Establishment and Characterization of 5-Fluorouracil-Resistant Human Colorectal Cancer Stem-Like Cells: Tumor Dynamics under Selection Pressure

Abstract
1. Introduction
2. Results
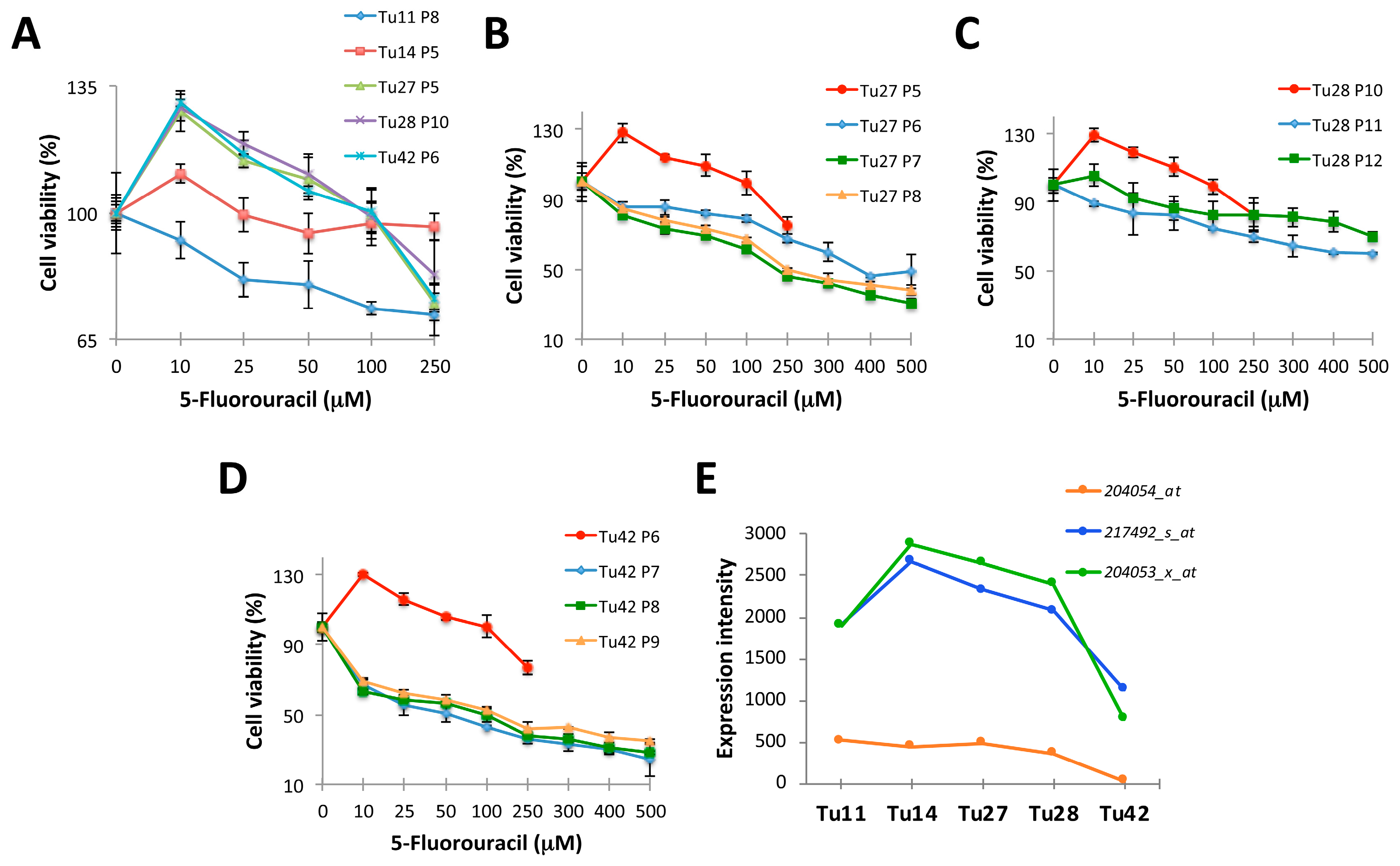
2.1. Establishment of 5-FU Resistant Human CRC Stem-Like Cells
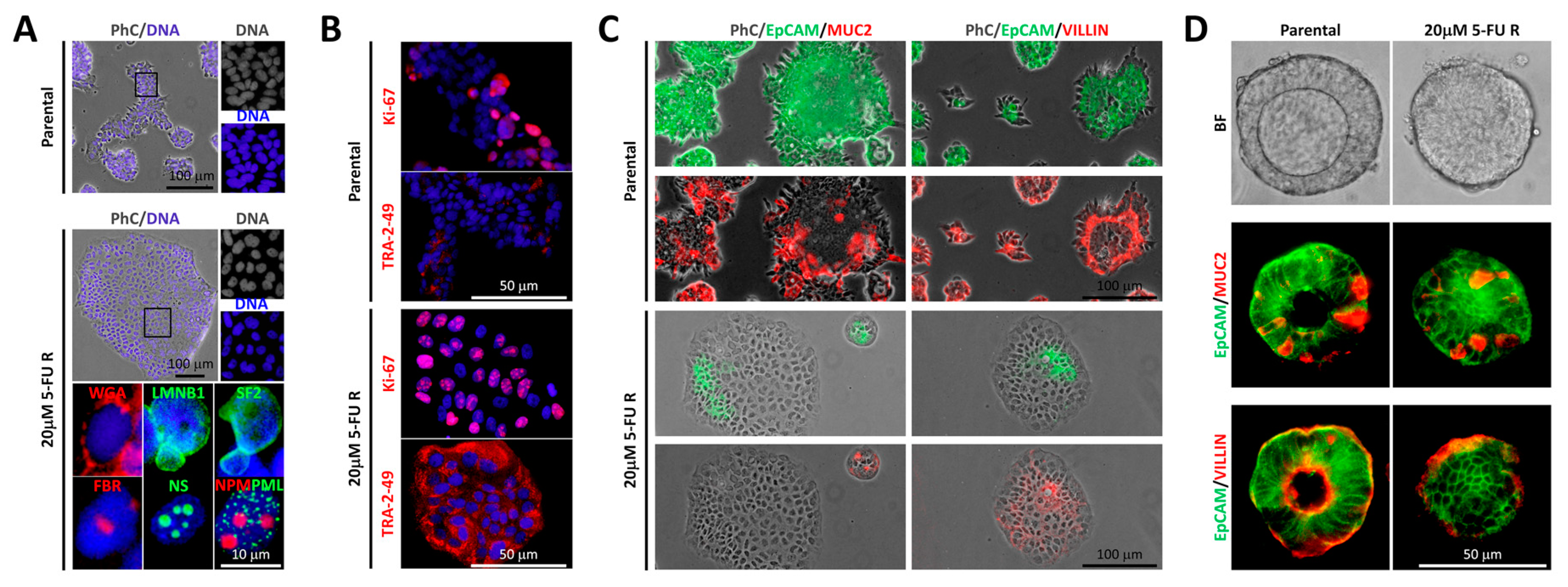
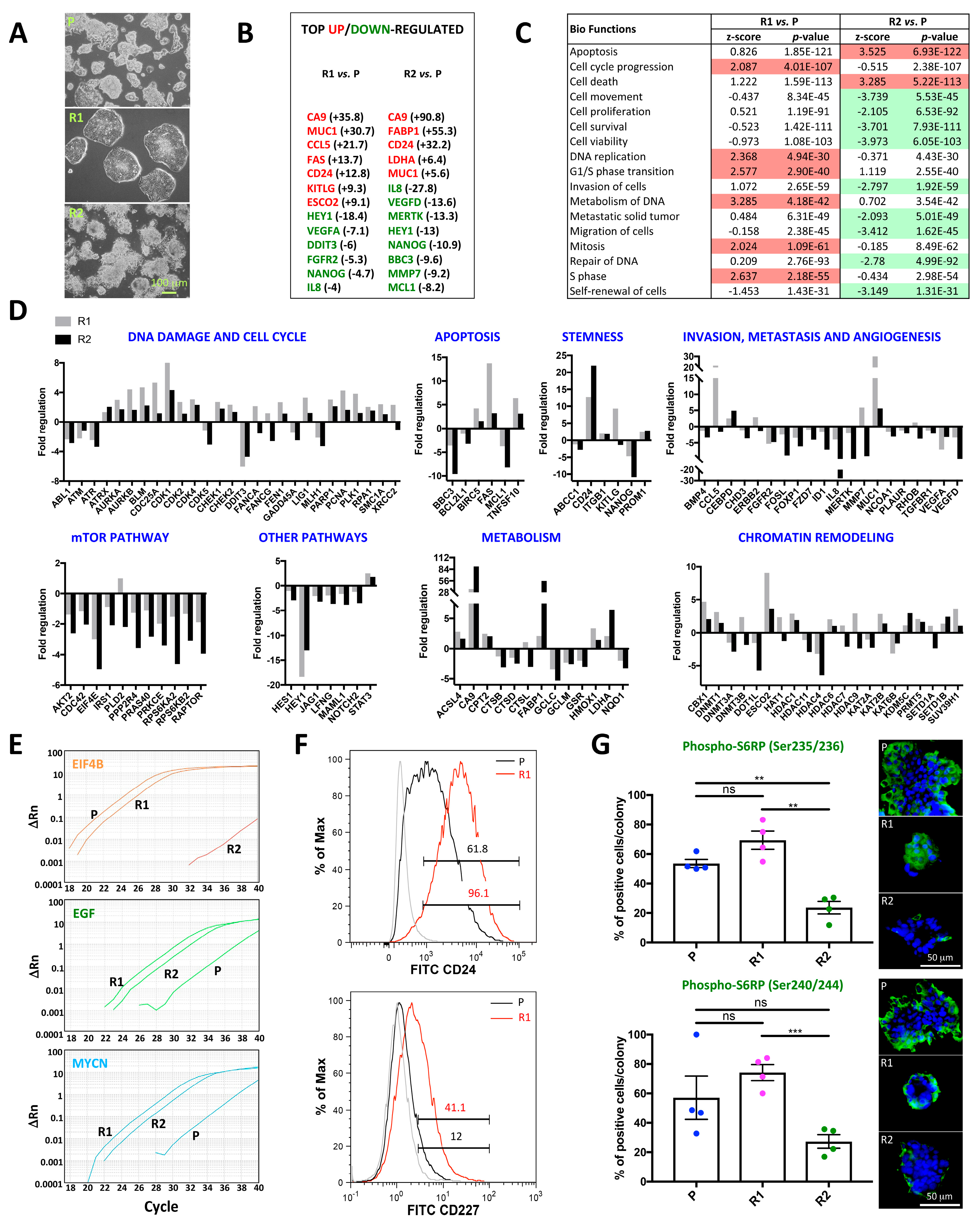
2.2. Differential Morphology, Gene Expression Profiles, and Biological Functions in 5-FU Resistant Tu42 CRC Stem-Like Cells as Compared to Parental Cells
2.2.1. DNA Damage and Cell Cycle
2.2.2. Apoptosis
2.2.3. Stemness
2.2.4. Invasion, Metastasis, and Angiogenesis
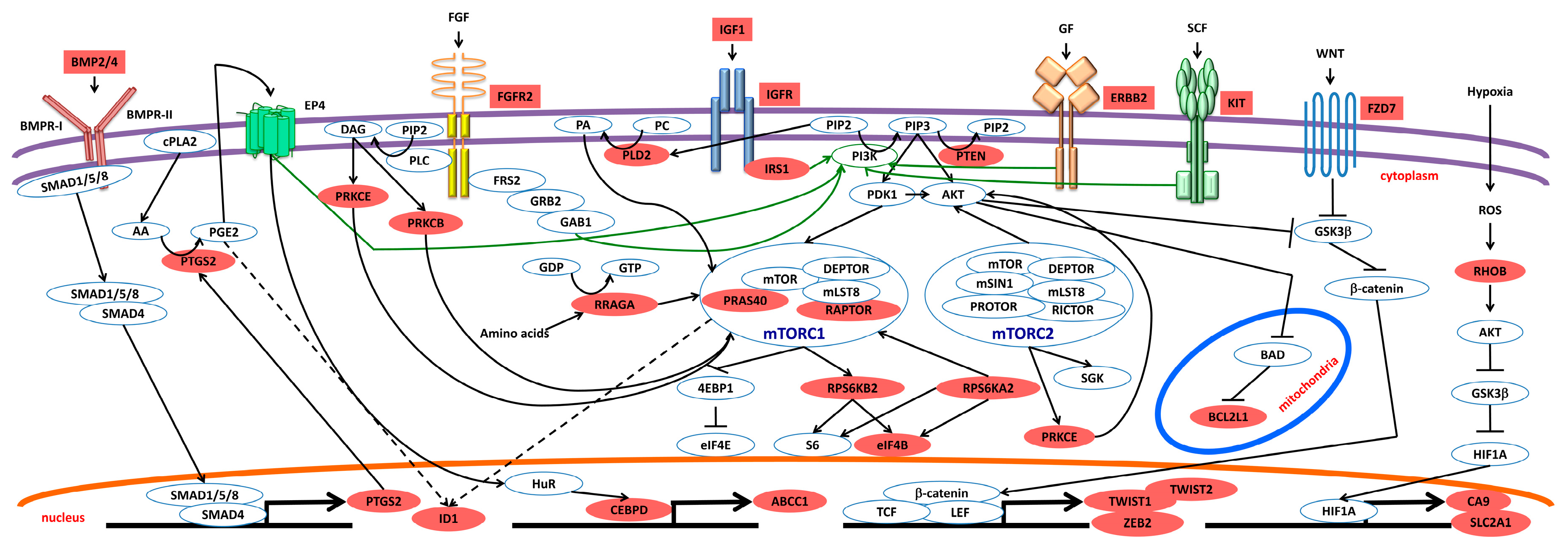
2.2.5. mTOR Pathway and Other Pathways
2.2.6. Metabolism
2.2.7. Chromatin Remodeling
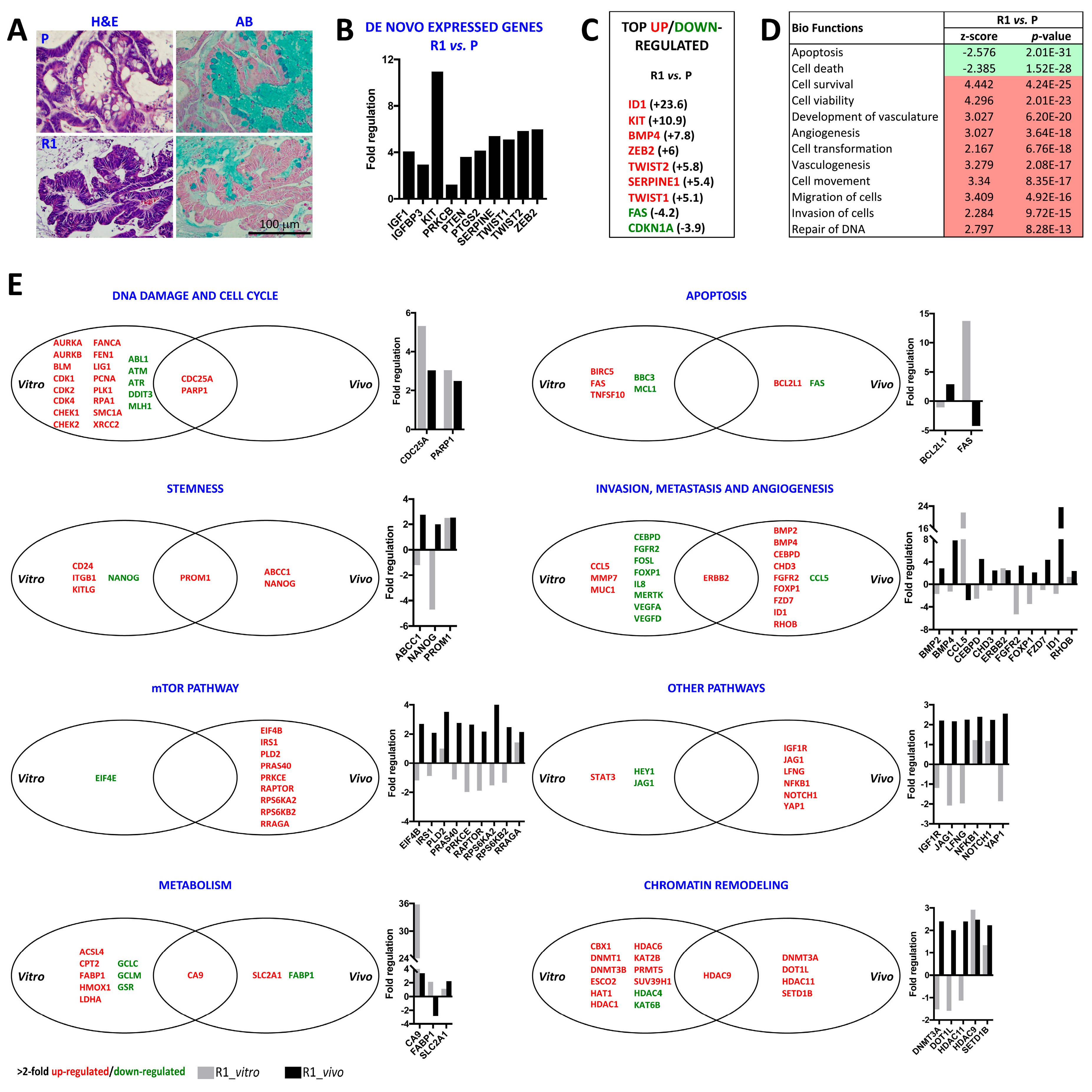
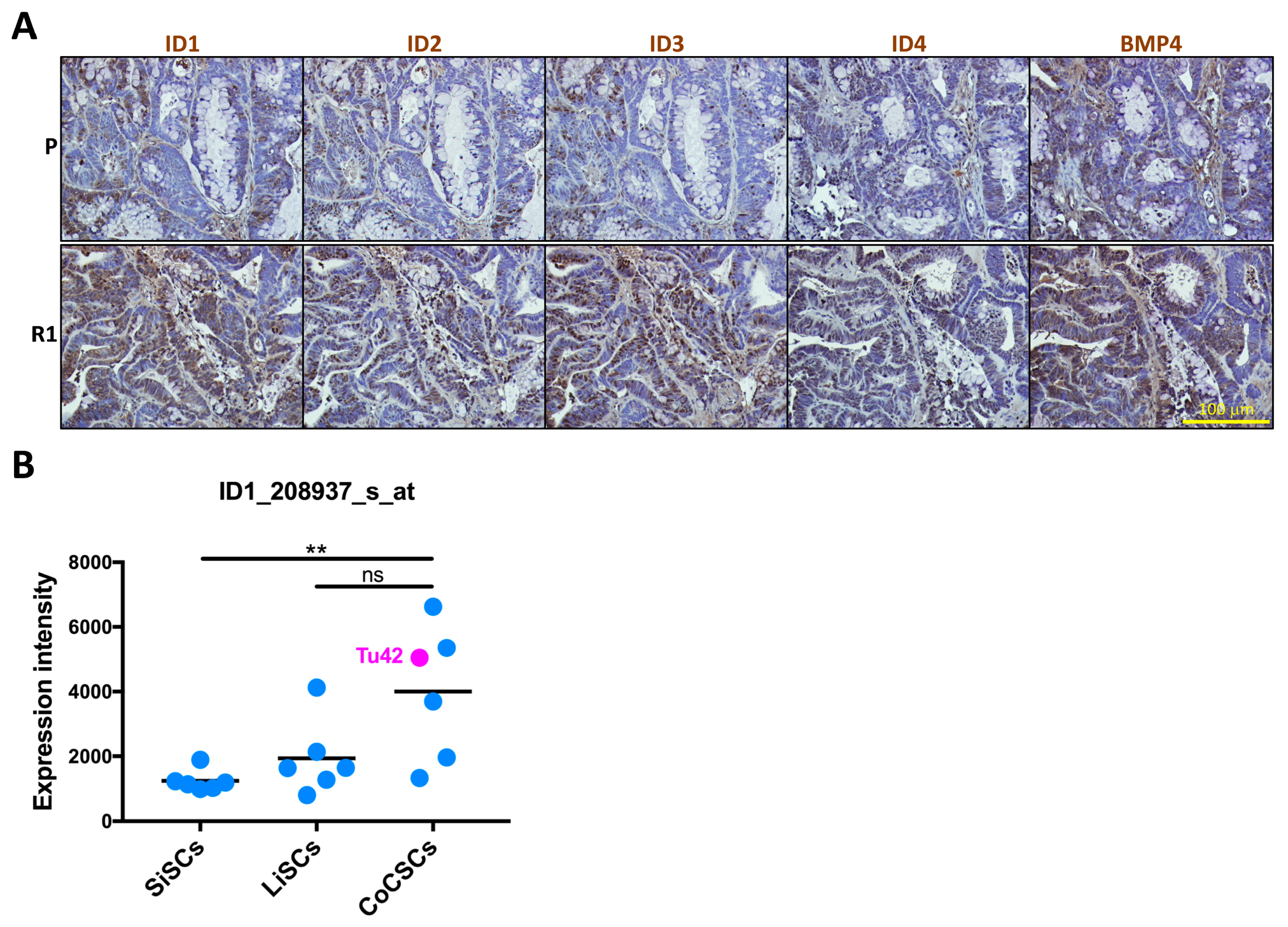
2.3. Differential Histology, Gene Expression Profiles, and Biological Functions of 5-FU Resistant Tu42 CRC Stem-Like Cells as Compared to Parental Cells In Vitro and In Vivo
2.4. ID1-Expressing Cell Enrichment Is a Feature of 5-FU Resistant Tu42 Cell-Derived Tumor Xenograft
3. Discussion
4. Materials and Methods
4.1. Specimens
4.2. Cell Culture and Determination of 5-FU IC50
4.3. Cytotoxicity Assay
4.4. Establishment of 5-FU Resistant Cell Lines
4.5. Organoid Preparation
4.6. Immunostains
4.7. RNA Extraction, cDNA Synthesis, PCR Arrays
4.8. Ingenuity Pathway Analysis (IPA)
4.9. Flow Cytometry
4.10. Animals
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef]
- Fulda, S. Evasion of apoptosis as a cellular stress response in cancer. Int. J. Cell Biol. 2010, 2010, 370835. [Google Scholar] [CrossRef] [PubMed]
- Mani, J.; Vallo, S.; Rakel, S.; Antonietti, P.; Gessler, F.; Blaheta, R.; Bartsch, G.; Michaelis, M.; Cinatl, J.; Haferkamp, A.; et al. Chemoresistance is associated with increased cytoprotective autophagy and diminished apoptosis in bladder cancer cells treated with the BH3 mimetic (-)-Gossypol (AT-101). BMC Cancer 2015, 15, 224. [Google Scholar] [CrossRef]
- Ghosal, G.; Chen, J. DNA damage tolerance: A double-edged sword guarding the genome. Transl. Cancer Res. 2013, 2, 107–129. [Google Scholar] [PubMed]
- Martinez-Outschoorn, U.E.; Peiris-Pages, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2016. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Greaves, M. Cancer stem cells: Back to Darwin? Semin. Cancer Biol. 2010, 20, 65–70. [Google Scholar] [CrossRef]
- Francipane, M.G.; Chandler, J.; Lagasse, E. Cancer Stem Cells: A Moving Target. Curr. Pathobiol. Rep. 2013, 1, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Francipane, M.G.; Lagasse, E. A Study of Cancer Heterogeneity: From Genetic Instability to Epigenetic Diversity in Colorectal Cancer. In Cancer Targeted Drug Delivery: An Elusive Dream; Bae, Y.H., Mrsny, R.J., Park, K., Eds.; Springer: New York, NY, USA, 2013; pp. 363–388. [Google Scholar] [CrossRef]
- Brooks, M.D.; Burness, M.L.; Wicha, M.S. Therapeutic Implications of Cellular Heterogeneity and Plasticity in Breast Cancer. Cell Stem Cell 2015, 17, 260–271. [Google Scholar] [CrossRef]
- Lagasse, E. Cancer stem cells with genetic instability: The best vehicle with the best engine for cancer. Gene Ther. 2008, 15, 136–142. [Google Scholar] [CrossRef]
