymphoid cells promote lung tissue homeostasis following acute influenza virus infection. Nat. Immunol. 2012, 12, 1045–1054. [Google Scholar] [CrossRef]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin—EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef]
- Zaiss, D.M.; Gause, W.C.; Osborne, L.C.; Artis, D. Emerging functions of amphiregulin in orchestrating immunity, inflammation and tissue repair. Immunity 2015, 42, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Kim, H.Y.; Albacker, A.L.; Baumgarth, N.; McKenzie, A.N.J.; Smith, E.D.; DeKruyff, R.H.; Umetsu, D.T. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat. Immunol. 2011, 12, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- Halim, T.Y.; Krauß, R.H.; Sun, A.C.; Takei, F. Lung Natural Helper Cells Are a Critical Source of Th2 Cell-Type Cytokines in Protease Allergen-Induced Airway Inflammation. Immunity 2012, 36, 451–463. [Google Scholar] [CrossRef]
- Kim, B.S.; Siracusa, M.C.; Saenz, S.A.; Noti, M.; Monticelli, L.A.; Sonnenberg, G.F.; Hepworth, M.R.; Van Voorhees, A.S.; Comeau, M.R.; Artis, D. TSLP elicits IL-33–independent innate lymphoid cell responses to promote skin inflammation. Sci. Transl. Med. 2013, 5, 170ra16. [Google Scholar] [CrossRef]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Maini, M.K.; Knolle, P. A Living in the liver: Hepatic infections. Nat. Rev. Immunol. 2012, 12, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Sebode, M.; Hartl, J.; Vergani, D.; The International Autoimmune Hepatitis Group (IAIHG); Lohse, A.W. Autoimmune hepatitis: From current knowledge and clinical practice to future research agenda. Liver Int. 2017, 38, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Lohse, A.W.; Vergani, D. Autoimmune hepatitis—Update 2015. J. Hepatol. 2015, 62, S100–S111. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Yang, Z.; Wei, X.; Yang, S.; Tian, Z. Type 1 innate lymphoid cells contribute to the pathogenesis of chronic hepatitis B. Innate Immun. 2015, 21, 665–673. [Google Scholar]
- Matsumoto, A.; Kanai, T.; Mikami, Y.; Chu, P.S.; Nakamoto, N.; Ebinuma, H.; Saito, H.; Sato, T.; Yagita, H.; Hibi, T. IL-22-Producing RORγt-Dependent Innate Lymphoid Cells Play a Novel Protective Role in Murine Acute Hepatitis. PLoS ONE 2013, 8, e62853. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Jie, Z.; Hou, L.; Aguilar-Valenzuela, R.; Vu, D.; Soong, L.; Sun, J. IL-33 induces nuocytes and modulates liver injury in viral hepatitis. J. Immunol. 2013, 190, 5666–5675. [Google Scholar] [CrossRef]
- Jie, Z.; Liang, Y.; Hou, L.; Dong, C.; Iwakura, Y.; Soong, L.; Cong, Y.; Sun, J. Intrahepatic Innate Lymphoid Cells Secrete IL-17A and IL-17F That Are Crucial for T Cell Priming in Viral Infection. J. Immunol. 2014, 192, 3289–3300. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, C. The Role of Innate Lymphoid Cells in Immune-Mediated Liver Diseases. Front. Immunol. 2017, 8, 695. [Google Scholar] [CrossRef]
- Klose, C.S.; Flach, M.; Möhle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of Type 1 ILCs from a Common Progenitor to All Helper-like Innate Lymphoid Cell Lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef]
- Verykokakis, M.; Krishnamoorthy, V.; Iavarone, A.; Lasorella, A.; Sigvardsson, M.; Kee, B.L. Essential functions for ID proteins at multiple checkpoints in invariant NKT cell development. J. Immunol. 2013, 191, 5973–5983. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, M.G.; McDonald, B.D.; Verhoef, P.A.; Bendelac, A. A committed precursor to innate lymphoid cells. Nature 2014, 508, 397–401. [Google Scholar] [CrossRef]
