SUMOylation in Glioblastoma: A Novel Therapeutic Target

 ,
,  , and
, and 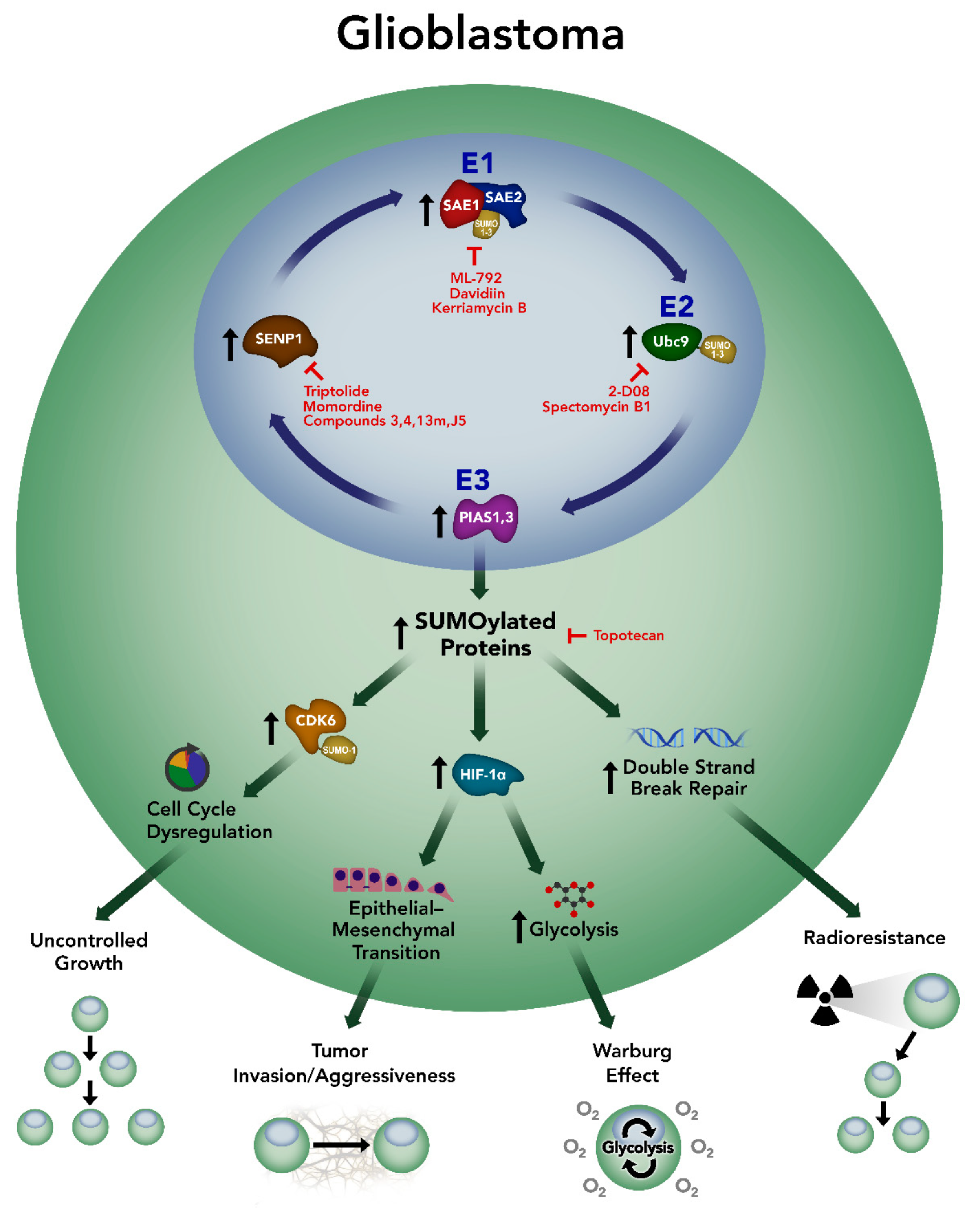{kind=link}
Abstract
1. Introduction
2. SUMOylation
3. SUMOylation and Cancer
4. SUMOylation in Glioblastoma
5. Targeting SUMOylation in Glioblastoma
6. Conclusions
Funding
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Polivka, J., Jr.; Polivka, J.; Holubec, L.; Kubikova, T.; Priban, V.; Hes, O.; Pivovarcikova, K.; Treskova, I. Advances in Experimental targeted therapy and immunotherapy for patients with glioblastoma multiforme. Anticancer Res. 2017, 37, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, I.A.; Vertegaal, A.C. A comprehensive compilation of SUMO proteomics. Nat. Rev. Mol. Cell Biol. 2016, 17, 581–595. [Google Scholar] [CrossRef] [PubMed]
- Eifler, K.; Vertegaal, A.C.O. SUMOylation-mediated regulation of cell cycle progression and cancer. Trends Biochem. Sci. 2015, 40, 779–793. [Google Scholar] [CrossRef]
- Bergink, S.; Jentsch, S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009, 458, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Sarangi, P.; Zhao, X. SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci. 2015, 40, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.; Arndt, A.; Metzger, S.; Mahajan, R.; Melchior, F.; Jaenicke, R.; Becker, J. Structure determination of the small ubiquitin-related modifier SUMO-1. J. Mol. Biol. 1998, 280, 275–286. [Google Scholar] [CrossRef]
- Shen, Z.; Pardington-Purtymun, P.E.; Comeaux, J.C.; Moyzis, R.K.; Chen, D.J. UBL1, a human ubiquitin-like protein associating with human RAD51/RAD52 proteins. Genomics 1996, 36, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Boddy, M.N.; Howe, K.; Etkin, L.D.; Solomon, E.; Freemont, P.S. PIC 1, a novel ubiquitin-like protein which interacts with the PML component of a multiprotein complex that is disrupted in acute promyelocytic leukaemia. Oncogene 1996, 13, 971–982. [Google Scholar] [PubMed]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Lapenta, V.; Chiurazzi, P.; van der Spek, P.; Pizzuti, A.; Hanaoka, F.; Brahe, C. SMT3A, a human homologue of the S. cerevisiae SMT3 gene, maps to chromosome 21qter and defines a novel gene family. Genomics 1997, 40, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Nguyen, H.P.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T. Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 3117–3120. [Google Scholar] [CrossRef] [PubMed]
- Kamitani, T.; Kito, K.; Nguyen, H.P.; Fukuda-Kamitani, T.; Yeh, E.T. Characterization of a second member of the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 1998, 273, 11349–11353. [Google Scholar] [CrossRef] [PubMed]
- Bohren, K.M.; Nadkarni, V.; Song, J.H.; Gabbay, K.H.; Owerbach, D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 2004, 279, 27233–27238. [Google Scholar] [CrossRef]
- Johnson, E.S. Protein modification by SUMO. Annu. Rev. Biochem. 2004, 73, 355–382. [Google Scholar] [CrossRef]
- Wang, L.; Wansleeben, C.; Zhao, S.; Miao, P.; Paschen, W.; Yang, W. SUMO2 is essential while SUMO3 is dispensable for mouse embryonic development. EMBO Rep. 2014, 15, 878–885. [Google Scholar] [CrossRef]
- Evdokimov, E.; Sharma, P.; Lockett, S.J.; Lualdi, M.; Kuehn, M.R. Loss of SUMO1 in mice affects RanGAP1 localization and formation of PML nuclear bodies, but is not lethal as it can be compensated by SUMO2 or SUMO3. J. Cell Sci. 2008, 121, 4106–4113. [Google Scholar] [CrossRef]
