Low-Luminance Blue Light-Enhanced Phototoxicity in A2E-Laden RPE Cell Cultures and Rats


Abstract
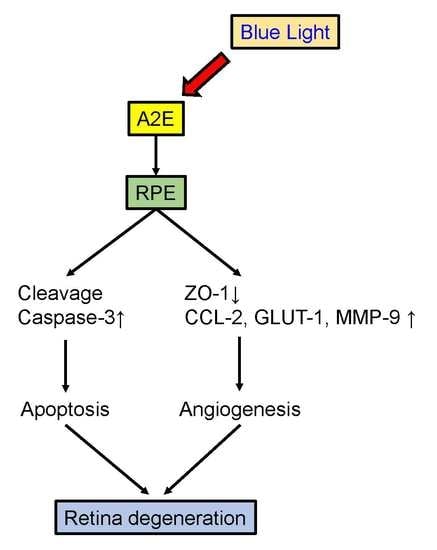
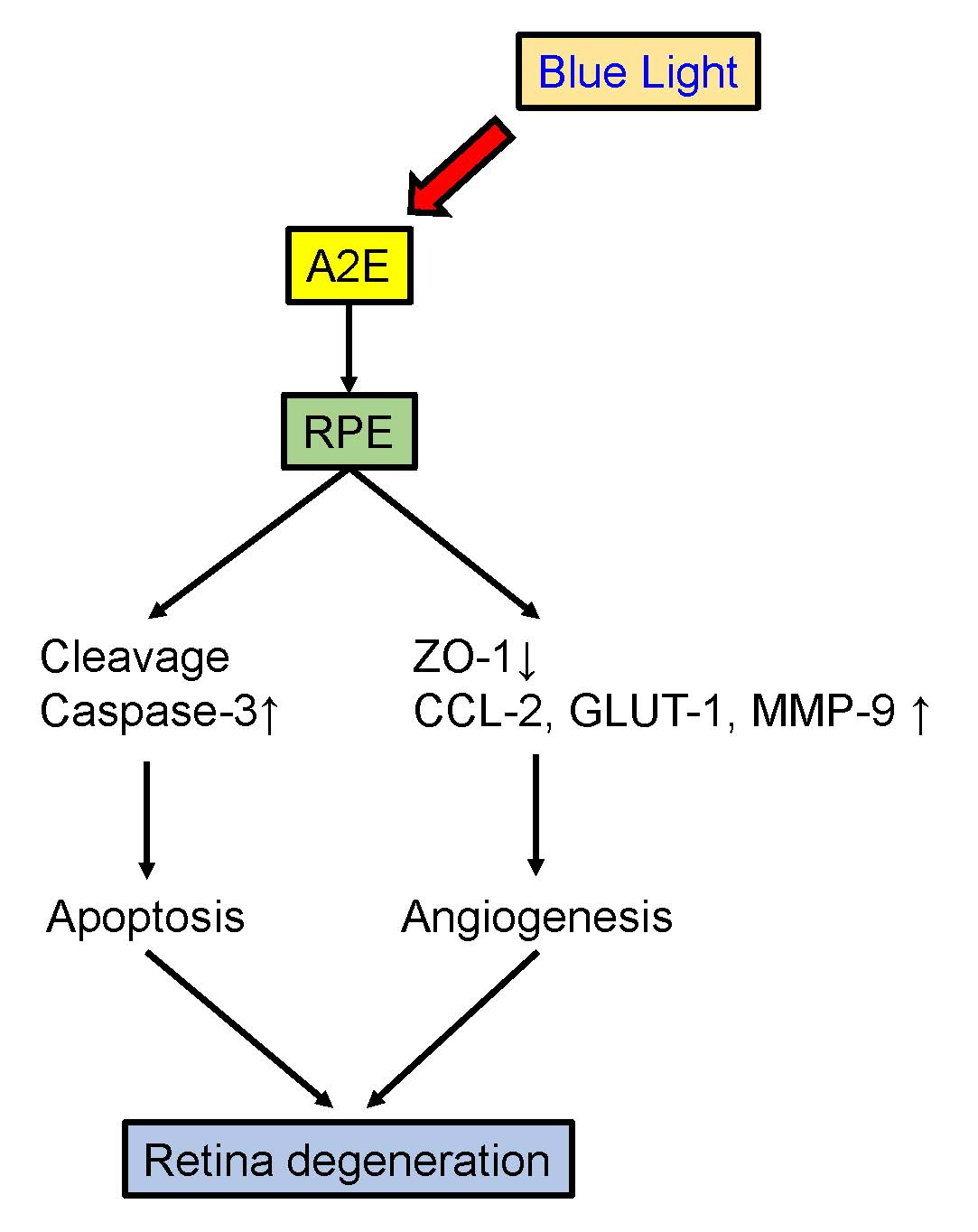