- Hoyler, T.; Klose, C.S.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The Transcription Factor GATA-3 Controls Cell Fate and Maintenance of Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef]
- Yang, Q.; Monticelli, L.A.; Saenz, S.A.; Chi, A.W.S.; Sonnenberg, G.F.; Tang, J.; De Obaldia, M.E.; Bailis, W.; Bryson, J.L.; Toscano, K.; et al. T Cell Factor 1 Is Required for Group 2 Innate Lymphoid Cell Generation. Immunity 2013, 38, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.; Bernink, J.; Golebski, K.; Karrich, J.J.; Peters, C.P.; Blom, B.; Velde, A.A.T.; Fokkens, W.J.; Van Drunen, C.M.; Spits, H. The Transcription Factor GATA3 Is Essential for the Function of Human Type 2 Innate Lymphoid Cells. Immunity 2012, 37, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Serafini, N.; Klein Wolterink, R.G.J.; Satoh-Takayama, N.; Xu, W.; Vosshenrich, C.A.J.; Hendriks, R.W.; Di Santo, J.P. Gata3 drives development of RORγt + group 3 innate lymphoid cells. J. Exp. Med. 2014, 211, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Yagi, R.; Zhong, C.; Northrup, D.L.; Yu, F.; Bouladoux, N.; Spencer, S.; Hu, G.; Barron, L.; Sharma, S.; Nakayama, T.; et al. The transcription factor GATA3 is critical for the development of all IL-7Rα-expressing innate lymphoid cells. Immunity 2014, 40, 378–388. [Google Scholar] [CrossRef]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORα is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef]
- Halim, T.Y.; MacLaren, A.; Romanish, M.T.; Gold, M.J.; McNagny, K.M.; Takei, F. Retinoic-Acid-Receptor-Related Orphan Nuclear Receptor Alpha Is Required for Natural Helper Cell Development and Allergic Inflammation. Immunity 2012, 37, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.I.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of TH2 cytokines by adipose tissue-associated c-Kit+Sca-1+lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.-A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1 + group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.-C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33–driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F.; E Paul, W. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential ‘inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2014, 16, 161–169. [Google Scholar] [CrossRef]
- Saenz, S.A.; Noti, M.; Artis, D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol. 2010, 31, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Price, A.E.; Liang, H.-E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13–expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [PubMed]
- Mirchandani, A.S.; Besnard, A.-G.; Yip, E.; Scott, C.; Bain, C.C.; Cerovic, V.; Salmond, R.J.; Liew, F.Y. Type 2 Innate Lymphoid Cells Drive CD4+ Th2 Cell Responses. J. Immunol. 2014, 192, 2442–2448. [Google Scholar] [CrossRef] [PubMed]
- Takai, T. TSLP Expression: Cellular Sources, Triggers, and Regulatory Mechanisms. Allergol. Int. 2012, 61, 3–17. [Google Scholar] [CrossRef]
- Howitt, M.R.; Lavoie, S.; Michaud, M.; Blum, A.M.; Tran, S.V.; Weinstock, J.V.; Gallini, C.A.; Redding, K.; Margolskee, R.F.; Osborne, L.C.; et al. Tuft cells, taste-chemosensory cells, orchestrate parasite type 2 immunity in the gut. Science 2016, 351, 1329–1333. [Google Scholar] [CrossRef]