- Zhang, F.P.; Mikkonen, L.; Toppari, J.; Palvimo, J.J.; Thesleff, I.; Janne, O.A. Sumo-1 function is dispensable in normal mouse development. Mol. Cell. Biol. 2008, 28, 5381–5390. [Google Scholar] [CrossRef]
- Mikkonen, L.; Hirvonen, J.; Janne, O.A. SUMO-1 regulates body weight and adipogenesis via PPARgamma in male and female mice. Endocrinology 2013, 154, 698–708. [Google Scholar] [CrossRef]
- Venteclef, N.; Jakobsson, T.; Ehrlund, A.; Damdimopoulos, A.; Mikkonen, L.; Ellis, E.; Nilsson, L.M.; Parini, P.; Janne, O.A.; Gustafsson, J.A.; et al. GPS2-dependent corepressor/SUMO pathways govern anti-inflammatory actions of LRH-1 and LXRbeta in the hepatic acute phase response. Genes Dev. 2010, 24, 381–395. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Yang, W.; Ye, D.G.; Shen, Y.; Pluchino, S.; Lee, Y.J.; Hallenbeck, J.M.; Paschen, W. SUMOylation in brain ischemia: Patterns, targets, and translational implications. J. Cereb. Blood Flow Metab. 2018, 38, 5–16. [Google Scholar] [CrossRef]
- Owerbach, D.; McKay, E.M.; Yeh, E.T.; Gabbay, K.H.; Bohren, K.M. A proline-90 residue unique to SUMO-4 prevents maturation and sumoylation. Biochem. Biophys. Res. Commun. 2005, 337, 517–520. [Google Scholar] [CrossRef]
- Wei, W.; Yang, P.; Pang, J.; Zhang, S.; Wang, Y.; Wang, M.H.; Dong, Z.; She, J.X.; Wang, C.Y. A stress-dependent SUMO4 sumoylation of its substrate proteins. Biochem. Biophys. Res. Commun. 2008, 375, 454–459. [Google Scholar] [CrossRef]
- Baczyk, D.; Audette, M.C.; Drewlo, S.; Levytska, K.; Kingdom, J.C. SUMO-4: A novel functional candidate in the human placental protein SUMOylation machinery. PLoS ONE 2017, 12, e0178056. [Google Scholar] [CrossRef]
- Hickey, C.M.; Wilson, N.R.; Hochstrasser, M. Function and regulation of SUMO proteases. Nat. Rev. Mol. Cell Biol. 2012, 13, 755–766. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Ye, D.; Smith, J.A.; Lee, Y.J.; Gessler, F.A.; Yasgar, A.; Kouznetsova, J.; Jadhav, A.; Wang, Z.; Pluchino, S.; et al. Quantitative high-throughput screening identifies cytoprotective molecules that enhance SUMO conjugation via the inhibition of SUMO-specific protease (SENP)2. FASEB J. 2018, 32, 1677–1691. [Google Scholar] [CrossRef]
- Gong, L.; Li, B.; Millas, S.; Yeh, E.T. Molecular cloning and characterization of human AOS1 and UBA2, components of the sentrin-activating enzyme complex. FEBS Lett. 1999, 448, 185–189. [Google Scholar] [CrossRef]
- Olsen, S.K.; Capili, A.D.; Lu, X.; Tan, D.S.; Lima, C.D. Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 2010, 463, 906–912. [Google Scholar] [CrossRef]
- Gong, L.; Kamitani, T.; Fujise, K.; Caskey, L.S.; Yeh, E.T. Preferential interaction of sentrin with a ubiquitin-conjugating enzyme, Ubc9. J. Biol. Chem. 1997, 272, 28198–28201. [Google Scholar] [CrossRef]
- Bouillot-Eimer, S.; Loiseau, H.; Vital, A. Subcutaneous tumoral seeding from a glioblastoma following stereotactic biopsy: Case report and review of the literature. Clin. Neuropathol. 2005, 24, 247–251. [Google Scholar]
- Desterro, J.M.; Thomson, J.; Hay, R.T. Ubch9 conjugates SUMO but not ubiquitin. FEBS Lett. 1997, 417, 297–300. [Google Scholar] [CrossRef]