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
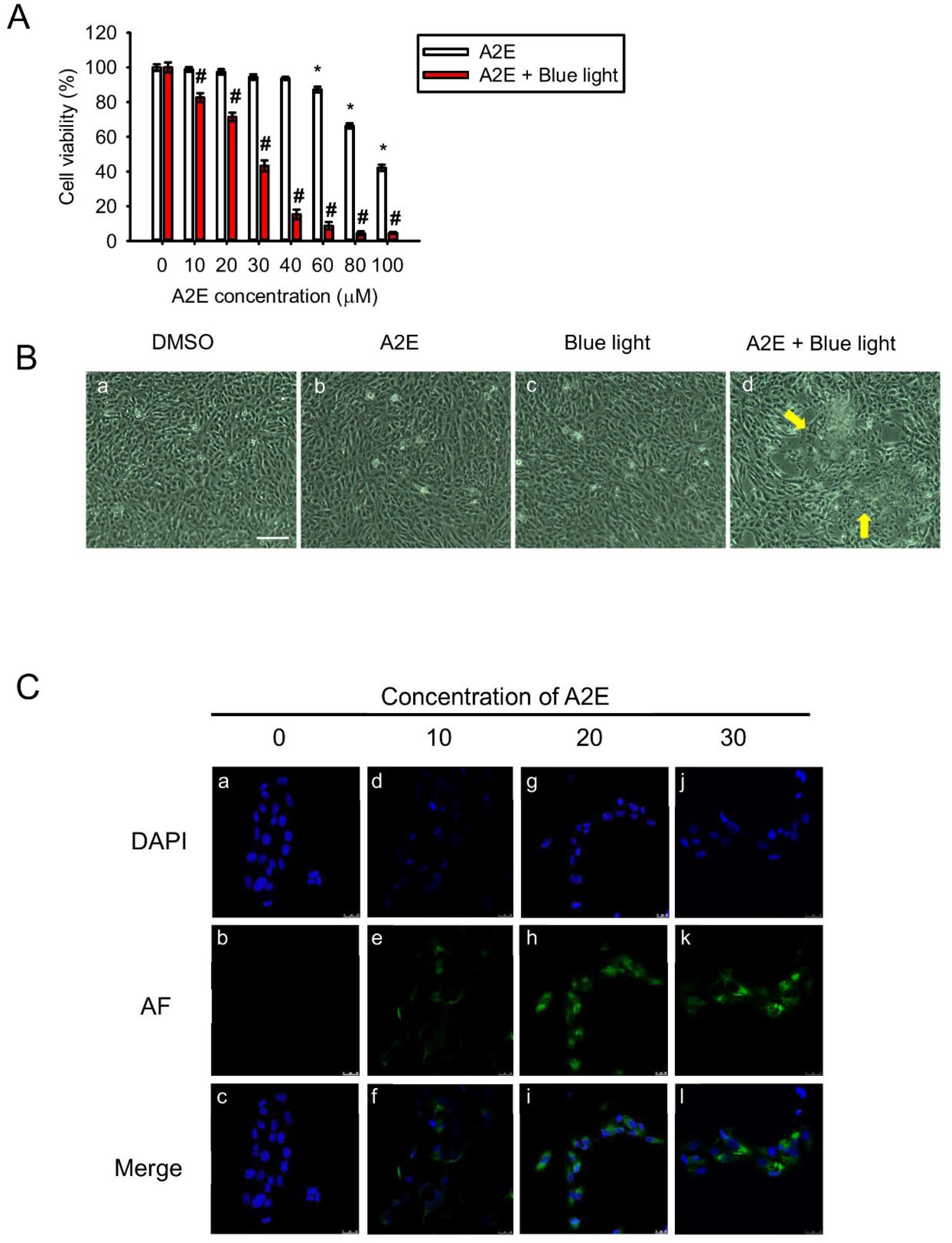
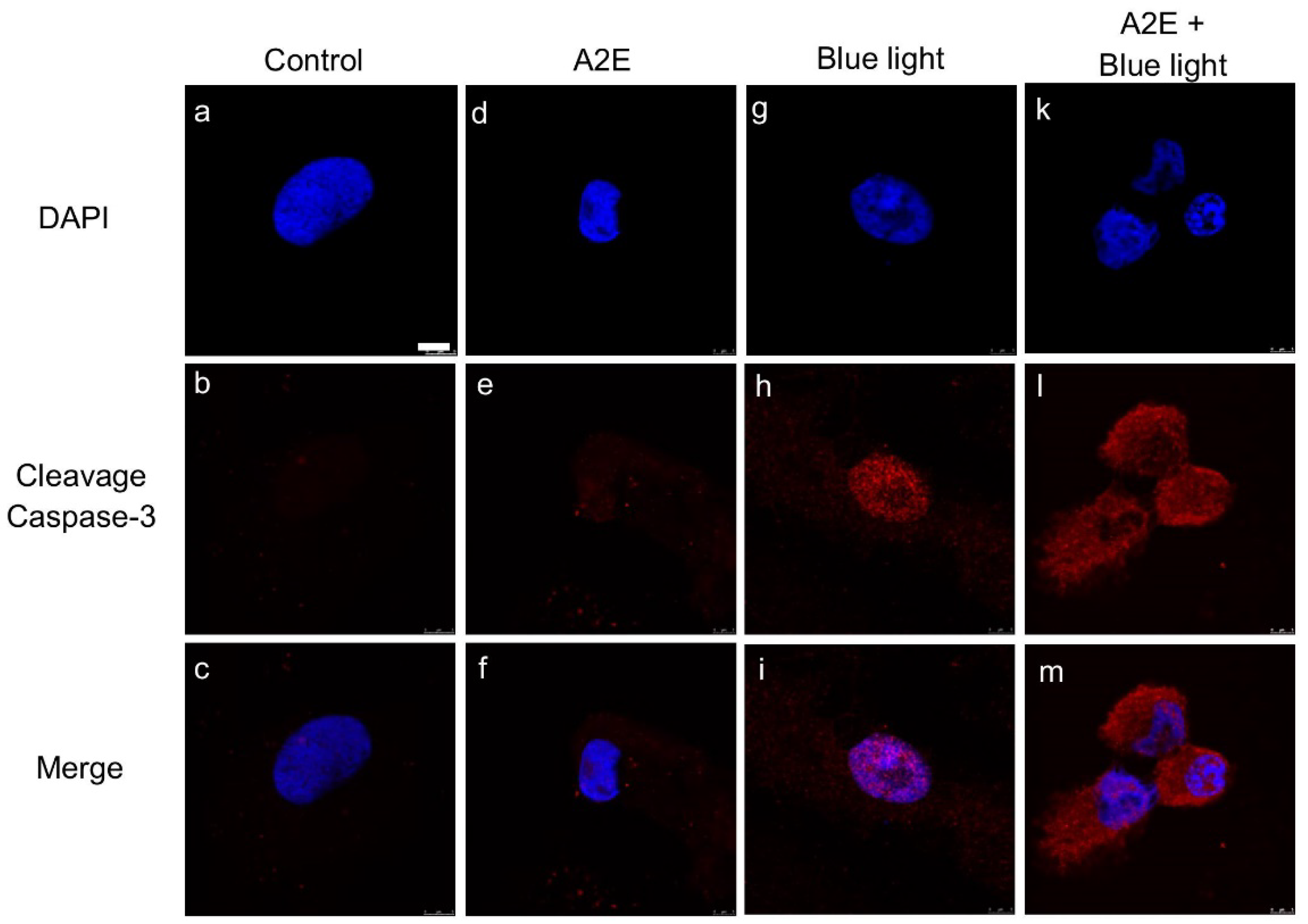
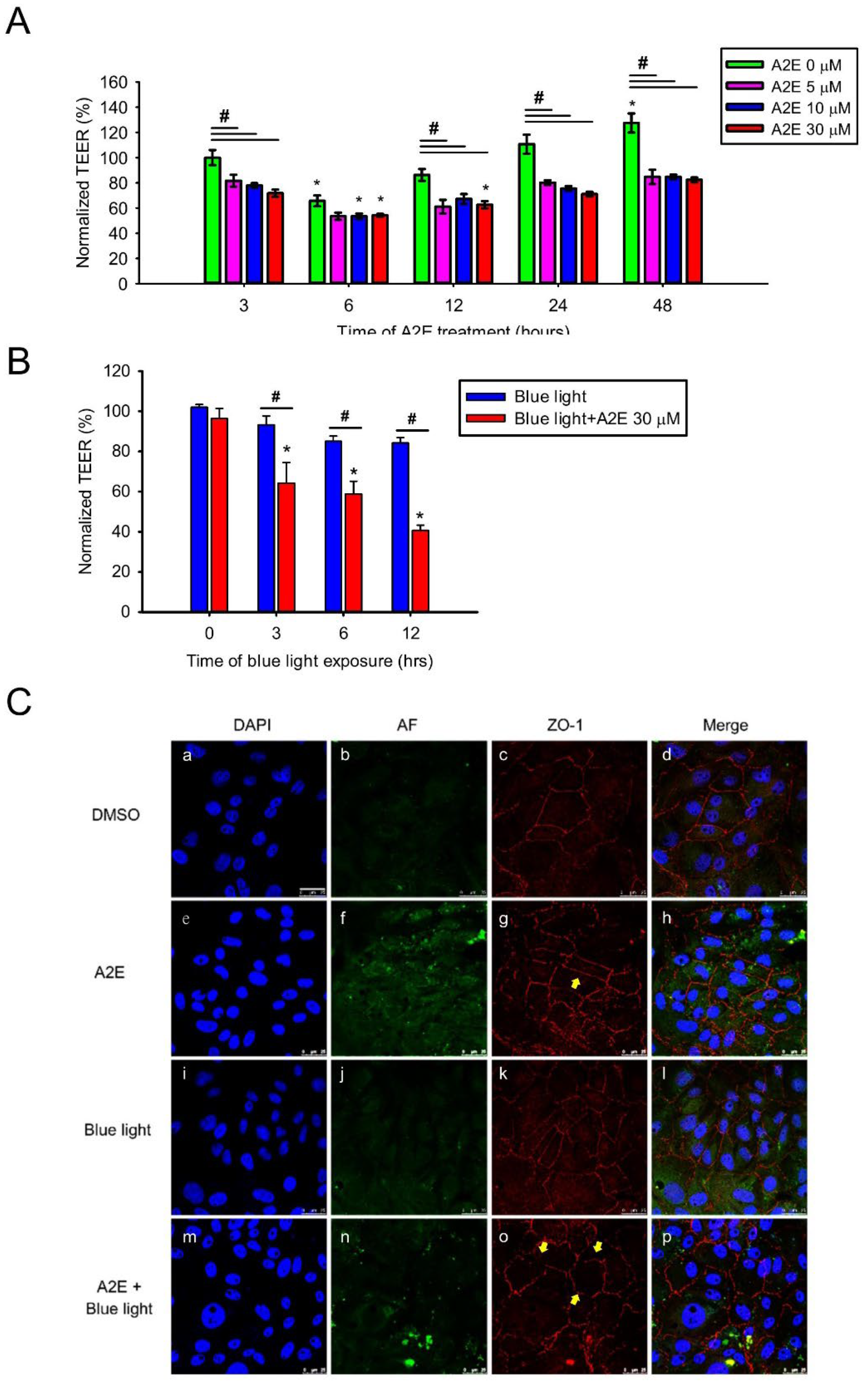