- Odoux, C.; Fohrer, H.; Hoppo, T.; Guzik, L.; Stolz, D.B.; Lewis, D.W.; Gollin, S.M.; Gamblin, T.C.; Geller, D.A.; Lagasse, E. A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 2008, 68, 6932–6941. [Google Scholar] [CrossRef] [PubMed]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef]
- Quin, J.E.; Devlin, J.R.; Cameron, D.; Hannan, K.M.; Pearson, R.B.; Hannan, R.D. Targeting the nucleolus for cancer intervention. Biochim. Biophys. Acta 2014, 1842, 802–816. [Google Scholar] [CrossRef]
- Starborg, M.; Gell, K.; Brundell, E.; Hoog, C. The murine Ki-67 cell proliferation antigen accumulates in the nucleolar and heterochromatic regions of interphase cells and at the periphery of the mitotic chromosomes in a process essential for cell cycle progression. J. Cell Sci. 1996, 109 Pt 1, 143–153. [Google Scholar]
- International Stem Cell, I.; Adewumi, O.; Aflatoonian, B.; Ahrlund-Richter, L.; Amit, M.; Andrews, P.W.; Beighton, G.; Bello, P.A.; Benvenisty, N.; Berry, L.S.; et al. Characterization of human embryonic stem cell lines by the International Stem Cell Initiative. Nat. Biotechnol. 2007, 25, 803–816. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef]
- Moll, R.; Robine, S.; Dudouet, B.; Louvard, D. Villin: A cytoskeletal protein and a differentiation marker expressed in some human adenocarcinomas. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1987, 54, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Jinno, S.; Suto, K.; Nagata, A.; Igarashi, M.; Kanaoka, Y.; Nojima, H.; Okayama, H. Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J. 1994, 13, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Kao, T.P.; Huang, H. CDK1 promotes cell proliferation and survival via phosphorylation and inhibition of FOXO1 transcription factor. Oncogene 2008, 27, 4733–4744. [Google Scholar] [CrossRef]
- Patel, D.S.; Misenko, S.M.; Her, J.; Bunting, S.F. BLM helicase regulates DNA repair by counteracting RAD51 loading at DNA double-strand break sites. J. Cell Biol. 2017, 216, 3521–3534. [Google Scholar] [CrossRef]
- Tsou, J.H.; Chang, K.C.; Chang-Liao, P.Y.; Yang, S.T.; Lee, C.T.; Chen, Y.P.; Lee, Y.C.; Lin, B.W.; Lee, J.C.; Shen, M.R.; et al. Aberrantly expressed AURKC enhances the transformation and tumourigenicity of epithelial cells. J. Pathol. 2011, 225, 243–254. [Google Scholar] [CrossRef]
- Jauhiainen, A.; Thomsen, C.; Strombom, L.; Grundevik, P.; Andersson, C.; Danielsson, A.; Andersson, M.K.; Nerman, O.; Rorkvist, L.; Stahlberg, A.; et al. Distinct cytoplasmic and nuclear functions of the stress induced protein DDIT3/CHOP/GADD153. PLoS ONE 2012, 7, e33208. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Fas and TRAIL ‘death receptors’ as initiators of inflammation: Implications for cancer. Semin. Cell Dev. Biol. 2015, 39, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Park, S.M.; Tumanov, A.V.; Hau, A.; Sawada, K.; Feig, C.; Turner, J.R.; Fu, Y.X.; Romero, I.L.; Lengyel, E.; et al. CD95 promotes tumour growth. Nature 2010, 465, 492–496. [Google Scholar] [CrossRef]