- Schwarz, S.E.; Matuschewski, K.; Liakopoulos, D.; Scheffner, M.; Jentsch, S. The ubiquitin-like proteins SMT3 and SUMO-1 are conjugated by the UBC9 E2 enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 560–564. [Google Scholar] [CrossRef]
- Lee, G.W.; Melchior, F.; Matunis, M.J.; Mahajan, R.; Tian, Q.; Anderson, P. Modification of Ran GTPase-activating protein by the small ubiquitin-related modifier SUMO-1 requires Ubc9, an E2-type ubiquitin-conjugating enzyme homologue. J. Biol. Chem. 1998, 273, 6503–6507. [Google Scholar] [CrossRef]
- Rodriguez, M.S.; Dargemont, C.; Hay, R.T. SUMO-1 conjugation in vivo requires both a consensus modification motif and nuclear targeting. J. Biol. Chem. 2001, 276, 12654–12659. [Google Scholar] [CrossRef]
- Sampson, D.A.; Wang, M.; Matunis, M.J. The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for SUMO-1 modification. J. Biol. Chem. 2001, 276, 21664–21669. [Google Scholar] [CrossRef]
- Johnson, E.S.; Gupta, A.A. An E3-like factor that promotes SUMO conjugation to the yeast septins. Cell 2001, 106, 735–744. [Google Scholar] [CrossRef]
- Rytinki, M.M.; Kaikkonen, S.; Pehkonen, P.; Jaaskelainen, T.; Palvimo, J.J. PIAS proteins: Pleiotropic interactors associated with SUMO. Cell. Mol. Life Sci. 2009, 66, 3029–3041. [Google Scholar] [CrossRef]
- Pichler, A.; Gast, A.; Seeler, J.S.; Dejean, A.; Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 2002, 108, 109–120. [Google Scholar] [CrossRef]
- Henley, J.M.; Craig, T.J.; Wilkinson, K.A. Neuronal SUMOylation: Mechanisms, physiology, and roles in neuronal dysfunction. Physiol. Rev. 2014, 94, 1249–1285. [Google Scholar] [CrossRef]
- Yeh, E.T. SUMOylation and De-SUMOylation: Wrestling with life’s processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Millas, S.; Maul, G.G.; Yeh, E.T. Differential regulation of sentrinized proteins by a novel sentrin-specific protease. J. Biol. Chem. 2000, 275, 3355–3359. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T.; Gong, L.; Kamitani, T. Ubiquitin-like proteins: New wines in new bottles. Gene 2000, 248, 1–14. [Google Scholar] [CrossRef]
- Schulz, S.; Chachami, G.; Kozaczkiewicz, L.; Winter, U.; Stankovic-Valentin, N.; Haas, P.; Hofmann, K.; Urlaub, H.; Ovaa, H.; Wittbrodt, J.; et al. Ubiquitin-specific protease-like 1 (USPL1) is a SUMO isopeptidase with essential, non-catalytic functions. EMBO Rep. 2012, 13, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.J.; Shin, H.M.; Nam, E.; Kim, W.S.; Kim, J.H.; Oh, B.H.; Yun, Y. DeSUMOylating isopeptidase: A second class of SUMO protease. EMBO Rep. 2012, 13, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Sternsdorf, T.; Jensen, K.; Will, H. Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 1997, 139, 1621–1634. [Google Scholar] [CrossRef]
- Rowley, J.D.; Golomb, H.M.; Dougherty, C. 15/17 translocation, a consistent chromosomal change in acute promyelocytic leukaemia. Lancet 1977, 1, 549–550. [Google Scholar] [CrossRef]
- De The, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Zhong, S.; Muller, S.; Ronchetti, S.; Freemont, P.S.; Dejean, A.; Pandolfi, P.P. Role of SUMO-1-modified PML in nuclear body formation. Blood 2000, 95, 2748–2752. [Google Scholar] [PubMed]