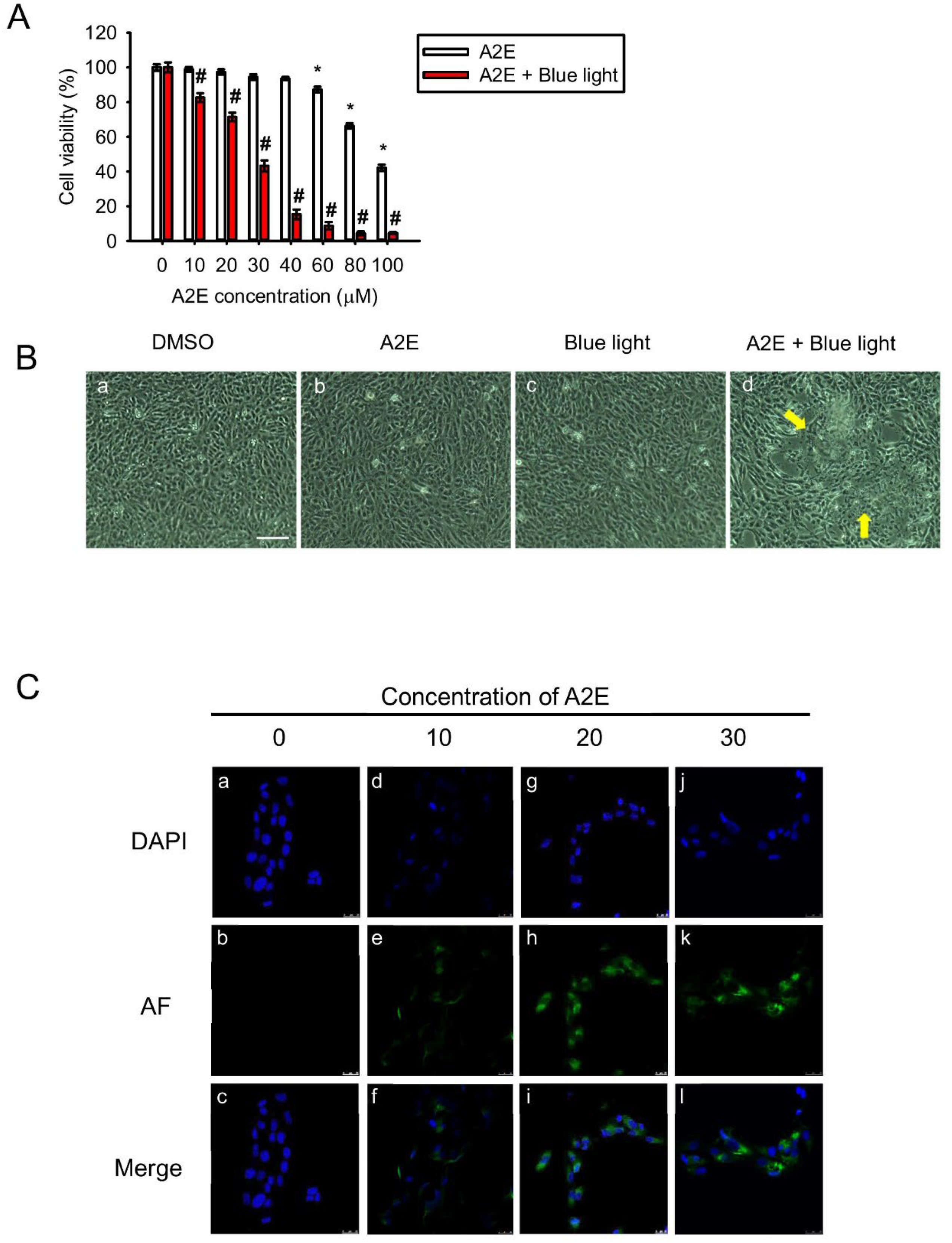
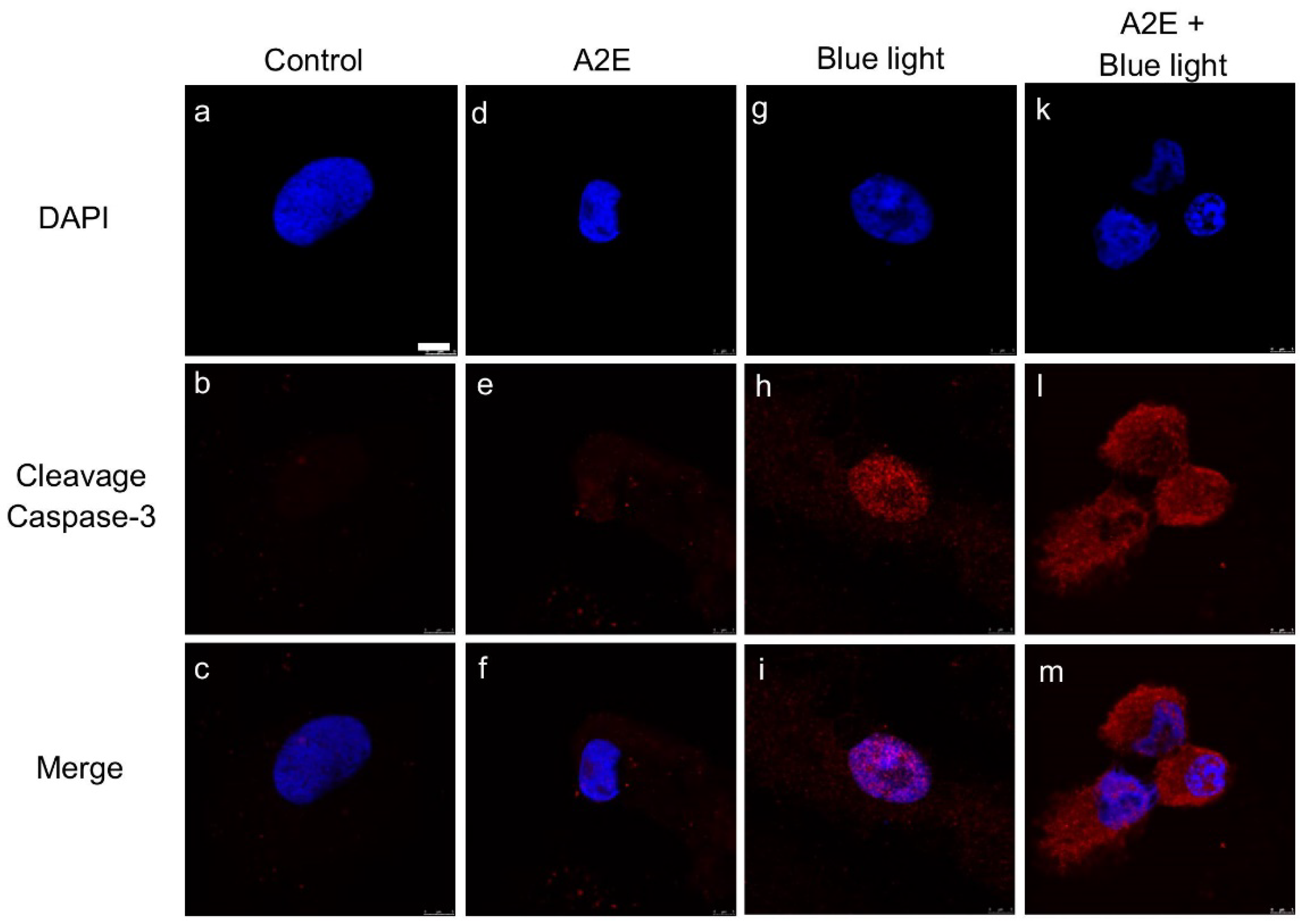