- Gerbe, F.; Sidot, E.; Smyth, D.J.; Ohmoto, M.; Matsumoto, I.; Dardalhon, V.; Cesses, P.; Garnier, L.; Pouzolles, M.; Brulin, B.; et al. Intestinal epithelial tuft cells initiate type 2 mucosal immunity to helminth parasites. Nature 2016, 529, 226–230. [Google Scholar] [CrossRef]
- Mchedlidze, T.; Waldner, M.; Zopf, S.; Walker, J.; Rankin, A.L.; Schuchmann, M.; Voehringer, D.; McKenzie, A.N.J.; Neurath, M.F.; Pflanz, S.; et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity 2013, 39, 357–371. [Google Scholar] [CrossRef]
- Neumann, K.; Karimi, K.; Meiners, J.; Voetlause, R.; Steinmann, S.; Dammermann, W.; Lüth, S.; Asghari, F.; Wegscheid, C.; Horst, A.K.; et al. A Proinflammatory Role of Type 2 Innate Lymphoid Cells in Murine Immune-Mediated Hepatitis. J. Immunol. 2017, 198, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Suzuki, N.; Hirata, Y.; Hikiba, Y.; Hayakawa, Y.; Kinoshita, H.; Ihara, S.; Uchino, K.; Nishikawa, Y.; Ijichi, H.; et al. Biliary epithelial injury-induced regenerative response by IL-33 promotes cholangiocarcinogenesis from peribiliary glands. Proc. Natl. Acad. Sci. USA 2017, 114, E3806–E3815. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, H.C.; McDowell, P.; Lutz, P.; Wawman, R.E.; Roberts, S.; Bagnall, C.; Birtwistle, J.; Adams, D.H.; Oo, Y.H. Human intrahepatic ILC2 are IL-13positiveamphiregulinpositiveand their frequency correlates with model of end stage liver disease score. PLoS ONE 2017, 12, e0188649. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; Berglin, L.; Kekäläinen, E.; Carlsson, A.; Svedin, E.; Michaëlsson, J.; Nagasawa, M.; Erjefält, J.S.; Mori, M.; Flodström-Tullberg, M.; et al. Composition and functionality of the intrahepatic innate lymphoid cell-compartment in human nonfibrotic and fibrotic livers. Eur. J. Immunol. 2017, 47, 1280–1294. [Google Scholar] [CrossRef] [PubMed]
- Vannella, K.M.; Ramalingam, T.R.; Borthwick, L.A.; Barron, L.; Hart, K.M.; Thompson, R.W.; Kindrachuk, K.N.; Cheever, A.W.; White, S.; Budelsky, A.L.; et al. Combinatorial targeting of TSLP, IL-25, and IL-33 in type 2 cytokine-driven inflammation and fibrosis. Sci. Transl. Med. 2016, 8, 337. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.M.; Trifari, S.; Crellin, N.K.; Peters, C.P.; Van Drunen, C.M.; Piet, B.; Fokkens, W.J.; Cupedo, T.; Spits, H. Human IL-25- and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat. Immunol. 2011, 12, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Rauber, S.; Luber, M.; Weber, S.; Maul, L.; Soare, A.; Wohlfahrt, T.; Lin, N.-Y.; Dietel, K.; Bozec, A.; Herrmann, M.; et al. Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat. Med. 2017, 23, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.-E.; Morrison, P.J.; Wilhelm, C.; Wilson, M.; Ahlfors, H.; Renauld, J.-C.; Panzer, U.; Helmby, H.; Stockinger, B. IL-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J. Exp. Med. 2013, 210, 2951–2965. [Google Scholar] [CrossRef]
- Shoyab, M.; McDonald, V.L.; Bradley, J.G.; Todaro, G.J. Amphiregulin: A bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc. Natl. Acad. Sci. USA 1988, 85, 6528–6532. [Google Scholar] [CrossRef] [PubMed]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.-L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. (BBA) Rev. 2011, 1816, 119–131. [Google Scholar] [CrossRef]