- Ehrhardt, H.; Fulda, S.; Schmid, I.; Hiscott, J.; Debatin, K.M.; Jeremias, I. TRAIL induced survival and proliferation in cancer cells resistant towards TRAIL-induced apoptosis mediated by NF-kappaB. Oncogene 2003, 22, 3842–3852. [Google Scholar] [CrossRef] [PubMed]
- Vequaud, E.; Desplanques, G.; Jezequel, P.; Juin, P.; Barille-Nion, S. Survivin contributes to DNA repair by homologous recombination in breast cancer cells. Breast Cancer Res. Treat. 2016, 155, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.; Lee, H.W.; Hur, K.Y.; Kim, J.J.; Park, G.S.; Jang, S.H.; Song, Y.S.; Jang, K.S.; Paik, S.S. Cancer stem cell markers CD133 and CD24 correlate with invasiveness and differentiation in colorectal adenocarcinoma. World J. Gastroenterol. 2009, 15, 2258–2264. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.E.; Eisenman, J.; Baird, A.; Rauch, C.; Van Ness, K.; March, C.J.; Park, L.S.; Martin, U.; Mochizuki, D.Y.; Boswell, H.S.; et al. Identification of a ligand for the c-kit proto-oncogene. Cell 1990, 63, 167–174. [Google Scholar] [CrossRef]
- Silva, J.; Nichols, J.; Theunissen, T.W.; Guo, G.; van Oosten, A.L.; Barrandon, O.; Wray, J.; Yamanaka, S.; Chambers, I.; Smith, A. Nanog is the gateway to the pluripotent ground state. Cell 2009, 138, 722–737. [Google Scholar] [CrossRef]
- Cole, S.P. Multidrug resistance protein 1 (MRP1, ABCC1), a “multitasking” ATP-binding cassette (ABC) transporter. J. Biol. Chem. 2014, 289, 30880–30888. [Google Scholar] [CrossRef]
- Long, H.; Xie, R.; Xiang, T.; Zhao, Z.; Lin, S.; Liang, Z.; Chen, Z.; Zhu, B. Autocrine CCL5 signaling promotes invasion and migration of CD133+ ovarian cancer stem-like cells via NF-kappaB-mediated MMP-9 upregulation. Stem Cells 2012, 30, 2309–2319. [Google Scholar] [CrossRef]
- Nath, S.; Mukherjee, P. MUC1: A multifaceted oncoprotein with a key role in cancer progression. Trends Mol. Med. 2014, 20, 332–342. [Google Scholar] [CrossRef]
- Nagase, H.; Woessner, J.F., Jr. Matrix metalloproteinases. J. Biol. Chem. 1999, 274, 21491–21494. [Google Scholar] [CrossRef]
- Kumara, H.; Bellini, G.A.; Caballero, O.L.; Herath, S.A.C.; Su, T.; Ahmed, A.; Njoh, L.; Cekic, V.; Whelan, R.L. P-Cadherin (CDH3) is overexpressed in colorectal tumors and has potential as a serum marker for colorectal cancer monitoring. Oncoscience 2017, 4, 139–147. [Google Scholar]
- Keating, A.K.; Salzberg, D.B.; Sather, S.; Liang, X.; Nickoloff, S.; Anwar, A.; Deryckere, D.; Hill, K.; Joung, D.; Sawczyn, K.K.; et al. Lymphoblastic leukemia/lymphoma in mice overexpressing the Mer (MerTK) receptor tyrosine kinase. Oncogene 2006, 25, 6092–6100. [Google Scholar] [CrossRef]
- Ling, M.T.; Wang, X.; Ouyang, X.S.; Xu, K.; Tsao, S.W.; Wong, Y.C. Id-1 expression promotes cell survival through activation of NF-kappaB signalling pathway in prostate cancer cells. Oncogene 2003, 22, 4498–4508. [Google Scholar] [CrossRef]
- Ueno, K.; Hazama, S.; Mitomori, S.; Nishioka, M.; Suehiro, Y.; Hirata, H.; Oka, M.; Imai, K.; Dahiya, R.; Hinoda, Y. Down-regulation of frizzled-7 expression decreases survival, invasion and metastatic capabilities of colon cancer cells. Br. J. Cancer 2009, 101, 1374–1381. [Google Scholar] [CrossRef]
- Qin, L.; Wu, Y.L.; Toneff, M.J.; Li, D.; Liao, L.; Gao, X.; Bane, F.T.; Tien, J.C.; Xu, Y.; Feng, Z.; et al. NCOA1 Directly Targets M-CSF1 Expression to Promote Breast Cancer Metastasis. Cancer Res. 2014, 74, 3477–3488. [Google Scholar] [CrossRef]