2.1. A2E Damages RPE Cells under Blue Light Exposure
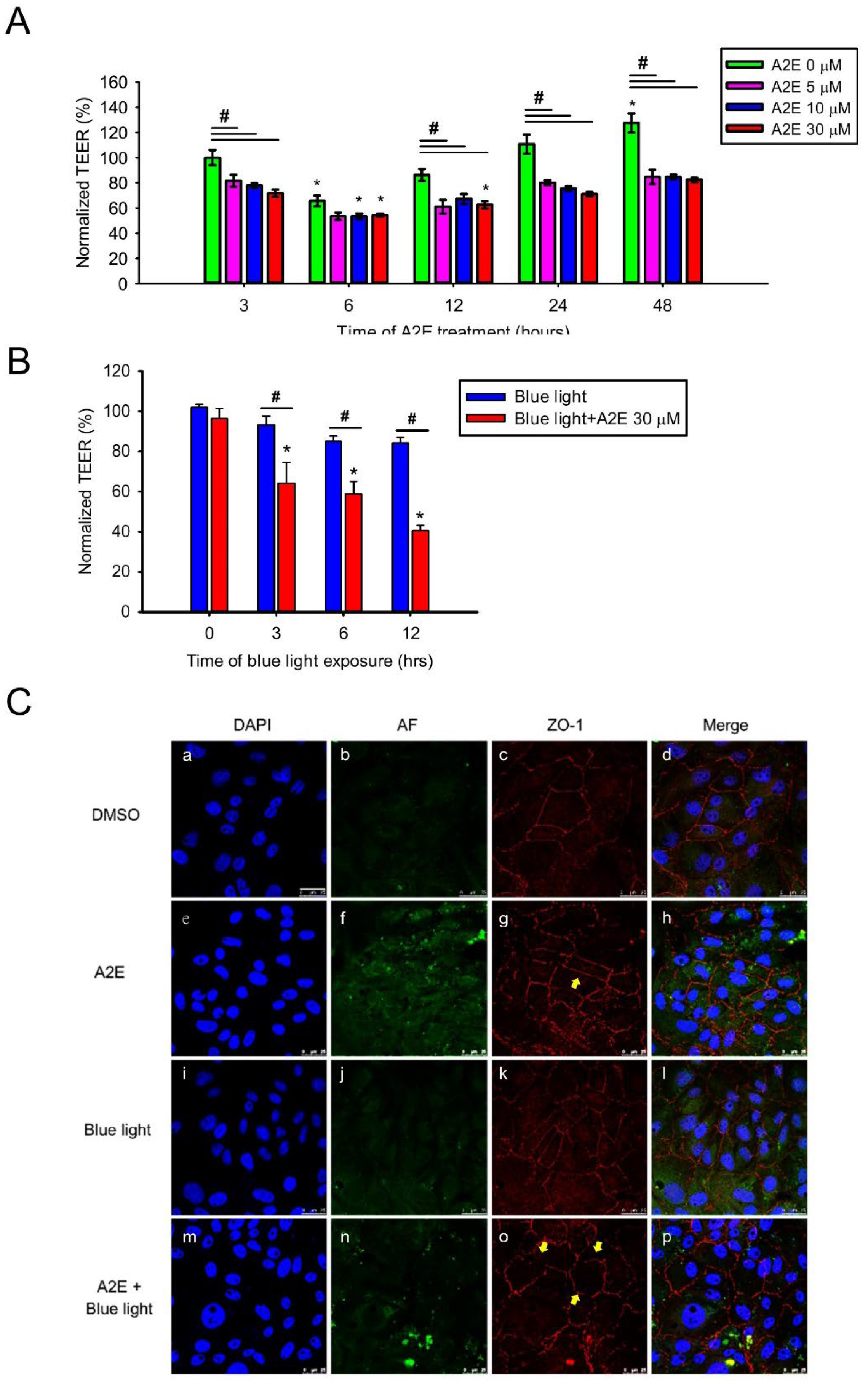
2.2. A2E Reduces Tight Junction Protein Expression and Increases