- Berasain, C.; García-Trevijano, E.R.; Castillo, J.; Erroba, E.; Lee, D.C.; Prieto, J.; Ávila, M.A. Amphiregulin: An early trigger of liver regeneration in mice. Gastroenterology 2005, 128, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, D.M.; Van Loosdregt, J.; Gorlani, A.; Bekker, C.P.; Grone, A.; Sibilia, M.; Henegouwen, P.M.P.V.B.E.; Roovers, R.C.; Coffer, P.J.; Sijts, A.J.A.M. Amphiregulin enhances regulatory T cell suppressive function via the epidermal growth factor receptor. Immunity 2013, 38, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Paclik, D.; Stehle, C.; Lahmann, A.; Hutloff, A.; Romagnani, C. ICOS regulates the pool of group 2 innate lymphoid cells under homeostatic and inflammatory conditions in mice. Eur. J. Immunol. 2015, 45, 2766–2772. [Google Scholar] [CrossRef]
- Maazi, H.; Patel, N.; Sankaranarayanan, I.; Suzuki, Y.; Rigas, D.; Soroosh, P.; Freeman, G.J.; Sharpe, A.H.; Akbari, O. ICOS: ICOS-Ligand interaction is required for type 2 innate lymphoid cell function, homeostasis and induction of airway hyperreactivity. Immunity 2015, 42, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Rigas, D.; Lewis, G.; Aron, J.L.; Wang, B.; Banie, H.; Sankaranarayanan, I.; Galle-Treger, L.; Maazi, H.; Lo, R.; Freeman, G.J.; et al. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator–inducible T-cell costimulator ligand interaction. J. Allergy Clin. Immunol. 2017, 139, 1468–1477.e2. [Google Scholar] [CrossRef] [PubMed]
- Sui, P.; Wiesner, D.L.; Xu, J.; Zhang, Y.; Lee, J.; Dyken, S. Pulmonary neuroendocrine cells amplify allergic asthma responses. Science 2018, 360, 8546. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, V.; Chesné, J.; Ribeiro, H.; García-Cassani, B.; Carvalho, T.; Bouchery, T.; Shah, K.; Barbosa-Morais, N.L.; Harris, N.; Veiga-Fernandes, H. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 2017, 549, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Abdulnour, R.-E.E.; Nyman, J.; Dionne, D.; Hofree, M.; Cuoco, M.S.; Rodman, C.; Farouq, D.; et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 2017, 549, 351–356. [Google Scholar]
- Klose, C.S.N.; Mahlakõiv, T.; Moeller, J.B.; Rankin, L.C.; Flamar, A.-L.; Kabata, H.; Monticelli, L.A.; Moriyama, S.; Putzel, G.G.; Rakhilin, N.; et al. The neuropeptide Neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 2017, 549, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.C.; Van Dyken, S.J.; Von Moltke, J.; Cheng, L.E.; Mohapatra, A.; Molofsky, A.B.; Thornton, E.E.; Krummel, M.F.; Chawla, A.; Liang, H.-E.; et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013, 502, 245–248. [Google Scholar] [CrossRef] [PubMed]
- Spencer, S.P.; Wilhelm, C.; Yang, Q.; Hall, J.A.; Bouladoux, N.; Boyd, A.; Nutman, T.B.; Urban, J.F.; Wang, J.; Ramalingam, T.R.; et al. Adaptation of innate lymphoid cells to a micronutrient deficiency promotes type 2 barrier immunity. Science 2014, 343, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, B.; Patil, S.U.; Shreffler, W.G. Vitamins A and D have antagonistic effects on expression of effector cytokines and gut-homing integrin in human innate lymphoid cells. Clin. Exp. 2015, 45, 1214–1225. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Kubo, T.; Rückert, B.; Ravindran, A.; Soyka, M.B.; Rinaldi, A.O.; Sugita, K.; Wawrzyniak, M.; Wawrzyniak, P.; Motomura, K.; et al. Induction of human regulatory innate lymphoid cells from group 2 innate lymphoid cells by retinoic acid. J. Clin. Immunol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Laffont, S.; Blanquart, E.; Savignac, M.; Cénac, C.; Laverny, G.; Metzger, D.; Girard, J.