- Li, D.; Wei, P.; Peng, Z.; Huang, C.; Tang, H.; Jia, Z.; Cui, J.; Le, X.; Huang, S.; Xie, K. The critical role of dysregulated FOXM1-PLAUR signaling in human colon cancer progression and metastasis. Clin. Cancer Res. 2013, 19, 62–72. [Google Scholar] [CrossRef]
- Xu, Y.; Pasche, B. TGF-beta signaling alterations and susceptibility to colorectal cancer. Hum. Mol. Genet. 2007, 16, R14–R20. [Google Scholar] [CrossRef]
- Siena, S.; Sartore-Bianchi, A.; Marsoni, S.; Hurwitz, H.I.; McCall, S.J.; Penault-Llorca, F.; Srock, S.; Bardelli, A.; Trusolino, L. Targeting the human epidermal growth factor receptor 2 (HER2) oncogene in colorectal cancer. Ann. Oncol. 2018, 29, 1108–1119. [Google Scholar] [CrossRef]
- Ackermann, S.; Kocak, H.; Hero, B.; Ehemann, V.; Kahlert, Y.; Oberthuer, A.; Roels, F.; Theissen, J.; Odenthal, M.; Berthold, F.; et al. FOXP1 inhibits cell growth and attenuates tumorigenicity of neuroblastoma. BMC Cancer 2014, 14, 840. [Google Scholar] [CrossRef]
- Nan, H.; Qureshi, A.A.; Hunter, D.J.; Han, J. Genetic variants in FGFR2 and FGFR4 genes and skin cancer risk in the Nurses’ Health Study. BMC Cancer 2009, 9, 172. [Google Scholar] [CrossRef]
- Kerbel, R.S. Tumor angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. [Google Scholar] [CrossRef]
- Jia, J.; Ye, T.; Cui, P.; Hua, Q.; Zeng, H.; Zhao, D. AP-1 transcription factor mediates VEGF-induced endothelial cell migration and proliferation. Microvasc. Res. 2016, 105, 103–108. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. mTOR pathway in colorectal cancer: An update. Oncotarget 2014, 5, 49–66. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Meng, R.D.; Shelton, C.C.; Li, Y.M.; Qin, L.X.; Notterman, D.; Paty, P.B.; Schwartz, G.K. gamma-Secretase inhibitors abrogate oxaliplatin-induced activation of the Notch-1 signaling pathway in colon cancer cells resulting in enhanced chemosensitivity. Cancer Res. 2009, 69, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Phan, L.M.; Yeung, S.C.; Lee, M.H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [PubMed]
- Benej, M.; Pastorekova, S.; Pastorek, J. Carbonic anhydrase IX: Regulation and role in cancer. Sub-Cell. Biochem. 2014, 75, 199–219. [Google Scholar]
- Wang, G.; Bonkovsky, H.L.; de Lemos, A.; Burczynski, F.J. Recent insights into the biological functions of liver fatty acid binding protein 1. J. Lipid Res. 2015, 56, 2238–2247. [Google Scholar] [CrossRef]
- Mizunoe, Y.; Sudo, Y.; Okita, N.; Hiraoka, H.; Mikami, K.; Narahara, T.; Negishi, A.; Yoshida, M.; Higashibata, R.; Watanabe, S.; et al. Involvement of lysosomal dysfunction in autophagosome accumulation and early pathologies in adipose tissue of obese mice. Autophagy 2017, 13, 642–653. [Google Scholar] [CrossRef]
- Tang, Y.; Zhou, J.; Hooi, S.C.; Jiang, Y.M.; Lu, G.D. Fatty acid activation in carcinogenesis and cancer development: Essential roles of long-chain acyl-CoA synthetases. Oncol. Lett. 2018, 16, 1390–1396. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione metabolism in cancer progression and treatment resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef]