- Muller, S.; Matunis, M.J.; Dejean, A. Conjugation with the ubiquitin-related modifier SUMO-1 regulates the partitioning of PML within the nucleus. EMBO J. 1998, 17, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Tatham, M.H.; Geoffroy, M.C.; Shen, L.; Plechanovova, A.; Hattersley, N.; Jaffray, E.G.; Palvimo, J.J.; Hay, R.T. RNF4 is a poly-SUMO-specific E3 ubiquitin ligase required for arsenic-induced PML degradation. Nat. Cell Biol. 2008, 10, 538–546. [Google Scholar] [CrossRef]
- Lallemand-Breitenbach, V.; Jeanne, M.; Benhenda, S.; Nasr, R.; Lei, M.; Peres, L.; Zhou, J.; Zhu, J.; Raught, B.; de The, H. Arsenic degrades PML or PML-RARalpha through a SUMO-triggered RNF4/ubiquitin-mediated pathway. Nat. Cell Biol. 2008, 10, 547–555. [Google Scholar] [CrossRef]
- Altmannova, V.; Kolesar, P.; Krejci, L. SUMO wrestles with recombination. Biomolecules 2012, 2, 350–375. [Google Scholar] [CrossRef]
- Cremona, C.A.; Sarangi, P.; Yang, Y.; Hang, L.E.; Rahman, S.; Zhao, X. Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol. Cell 2012, 45, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Silver, H.R.; Nissley, J.A.; Reed, S.H.; Hou, Y.M.; Johnson, E.S. A role for SUMO in nucleotide excision repair. DNA Repair 2011, 10, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Maeda, D.; Seki, M.; Onoda, F.; Branzei, D.; Kawabe, Y.; Enomoto, T. Ubc9 is required for damage-tolerance and damage-induced interchromosomal homologous recombination in S. cerevisiae. DNA Repair 2004, 3, 335–341. [Google Scholar] [CrossRef]
- Morris, J.R.; Boutell, C.; Keppler, M.; Densham, R.; Weekes, D.; Alamshah, A.; Butler, L.; Galanty, Y.; Pangon, L.; Kiuchi, T.; et al. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 2009, 462, 886–890. [Google Scholar] [CrossRef]
- Luo, K.; Zhang, H.; Wang, L.; Yuan, J.; Lou, Z. Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 2012, 31, 3008–3019. [Google Scholar] [CrossRef]
- Bonne-Andrea, C.; Kahli, M.; Mechali, F.; Lemaitre, J.M.; Bossis, G.; Coux, O. SUMO2/3 modification of cyclin E contributes to the control of replication origin firing. Nat. Commun. 2013, 4, 1850. [Google Scholar] [CrossRef]
- Schimmel, J.; Eifler, K.; Sigurethsson, J.O.; Cuijpers, S.A.; Hendriks, I.A.; Verlaan-de Vries, M.; Kelstrup, C.D.; Francavilla, C.; Medema, R.H.; Olsen, J.V.; et al. Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol. Cell 2014, 53, 1053–1066. [Google Scholar] [CrossRef]
- Hendriks, I.A.; D’Souza, R.C.; Yang, B.; Verlaan-de Vries, M.; Mann, M.; Vertegaal, A.C. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat. Struct. Mol. Biol. 2014, 21, 927–936. [Google Scholar] [CrossRef]
- Bellail, A.C.; Olson, J.J.; Hao, C. SUMO1 modification stabilizes CDK6 protein and drives the cell cycle and glioblastoma progression. Nature Commun. 2014, 5, 4234. [Google Scholar] [CrossRef]
- Carter, S.; Bischof, O.; Dejean, A.; Vousden, K.H. C-terminal modifications regulate MDM2 dissociation and nuclear export of p53. Nat. Cell Biol. 2007, 9, 428–435. [Google Scholar] [CrossRef]
- Ledl, A.; Schmidt, D.; Muller, S. Viral oncoproteins E1A and E7 and cellular LxCxE proteins repress SUMO modification of the retinoblastoma tumor suppressor. Oncogene 2005, 24, 3810–3818. [Google Scholar] [CrossRef]
- Gonzalez-Prieto, R.; Cuijpers, S.A.; Kumar, R.; Hendriks, I.A.; Vertegaal, A.C. c-Myc is targeted to the proteasome for degradation in a SUMOylation-dependent manner, regulated by PIAS1, SENP7 and RNF4. Cell Cycle 2015, 14, 1859–1872. [Google Scholar] [CrossRef]