-P.; Belz, G.T.; Pelletier, L.; Seillet, C.; et al. Androgen signaling negatively controls group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.E.V.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; He, Z.; Chen, W.; Holzman, I.R.; Lin, J. Effects of Butyrate on Intestinal Barrier Function in a Caco-2 Cell Monolayer Model of Intestinal Barrier. Pediatr. Res. 2007, 61, 37–41. [Google Scholar] [CrossRef]
- Peng, L.; Li, Z.-R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate Enhances the Intestinal Barrier by Facilitating Tight Junction Assembly via Activation of AMP-Activated Protein Kinase in Caco-2 Cell Monolayers12. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef]
- Weiner, H.L.; da Cunha, A.P.; Quintana, F.; Wu, H. Oral tolerance. Immunol. Rev. 2011, 241, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Izcue, A.; Coombes, J.L.; Powrie, F. Regulatory T cells suppress systemic and mucosal immune activation to control intestinal inflammation. Immunol. Rev. 2006, 212, 256–271. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef]
- Bertolino, P.; Trescol-Biémont, M.C.; Rabourdin-Combe, C. Hepatocytes induce functional activation of naive CD8+T lymphocytes but fail to promote survival. Eur. J. Immunol. 1998, 28, 221–236. [Google Scholar] [CrossRef]
- Herkel, J.; Jagemann, B.; Wiegard, C.; Garcia Lazaro, J.F.; Lueth, S.; Kanzler, S.; Blessing, M.; Schmitt, E.; Lohse, A.W. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocytes. Hepatology 2003, 37, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, S.; Claass, B.; Erhardt, A.; Karimi, K.; Tiegs, G. Hepatocytes induce Foxp3 + regulatory T cells by Notch signaling. J. Leukoc. Biol. 2014, 96, 571–577. [Google Scholar] [CrossRef]
- Ebrahimkhani, M.R.; Mohar, I.; Crispe, I.N. Cross-presentation of antigen by diverse subsets of murine liver cells. Hepatology 2011, 54, 1379–1387. [Google Scholar] [CrossRef] [PubMed]
- Knolle, P.A.; Gerken, G. Local control of the immune response in the liver. Immunol. Rev. 2000, 174, 21–34. [Google Scholar] [CrossRef]
- Doherty, D.G. Immunity, tolerance and autoimmunity in the liver: A comprehensive review. J. Autoimmun. 2016, 66, 60–75. [Google Scholar] [CrossRef]
- Milosevic, I.; Vujovic, A.; Barac, A.; Djelic, M.; Korac, M.; Radovanovic Spurnic, A.; Gmizic, I.; Stevanovic, O.; Djordjevic, V.; Lekic, N.; et al. Gut-Liver Axis, Gut Microbiota, and Its Modulation in the Management of Liver Diseases: A Review of the Literature. Int. J. Mol. Sci. 2019, 20, 395. [Google Scholar] [CrossRef] [PubMed]
- Fousekis, F.S.; Theopistos, V.I.; Mitselos, I.V.; Skamnelos, A.; Kavvadias, A.; Katsanos, K.H.; Christodoulou, D.K. Specific Features of Patients with Inflammatory Bowel Disease and Primary Sclerosing Cholangitis. J. Clin. Med. Res. 2019, 11, 81–88. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kramer, B.; Goeser, F.; Lutz, P.; Glässner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef]
- Johansson, M.E.V.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjovall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Bergstrom, K.S.B.; Kissoon-Singh, V.; Gibson, D.L.; Ma, C.; Montero, M.; Sham, H.P.; Ryz, N.; Huang, T.; Velcich, A.; Finlay, B.B.; et al. Muc2 Protects against Lethal Infectious Colitis by Disassociating Pathogenic and Commensal Bacteria from the Colonic Mucosa. PLoS Pathog. 2010, 6, e1000902. [Google Scholar] [CrossRef] [PubMed]