- Poss, K.D.; Tonegawa, S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. USA 1997, 94, 10925–10930. [Google Scholar] [CrossRef] [PubMed]
- Whelan, G.; Kreidl, E.; Wutz, G.; Egner, A.; Peters, J.M.; Eichele, G. Cohesin acetyltransferase Esco2 is a cell viability factor and is required for cohesion in pericentric heterochromatin. EMBO J. 2012, 31, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Mattout, A.; Aaronson, Y.; Sailaja, B.S.; Raghu Ram, E.V.; Harikumar, A.; Mallm, J.P.; Sim, K.H.; Nissim-Rafinia, M.; Supper, E.; Singh, P.B.; et al. Heterochromatin Protein 1beta (HP1beta) has distinct functions and distinct nuclear distribution in pluripotent versus differentiated cells. Genome Biol. 2015, 16, 213. [Google Scholar] [CrossRef]
- Yokoyama, Y.; Hieda, M.; Nishioka, Y.; Matsumoto, A.; Higashi, S.; Kimura, H.; Yamamoto, H.; Mori, M.; Matsuura, S.; Matsuura, N. Cancer-associated upregulation of histone H3 lysine 9 trimethylation promotes cell motility in vitro and drives tumor formation in vivo. Cancer Sci. 2013, 104, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Nasser, S.; Murray, L.B.; Ayyanar, K.; Arango, D.; Figueroa, M.; Melnick, A.; Kao, G.D.; Augenlicht, L.H.; et al. HDAC4 promotes growth of colon cancer cells via repression of p21. Mol. Biol. Cell 2008, 19, 4062–4075. [Google Scholar] [CrossRef]
- Kim, W.; Kim, R.; Park, G.; Park, J.W.; Kim, J.E. Deficiency of H3K79 histone methyltransferase Dot1-like protein (DOT1L) inhibits cell proliferation. J. Biol. Chem. 2012, 287, 5588–5599. [Google Scholar] [CrossRef]
- Shahbazian, D.; Parsyan, A.; Petroulakis, E.; Topisirovic, I.; Martineau, Y.; Gibbs, B.F.; Svitkin, Y.; Sonenberg, N. Control of cell survival and proliferation by mammalian eukaryotic initiation factor 4B. Mol. Cell. Biol. 2010, 30, 1478–1485. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Takatori, A.; Nakamura, Y.; Suenaga, Y.; Kamijo, T.; Nakagawara, A. NLRR1 enhances EGF-mediated MYCN induction in neuroblastoma and accelerates tumor growth in vivo. Cancer Res. 2012, 72, 4587–4596. [Google Scholar] [CrossRef]
- Aland, S.; Hatzikirou, H.; Lowengrub, J.; Voigt, A. A Mechanistic Collective Cell Model for Epithelial Colony Growth and Contact Inhibition. Biophys. J. 2015, 109, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Dey-Guha, I.; Wolfer, A.; Yeh, A.C.; Albeck, J.G.; Darp, R.; Leon, E.; Wulfkuhle, J.; Petricoin, E.F., 3rd; Wittner, B.S.; Ramaswamy, S. Asymmetric cancer cell division regulated by AKT. Proc. Natl. Acad. Sci. USA 2011, 108, 12845–12850. [Google Scholar] [CrossRef] [PubMed]
- Foster, B.M.; Zaidi, D.; Young, T.R.; Mobley, M.E.; Kerr, B.A. CD117/c-kit in Cancer Stem Cell-Mediated Progression and Therapeutic Resistance. Biomedicines 2018, 6, 31. [Google Scholar] [CrossRef]
- Rakic, J.M.; Maillard, C.; Jost, M.; Bajou, K.; Masson, V.; Devy, L.; Lambert, V.; Foidart, J.M.; Noel, A. Role of plasminogen activator-plasmin system in tumor angiogenesis. Cell. Mol. Life Sci. 2003, 60, 463–473. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Hanrahan, K.; O’Neill, A.; Prencipe, M.; Bugler, J.; Murphy, L.; Fabre, A.; Puhr, M.; Culig, Z.; Murphy, K.; Watson, R.W. The role of epithelial-mesenchymal transition drivers ZEB1 and ZEB2 in mediating docetaxel-resistant prostate cancer. Mol. Oncol. 2017, 11, 251–265. [Google Scholar] [CrossRef]