- Ding, B.; Sun, Y.; Huang, J. Overexpression of SKI oncoprotein leads to p53 degradation through regulation of MDM2 protein sumoylation. J. Biol. Chem. 2012, 287, 14621–14630. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, H.J.; Kim, B.; Kim, M.H.; Lee, J.M.; Kim, I.S.; Lee, M.H.; Choi, S.J.; Kim, K.I.; Kim, S.I.; et al. Roles of sumoylation of a reptin chromatin-remodelling complex in cancer metastasis. Nature Cell Biol. 2006, 8, 631–639. [Google Scholar] [CrossRef]
- Yang, W.; Wang, L.; Roehn, G.; Pearlstein, R.D.; Ali-Osman, F.; Pan, H.; Goldbrunner, R.; Krantz, M.; Harms, C.; Paschen, W. Small ubiquitin-like modifier 1-3 conjugation [corrected] is activated in human astrocytic brain tumors and is required for glioblastoma cell survival. Cancer Sci. 2013, 104, 70–77. [Google Scholar] [CrossRef]
- Li, H.; Niu, H.; Peng, Y.; Wang, J.; He, P. Ubc9 promotes invasion and metastasis of lung cancer cells. Oncol. Rep. 2013, 29, 1588–1594. [Google Scholar] [CrossRef]
- Shao, D.F.; Wang, X.H.; Li, Z.Y.; Xing, X.F.; Cheng, X.J.; Guo, T.; Du, H.; Hu, Y.; Dong, B.; Ding, N.; et al. High-level SAE2 promotes malignant phenotype and predicts outcome in gastric cancer. Am. J. Cancer Res. 2015, 5, 140–154. [Google Scholar]
- Zhang, H.; Kuai, X.; Ji, Z.; Li, Z.; Shi, R. Over-expression of small ubiquitin-related modifier-1 and sumoylated p53 in colon cancer. Cell Biochem. Biophys. 2013, 67, 1081–1087. [Google Scholar] [CrossRef]
- Guo, W.H.; Yuan, L.H.; Xiao, Z.H.; Liu, D.; Zhang, J.X. Overexpression of SUMO-1 in hepatocellular carcinoma: A latent target for diagnosis and therapy of hepatoma. J. Cancer Res. Clin. Oncol. 2011, 137, 533–541. [Google Scholar] [CrossRef]
- Chien, W.; Lee, K.L.; Ding, L.W.; Wuensche, P.; Kato, H.; Doan, N.B.; Poellinger, L.; Said, J.W.; Koeffler, H.P. PIAS4 is an activator of hypoxia signalling via VHL suppression during growth of pancreatic cancer cells. Br. J. Cancer 2013, 109, 1795–1804. [Google Scholar] [CrossRef][Green Version]
- Moschos, S.J.; Jukic, D.M.; Athanassiou, C.; Bhargava, R.; Dacic, S.; Wang, X.; Kuan, S.F.; Fayewicz, S.L.; Galambos, C.; Acquafondata, M.; et al. Expression analysis of Ubc9, the single small ubiquitin-like modifier (SUMO) E2 conjugating enzyme, in normal and malignant tissues. Hum. Pathol. 2010, 41, 1286–1298. [Google Scholar] [CrossRef]
- Oliveira Alves, M.G.; da Mota Delgado, A.; Balducci, I.; Carvalho, Y.R.; Cavalcante, A.S.; Almeida, J.D. Study of MDM2 and SUMO-1 expression in actinic cheilitis and lip cancer. Arch. Dermatol. Res. 2014, 306, 837–841. [Google Scholar] [CrossRef]
- Hoellein, A.; Fallahi, M.; Schoeffmann, S.; Steidle, S.; Schaub, F.X.; Rudelius, M.; Laitinen, I.; Nilsson, L.; Goga, A.; Peschel, C.; et al. Myc-induced SUMOylation is a therapeutic vulnerability for B-cell lymphoma. Blood 2014, 124, 2081–2090. [Google Scholar] [CrossRef]
- Driscoll, J.J.; Pelluru, D.; Lefkimmiatis, K.; Fulciniti, M.; Prabhala, R.H.; Greipp, P.R.; Barlogie, B.; Tai, Y.T.; Anderson, K.C.; Shaughnessy, J.D., Jr.; et al. The sumoylation pathway is dysregulated in multiple myeloma and is associated with adverse patient outcome. Blood 2010, 115, 2827–2834. [Google Scholar] [CrossRef]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef]