- Camelo, A.; Barlow, J.L.; Drynan, L.F.; Neill, D.R.; Ballantyne, S.J.; Wong, S.H.; Pannell, R.; Gao, W.; Wrigley, K.; Sprenkle, J.; et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J. Gastroenterol. 2012, 47, 1198–1211. [Google Scholar] [CrossRef] [PubMed]
- Mankertz, J.; Schulzke, J.-D. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mao, K.; Chen, X.; Sun, M.A.; Kawabe, T.; Li, W.; Usher, N.; Zhu, J.; Urban, J.F.; Paul, W.E.; et al. S1P-dependent interorgan trafficking of group 2 innate lymphoid cells supports host defense. Science 2018, 359, 114–119. [Google Scholar] [CrossRef]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Louis, H.; Le Moine, A.; Flamand, V.; Nagy, N.; Quertinmont, E.; Paulart, F.; Abramowicz, D.; Le Moine, O.; Goldman, M.; Devière, J. Critical role of interleukin 5 and eosinophils in concanavalin A–induced hepatitis in mice. Gastroenterology 2002, 122, 2001–2010. [Google Scholar] [CrossRef] [PubMed]
- Volarevic, V.; Mitrovic, M.; Milovanovic, M.; Zelen, I.; Nikolic, I.; Mitrovic, S.; Pejnovic, N.; Arsenijevic, N.; Lukic, M.L. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J. Hepatol. 2012, 56, 26–33. [Google Scholar] [CrossRef]
- Besnard, A.-G.; Guabiraba, R.; Niedbala, W.; Palomo, J.; Reverchon, F.; Shaw, T.N.; Couper, K.N.; Ryffel, B.; Liew, F.Y. IL-33-Mediated Protection against Experimental Cerebral Malaria Is Linked to Induction of Type 2 Innate Lymphoid Cells, M2 Macrophages and Regulatory T Cells. PLoS Pathog. 2015, 11, e1004607. [Google Scholar] [CrossRef]
- Martinez-Gonzalez, I.; Mathä, L.; Steer, C.A.; Ghaedi, M.; Poon, G.F.; Takei, F. Allergen-Experienced Group 2 Innate Lymphoid Cells Acquire Memory-like Properties and Enhance Allergic Lung Inflammation. Immunity 2016, 45, 198–208. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Perugorria, M.J.; Latasa, M.U.; Nicou, A.; Cartagena-Lirola, H.; Castillo, J.; Goñi, S.; Vespasiani-Gentilucci, U.; Zagami, M.G.; Lotersztajn, S.; Prieto, J.; et al. The epidermal growth factor receptor ligand amphiregulin participates in the development of mouse liver fibrosis. Hepatology 2008, 48, 1251–1261. [Google Scholar] [CrossRef]
- McKee, C.; Sigala, B.; Soeda, J.; Mouralidarane, A.; Morgan, M.; Mazzoccoli, G.; Rappa, F.; Cappello, F.; Cabibi, D.; Pazienza, V.; et al. Amphiregulin activates human hepatic stellate cells and is upregulated in non alcoholic steatohepatitis. Sci. Rep. 2015, 5, 8812. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 18, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Hikiba, Y.; Hirata, Y.; Font-Burgada, J.; Sakamoto, K.; Hayakawa, Y.; Taniguchi, K.; Umemura, A.; Kinoshita, H.; Sakitani, K.; et al. Loss of liver E-cadherin induces sclerosing cholangitis and promotes carcinogenesis. Proc. Natl. Acad. Sci. USA 2014, 111, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Berasain, C.; Castillo, J.; Perugorria, M.; Prieto, J.; Avila, M. Amphiregulin: A new growth factor in hepatocarcinogenesis. Cancer Lett. 2007, 254, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Erroba, E.; Perugorria, M.J.; Santamaria, M.; Lee, D.C.; Prieto, J.; Ávila, M.A.; Berasain, C. Amphiregulin Contributes to the Transformed Phenotype of Human Hepatocellular Carcinoma Cells. Cancer Res. 2006, 66, 6129–6138. [Google Scholar] [CrossRef] [PubMed]