- Francipane, M.G.; Lagasse, E. Selective targeting of human colon cancer stem-like cells by the mTOR inhibitor Torin-1. Oncotarget 2013, 4, 1948–1962. [Google Scholar] [CrossRef]
- Cramer, J.M.; Thompson, T.; Geskin, A.; LaFramboise, W.; Lagasse, E. Distinct human stem cell populations in small and large intestine. PLoS ONE 2015, 10, e0118792. [Google Scholar] [CrossRef]
- Hollnagel, A.; Oehlmann, V.; Heymer, J.; Ruther, U.; Nordheim, A. Id genes are direct targets of bone morphogenetic protein induction in embryonic stem cells. J. Biol. Chem. 1999, 274, 19838–19845. [Google Scholar] [CrossRef]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel) 2017, 9, 52. [Google Scholar]
- Fabregat, I.; Malfettone, A.; Soukupova, J. New Insights into the Crossroads between EMT and Stemness in the Context of Cancer. J. Clin. Med. 2016, 5, 37. [Google Scholar] [CrossRef]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kaminska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354. [Google Scholar] [CrossRef]
- Zhang, N.; Yantiss, R.K.; Nam, H.S.; Chin, Y.; Zhou, X.K.; Scherl, E.J.; Bosworth, B.P.; Subbaramaiah, K.; Dannenberg, A.J.; Benezra, R. ID1 is a functional marker for intestinal stem and progenitor cells required for normal response to injury. Stem Cell Rep. 2014, 3, 716–724. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Official Symbol | Official Full Name |
|---|---|
| ABCC1 | ATP binding cassette subfamily C member 1 |
| ABL1 | ABL proto-oncogene 1, non-receptor tyrosine kinase |
| ACSL4 | Acyl-CoA synthetase long chain family member 4 |
| AKT2 | AKT serine/threonine kinase 2 |
| ATM | ATM serine/threonine kinase |
| ATR | ATR serine/threonine kinase |
| ATRX | ATRX, chromatin remodeler |
| AURKA | Aurora kinase A |
| AURKB | Aurora kinase B |
| BBC3 | BCL2 binding component 3 |
| BCL2L1 | BCL2 like 1, Bcl-xl |
| BIRC5 | Baculoviral IAP repeat containing 5, Survivin |
| BLM | BLM RecQ like helicase |
| BMP4 | Bone morphogenetic protein 4 |
| CA9 | Carbonic anhydrase 9 |
| CBX1 | Chromobox 1 |
| CCL5 | C-C motif chemokine ligand 5 |
| CD24 | CD24 molecule |
| CDC25A | Cell division cycle 25A |
| CDC42 | Cell division cycle 42 |
| CDK1 | Cyclin dependent kinase 1 |
| CDK2 | Cyclin dependent kinase 2 |
| CDK4 | Cyclin dependent kinase 4 |
| CDK5 | Cyclin dependent kinase 5 |
| CEBPD | CCAAT enhancer binding protein delta |
| CHD3 | Chromodomain helicase DNA binding protein 3 |
| CHEK1 | Checkpoint kinase 1 |
| CHEK2 | Checkpoint kinase 2 |
| CPT2 | Carnitine palmitoyltransferase 2 |
| CTSB | Cathepsin B |
| CTSD | Cathepsin D |
| CTSL | Cathepsin L |
| DDIT3 | DNA-damage inducible transcript 3 |
| DNMT1 | DNA methyltransferase 1 |
| DNMT3A | DNA methyltransferase 3 alpha |
| DNMT3B | DNA methyltransferase 3 beta |
| DOT1L | DOT1 like histone lysine methyltransferase |
| EGF | Epidermal growth factor |
| EIF4B | Eukaryotic translation initiation factor 4B |
| EIF4E | Eukaryotic translation initiation factor 4E |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 |
| ESCO2 | Establishment of sister chromatid cohesion N-acetyltransferase 2 |