- Renner, F.; Moreno, R.; Schmitz, M.L. SUMOylation-dependent localization of IKKepsilon in PML nuclear bodies is essential for protection against DNA-damage-triggered cell death. Mol. Cell 2010, 37, 503–515. [Google Scholar] [CrossRef]
- Potts, P.R.; Yu, H. The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat. Struct. Mol. Biol. 2007, 14, 581–590. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Long, X.D.; Wang, W.; Jiao, H.K.; Mei, Z.; Yin, Q.Q.; Ma, L.N.; Zhou, A.W.; Wang, L.S.; et al. Cbx4 governs HIF-1alpha to potentiate angiogenesis of hepatocellular carcinoma by its SUMO E3 ligase activity. Cancer Cell 2014, 25, 118–131. [Google Scholar] [CrossRef]
- Cashman, R.; Cohen, H.; Ben-Hamo, R.; Zilberberg, A.; Efroni, S. SENP5 mediates breast cancer invasion via a TGFbetaRI SUMOylation cascade. Oncotarget 2014, 5, 1071–1082. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Ye, D.G.; Lee, Y.J.; Gessler, F.; Friedman, G.K.; Zheng, W.; Hallenbeck, J.M. Drugging SUMOylation for neuroprotection and oncotherapy. Neural Regen. Res. 2018, 13, 415–416. [Google Scholar] [CrossRef]
- Mao, H.; Lebrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kogel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci. Rep. 2017, 7, 7425. [Google Scholar] [CrossRef]
- Soares, I.N.; Caetano, F.A.; Pinder, J.; Rodrigues, B.R.; Beraldo, F.H.; Ostapchenko, V.G.; Durette, C.; Pereira, G.S.; Lopes, M.H.; Queiroz-Hazarbassanov, N.; et al. Regulation of stress-inducible phosphoprotein 1 nuclear retention by protein inhibitor of activated STAT PIAS1. Mol. Cell Proteom. 2013, 12, 3253–3270. [Google Scholar] [CrossRef]
- Wang, L.; Banerjee, S. Differential PIAS3 expression in human malignancy. Oncol. Rep. 2004, 11, 1319–1324. [Google Scholar] [CrossRef]
- Xia, W.; Tian, H.; Cai, X.; Kong, H.; Fu, W.; Xing, W.; Wang, Y.; Zou, M.; Hu, Y.; Xu, D. Inhibition of SUMO-specific protease 1 induces apoptosis of astroglioma cells by regulating NF-kappaB/Akt pathways. Gene 2016, 595, 175–179. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Tang, S.; Huang, G.; Tong, X.; Xu, L.; Cai, R.; Li, J.; Zhou, X.; Song, S.; Huang, C.; Cheng, J. Role of SUMO-specific protease 2 in reprogramming cellular glucose metabolism. PLoS ONE 2013, 8, e63965. [Google Scholar] [CrossRef]
- Agbor, T.A.; Cheong, A.; Comerford, K.M.; Scholz, C.C.; Bruning, U.; Clarke, A.; Cummins, E.P.; Cagney, G.; Taylor, C.T. Small ubiquitin-related modifier (SUMO)-1 promotes glycolysis in hypoxia. J. Biol. Chem. 2011, 286, 4718–4726. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro. Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef]
- Xu, H.; Rahimpour, S.; Nesvick, C.L.; Zhang, X.; Ma, J.; Zhang, M.; Zhang, G.; Wang, L.; Yang, C.; Hong, C.S.; et al. Activation of hypoxia signaling induces phenotypic transformation of glioma cells: Implications for bevacizumab antiangiogenic therapy. Oncotarget 2015, 6, 11882–11893. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef]
- Minata, M.; Audia, A.; Shi, J.; Lu, S.; Bernstock, J.; Pavlyukov, M.S.; Das, A.; Kim, S.H.; Shin, Y.J.; Lee, Y.; et al. Phenotypic plasticity of invasive edge glioma stem-like cells in response to ionizing radiation. Cell Rep. 2019, 26, 1893.e7–1905.e7. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, Z.; Wang, X.; Zhao, X.; Sheng, Z.; Ye, Y.; He, G.; Zhou, L.; Zhu, H.; Xu, N.; et al. Small-molecule inhibitors targeting protein sumoylation as novel anticancer compounds. Mol. Pharmcol. 2018, 94, 885–894. [Google Scholar] [CrossRef]