- Dai, K.; Huang, L.; Chen, J.; Yang, L.; Gong, Z. Amphiregulin promotes the immunosuppressive activity of intrahepatic CD4 + regulatory T cells to impair CD8 + T cell immunity against hepatitis B virus infection. Immunology 2014, 144, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and non-lymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef]
- Moro, K.; Kabata, H.; Tanabe, M.; Koga, S.; Takeno, N.; Mochizuki, M.; Fukunaga, K.; Asano, K.; Betsuyaku, T.; Koyasu, S. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat. Immunol. 2016, 17, 76–86. [Google Scholar] [CrossRef]
- Erhardt, A.; Biburger, M.; Papadopoulos, T.; Tiegs, G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 2007, 45, 475–485. [Google Scholar] [CrossRef]
- Watanabe, S.; Ohnuki, K.; Hara, Y.; Ishida, Y.; Ikarashi, Y.; Ogawa, S.; Kishimoto, H.; Tanabe, K.; Abe, R. Suppression of Con A-induced hepatitis induction in ICOS-deficient mice. Immunol. Lett. 2010, 128, 51–58. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Function | Expressed on | Demonstrated on | References |
|---|---|---|---|---|
| ICOS | Maturation and maintenance | ILC2s | murine and human ILC2s | [63,64] |
| ICOS-L | Negative regulator of ILC2 activity by ligation of ICOS+ iTregs | ILC2s | murine ILC2s | [65] |
| KLRG1 | Regulates phosphorylation of STAT5 to enhance proliferation and effector cytokine production | ILC2s | murine and human ILC2s | [41,42] |
| E-cadherin | Impairs ILC2 function by ligation of KLRG1 | Epithelial cells | human ILC2s | [42] |
| Programmed death (PD)-1 | Negative regulator of KLRG1 signaling, impairs phosphorylation of STAT5 | ILC2s | murine and human ILC2s | [41] |
| Neuropeptides | ||||
| Calcitonin gene-related peptide (CGRP) | Amplifies effector of lung ILC2s | murine ILC2s | [66] | |
| Neuromedin U (NMU) | Induces ILC2 effector function | murine ILC2s | [67,68,69] | |
| Vasoactive intestinal peptide (VIP) | regulates IL-5 production in ILC2s, confers tissue homeostasis by eosinophil-recruitment | murine ILC2s | [70] | |
| Nutrients | ||||
| Retinoic acid (RA) | Downregulates expression of IL-7Rα and thus, limits cellular maintenance of murine ILC2s. | murine ILC2s | [71] | |
| Enhances expression of ILC2-derived effector cytokines on human ILC2s. | human ILC2s | [72] | ||
| converts ILC2s into a regulatory phenotype (ILC2reg) | human ILC2s | [73] | ||
| Hormones | ||||
| Androgens | Negative regulator of ILC2 effector function | murine ILC2s | [74] | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ochel, A.; Tiegs, G.; Neumann, K. Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives. Int. J. Mol. Sci. 2019, 20, 1896. https://doi.org/10.3390/ijms20081896
Ochel A, Tiegs G, Neumann K. Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives. International Journal of Molecular Sciences. 2019; 20(8):1896. https://doi.org/10.3390/ijms20081896
Chicago/Turabian StyleOchel, Aaron, Gisa Tiegs, and Katrin Neumann. 2019. "Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives" International Journal of Molecular Sciences 20, no. 8: 1896. https://doi.org/10.3390/ijms20081896
APA StyleOchel, A., Tiegs, G., & Neumann, K. (2019). Type 2 Innate Lymphoid Cells in Liver and Gut: From Current Knowledge to Future Perspectives. International Journal of Molecular Sciences, 20(8), 1896. https://doi.org/10.3390/ijms20081896