| FABP1 | Fatty acid binding protein 1 |
| FANCA | FA complementation group A |
| FANCG | FA complementation group G |
| FAS | Fas (TNF receptor superfamily member 6) |
| FEN1 | Flap structure-specific endonuclease 1 |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FOSL1 | FOS like 1, AP-1 transcription factor subunit |
| FOXP1 | Forkhead box P1 |
| FZD7 | Frizzled class receptor 7 |
| GADD45A | Growth arrest and DNA damage inducible alpha |
| GCLC | Glutamate-cysteine ligase catalytic subunit |
| GCLM | Glutamate-cysteine ligase, modifier subunit |
| GSR | Glutathione-disulfide reductase |
| HAT1 | Histone acetyltransferase 1 |
| HDAC1 | Histone deacetylase 1 |
| HDAC11 | Histone deacetylase 11 |
| HDAC4 | Histone deacetylase 4 |
| HDAC6 | Histone deacetylase 6 |
| HDAC7 | Histone deacetylase 7 |
| HDAC9 | Histone deacetylase 9 |
| HES1 | Hes family bHLH transcription factor 1 |
| HEY1 | Hairy/enhancer-of-split related with YRPW motif 1 |
| HMOX1 | Heme oxygenase 1 |
| ID1 | Inhibitor of DNA binding 1, HLH protein |
| IL8 | Interleukin 8 |
| IRS1 | Insulin receptor substrate 1 |
| ITGB1 | Integrin Subunit Beta 1 |
| JAG1 | Jagged 1 |
| KAT2A | Lysine acetyltransferase 2A |
| KAT2B | Lysine acetyltransferase 2B |
| KAT6B | Lysine acetyltransferase 6B |
| KDM5C | Lysine demethylase 5C |
| KITLG | KIT ligand |
| LDHA | Lactate dehydrogenase A |
| LFNG | LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase |
| LIG1 | DNA ligase 1 |
| MAML1 | Mastermind like transcriptional coactivator 1 |
| MCL1 | MCL1, BCL2 family apoptosis regulator |
| MERTK | MER proto-oncogene, tyrosine kinase |
| MLH1 | MutL homolog 1 |
| MMP7 | Matrix metallopeptidase 7 |
| MUC1 | Mucin 1 |
| MYCN | MYCN proto-oncogene, bHLH transcription factor |
| NANOG | Nanog |
| NCOA1 | Nuclear receptor coactivator 1 |
| NOTCH2 | Notch 2 |
| NQO1 | NAD(P)H quinone dehydrogenase 1 |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| PCNA | Proliferating cell nuclear antigen |
| PLAUR | Plasminogen activator, urokinase receptor |
| PLD2 | Phospholipase D2 |
| PLK1 | Polo like kinase 1 |
| PPP2R4 | Phosphotyrosyl phosphatase activator |
| PRAS40 | Proline-rich Akt substrate 40 kDa |
| PRKCE | Protein kinase C epsilon |
| PRMT5 | Protein arginine methyltransferase 5 |
| PROM1 | Prominin 1, CD133 |
| RAPTOR | Regulatory associated protein of mTOR |
| RHOB | Ras homolog family member B |
| RPA1 | Replication protein A1 |
| RPS6KA2 | Ribosomal protein S6 kinase A2 |
| RPS6KB2 | Ribosomal protein S6 kinase B2 |
| SETD1A | SET domain containing 1A |
| SETD1B | SET domain containing 1B |
| SMC1A | Structural maintenance of chromosomes 1A |
| STAT3 | Signal transducer and activator of transcription 3 |
| SUV39H1 | Suppressor of variegation 3-9 homolog 1 |
| TGFBR1 | Transforming growth factor beta receptor 1 |
| TNFSF10 | TNF superfamily member 10, TRAIL |
| VEGFA | Vascular endothelial growth factor A |
| VEGFD | Vascular endothelial growth factor D |
| XRCC2 | X-ray repair cross complementing 2 |
| Official Symbol | Official Full Name |
|---|---|
| ABCC1 | ATP binding cassette subfamily C member 1 |
| BCL2L1 | BCL2 like 1, Bcl-xl |
| BMP2 | Bone morphogenetic protein 2 |
| BMP4 | Bone morphogenetic protein 4 |