- Huang, W.; He, T.; Chai, C.; Yang, Y.; Zheng, Y.; Zhou, P.; Qiao, X.; Zhang, B.; Liu, Z.; Wang, J.J.; et al. Triptolide inhibits the proliferation of prostate cancer cells and down-regulates SUMO-specific protease 1 expression. PLoS ONE 2012, 7, e37693. [Google Scholar] [CrossRef]
- Uno, M.; Koma, Y.; Ban, H.S.; Nakamura, H.J.B. Discovery of 1-[4-(N-benzylamino) phenyl]-3-phenylurea derivatives as non-peptidic selective SUMO-sentrin specific protease (SENP) 1 inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5169–5173. [Google Scholar] [CrossRef]
- Xie, W.; Wang, Z.; Zhang, J.; Wang, L.; Zhao, Y.; Zhou, H. Development and evaluation of a highly reliable assay for SUMO-specific protease inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 2124–2128. [Google Scholar] [CrossRef]
- Chen, Y.; Wen, D.; Huang, Z.; Huang, M.; Luo, Y.; Liu, B.; Lu, H.; Wu, Y.; Peng, Y.; Zhang, J. 2-(4-Chlorophenyl)-2-oxoethyl 4-benzamidobenzoate derivatives, a novel class of SENP1 inhibitors: Virtual screening, synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 6867–6870. [Google Scholar] [CrossRef]
- Brave, M.; Dagher, R.; Farrell, A.; Abraham, S.; Ramchandani, R.; Gobburu, J.; Booth, B.; Jiang, X.; Sridhara, R.; Justice, R. Topotecan in combination with cisplatin for the treatment of stage IVB, recurrent, or persistent cervical cancer. J. Oncol. 2006, 20, 1401–1404. [Google Scholar]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. J. Nat. Rev. Cancer 2006, 6, 789. [Google Scholar] [CrossRef]
- Ling, Y.-H.; Donato, N.J.; Perez-Soler, R. Sensitivity to topoisomerase I inhibitors and cisplatin is associated with epidermal growth factor receptor expression in human cervical squamous carcinoma ME180 sublines. J. Cancer Chemother. Pharmacol. 2001, 47, 473–480. [Google Scholar]
- Mo, Y.-Y.; Yu, Y.; Shen, Z.; Beck, W.T. Nucleolar delocalization of human topoisomerase I in response to topotecan correlates with sumoylation of the protein. J. Biol. Chem. 2002, 277, 2958–2964. [Google Scholar] [CrossRef]
- Rapisarda, A.; Uranchimeg, B.; Scudiero, D.A.; Selby, M.; Sausville, E.A.; Shoemaker, R.H.; Melillo, G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. J. Cancer Res. 2002, 62, 4316–4324. [Google Scholar]
- Hirohama, M.; Kumar, A.; Fukuda, I.; Matsuoka, S.; Igarashi, Y.; Saitoh, H.; Takagi, M.; Shin-ya, K.; Honda, K.; Kondoh, Y.; et al. Spectomycin B1 as a novel SUMOylation inhibitor that directly binds to SUMO E2. ACS Chem. Biol. 2013, 8, 2635–2642. [Google Scholar] [CrossRef]
- Kho, C.; Lee, A.; Jeong, D.; Oh, J.G.; Gorski, P.A.; Fish, K.; Sanchez, R.; DeVita, R.J.; Christensen, G.; Dahl, R.; et al. Small-molecule activation of SERCA2a SUMOylation for the treatment of heart failure. Nat. Commun. 2015, 6, 7229. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Lee, Y.-j.; Peruzzotti-Jametti, L.; Southall, N.; Johnson, K.R.; Maric, D.; Volpe, G.; Kouznetsova, J.; Zheng, W.; Pluchino, S.; et al. A novel quantitative high-throughput screen identifies drugs that both activate SUMO conjugation via the inhibition of microRNAs 182 and 183 and facilitate neuroprotection in a model of oxygen and glucose deprivation. J. Cereb. Blood Flow Metab. 2015, 36, 426–441. [Google Scholar] [CrossRef]
- Schneekloth, J.S., Jr. Controlling protein SUMOylation. Nat. Chem. Biol. 2017, 13, 1141. [Google Scholar] [CrossRef]
- He, X.; Riceberg, J.; Soucy, T.; Koenig, E.; Minissale, J.; Gallery, M.; Bernard, H.; Yang, X.; Liao, H.; Rabino, C.; et al. Probing the roles of SUMOylation in cancer cell biology by using a selective SAE inhibitor. Nat. Chem. Biol. 2017, 13, 1164–1171. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Hirai, G.; Nishimura, S.; Kawasaki, H.; Saitoh, H.; Kimura, K.; Sodeoka, M.; Yoshida, M. Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 2009, 16, 133–140. [Google Scholar] [CrossRef]
- Fukuda, I.; Ito, A.; Uramoto, M.; Saitoh, H.; Kawasaki, H.; Osada, H.; Yoshida, M. Kerriamycin B inhibits protein SUMOylation. J. Antibiot. 2009, 62, 221. [Google Scholar] [CrossRef]
- Takemoto, M.; Kawamura, Y.; Hirohama, M.; Yamaguchi, Y.; Handa, H.; Saitoh, H.; Nakao, Y.; Kawada, M.; Khalid, K.; Koshino, H.; et al. Inhibition of protein SUMOylation by davidiin, an ellagitannin from Davidia involucrata. J. Antibiot. 2014, 67, 335. [Google Scholar] [CrossRef]
- Suzawa, M.; Miranda, D.A.; Ramos, K.A.; Ang, K.K.; Faivre, E.J.; Wilson, C.G.; Caboni, L.; Arkin, M.R.; Kim, Y.S.; Fletterick, R.J.; et al. A gene-expression screen identifies a non-toxic sumoylation inhibitor that mimics SUMO-less human LRH-1 in liver. eLife 2015, 4, e09003. [Google Scholar] [CrossRef]
- Wu, J.; Lei, H.; Zhang, J.; Chen, X.; Tang, C.; Wang, W.; Xu, H.; Xiao, W.; Gu, W.; Wu, Y. Momordin Ic, a new natural SENP1 inhibitor, inhibits prostate cancer cell proliferation. Oncotarget 2016, 7, 58995–59005. [Google Scholar] [CrossRef]
- Schulman, B.A.; Wade Harper, J. Ubiquitin-like protein activation by E1 enzymes: The apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 2009, 10, 319. [Google Scholar] [CrossRef]
- Zhao, B.; Villhauer, E.B.; Bhuripanyo, K.; Kiyokawa, H.; Schindelin, H.; Yin, J. SUMO-mimicking peptides inhibiting protein SUMOylation. ChemBioChem Eur. J. Chem. Biol. 2014, 15, 2662–2666. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fox, B.M.; Janssen, A.; Estevez-Ordonez, D.; Gessler, F.; Vicario, N.; Chagoya, G.; Elsayed, G.; Sotoudeh, H.; Stetler, W.; Friedman, G.K.; et al. SUMOylation in Glioblastoma: A Novel Therapeutic Target. Int. J. Mol. Sci. 2019, 20, 1853. https://doi.org/10.3390/ijms20081853
Fox BM, Janssen A, Estevez-Ordonez D, Gessler F, Vicario N, Chagoya G, Elsayed G, Sotoudeh H, Stetler W, Friedman GK, et al. SUMOylation in Glioblastoma: A Novel Therapeutic Target. International Journal of Molecular Sciences. 2019; 20(8):1853. https://doi.org/10.3390/ijms20081853
Chicago/Turabian StyleFox, Brandon M., Andrew Janssen, Dagoberto Estevez-Ordonez, Florian Gessler, Nunzio Vicario, Gustavo Chagoya, Galal Elsayed, Houman Sotoudeh, William Stetler, Gregory K. Friedman, and et al. 2019. "SUMOylation in Glioblastoma: A Novel Therapeutic Target" International Journal of Molecular Sciences 20, no. 8: 1853. https://doi.org/10.3390/ijms20081853
APA StyleFox, B. M., Janssen, A., Estevez-Ordonez, D., Gessler, F., Vicario, N., Chagoya, G., Elsayed, G., Sotoudeh, H., Stetler, W., Friedman, G. K., & Bernstock, J. D. (2019). SUMOylation in Glioblastoma: A Novel Therapeutic Target. International Journal of Molecular Sciences, 20(8), 1853. https://doi.org/10.3390/ijms20081853