| CA9 | Carbonic anhydrase 9 |
| CEBPD | CCAAT enhancer binding protein delta |
| CDC25A | Cell division cycle 25A |
| CHD3 | Chromodomain helicase DNA binding protein 3 |
| DNMT3A | DNA methyltransferase 3 alpha |
| DOT1L | DOT1 like histone lysine methyltransferase |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 |
| EIF4B | Eukaryotic translation initiation factor 4B |
| FAS | Fas (TNF receptor superfamily member 6) |
| FABP1 | Fatty acid binding protein 1 |
| FGFR2 | Fibroblast growth factor receptor 2 |
| FOXP1 | Forkhead box P1 |
| FZD7 | Frizzled class receptor 7 |
| HDAC11 | Histone deacetylase 11 |
| HDAC9 | Histone deacetylase 9 |
| ID1 | Inhibitor of DNA binding 1, HLH protein |
| IGF1 | Insulin like growth factor 1 |
| IGF1R | Insulin like growth factor 1 receptor |
| IGFBP3 | insulin like growth factor binding protein 3 |
| IRS1 | Insulin receptor substrate 1 |
| JAG1 | Jagged 1 |
| KIT | KIT proto-oncogene receptor tyrosine kinase |
| LFNG | LFNG O-fucosylpeptide 3-beta-N-acetylglucosaminyltransferase |
| NANOG | Nanog |
| NOTCH1 | Notch 1 |
| NFKB1 | Nuclear factor kappa B subunit 1 |
| PTEN | Phosphatase and tensin homolog |
| PLD2 | Phospholipase D2 |
| PARP1 | Poly(ADP-ribose) polymerase 1 |
| PRAS40 | Proline-rich Akt substrate 40 kDa |
| PROM1 | Prominin 1, CD133 |
| PTGS2 | Prostaglandin-endoperoxide synthase 2 |
| PRKCB | Protein kinase C beta |
| PRKCE | Protein kinase C epsilon |
| RHOB | Ras homolog family member B |
| RRAGA | Ras related GTP binding A |
| RAPTOR | Regulatory associated protein of mTOR |
| RPS6KA2 | Ribosomal protein S6 kinase A2 |
| RPS6KB2 | Ribosomal protein S6 kinase B2 |
| SERPINE1 | Serpin family E member 1 |
| SETD1B | SET domain containing 1B |
| SLC2A1 | Solute carrier family 2 member 1, GLUT-1 |
| TWIST1 | Twist family bHLH transcription factor 1 |
| TWIST2 | Twist family bHLH transcription factor 2 |
| YAP1 | Yes associated protein 1 |
| ZEB2 | Zinc finger E-box binding homeobox 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francipane, M.G.; Bulanin, D.; Lagasse, E. Establishment and Characterization of 5-Fluorouracil-Resistant Human Colorectal Cancer Stem-Like Cells: Tumor Dynamics under Selection Pressure. Int. J. Mol. Sci. 2019, 20, 1817. https://doi.org/10.3390/ijms20081817
Francipane MG, Bulanin D, Lagasse E. Establishment and Characterization of 5-Fluorouracil-Resistant Human Colorectal Cancer Stem-Like Cells: Tumor Dynamics under Selection Pressure. International Journal of Molecular Sciences. 2019; 20(8):1817. https://doi.org/10.3390/ijms20081817
Chicago/Turabian StyleFrancipane, Maria Giovanna, Denis Bulanin, and Eric Lagasse. 2019. "Establishment and Characterization of 5-Fluorouracil-Resistant Human Colorectal Cancer Stem-Like Cells: Tumor Dynamics under Selection Pressure" International Journal of Molecular Sciences 20, no. 8: 1817. https://doi.org/10.3390/ijms20081817
APA StyleFrancipane, M. G., Bulanin, D., & Lagasse, E. (2019). Establishment and Characterization of 5-Fluorouracil-Resistant Human Colorectal Cancer Stem-Like Cells: Tumor Dynamics under Selection Pressure. International Journal of Molecular Sciences, 20(8), 1817. https://doi.org/10.3390/ijms20081817