Paracellular Permeability in RPE Cells
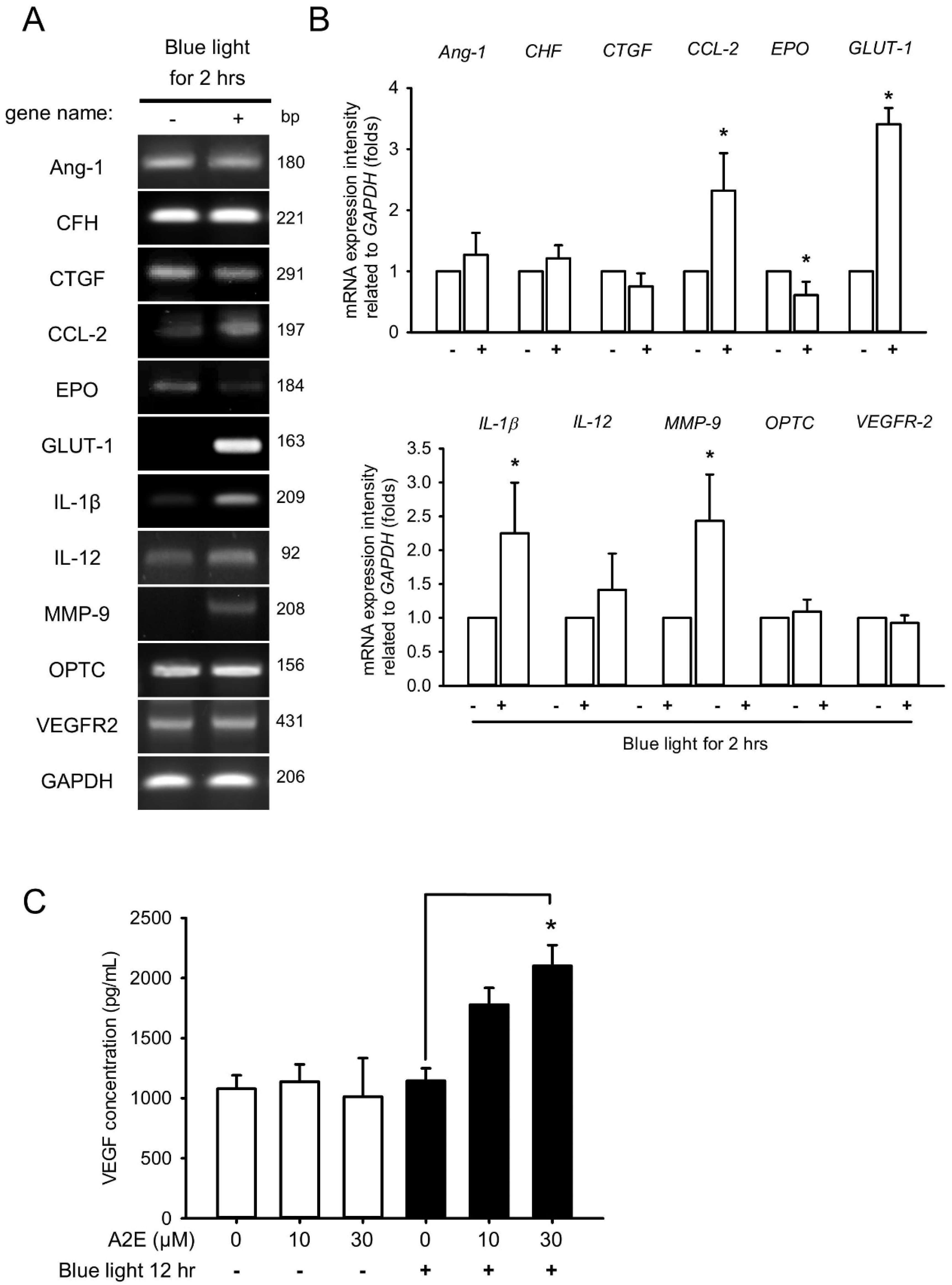
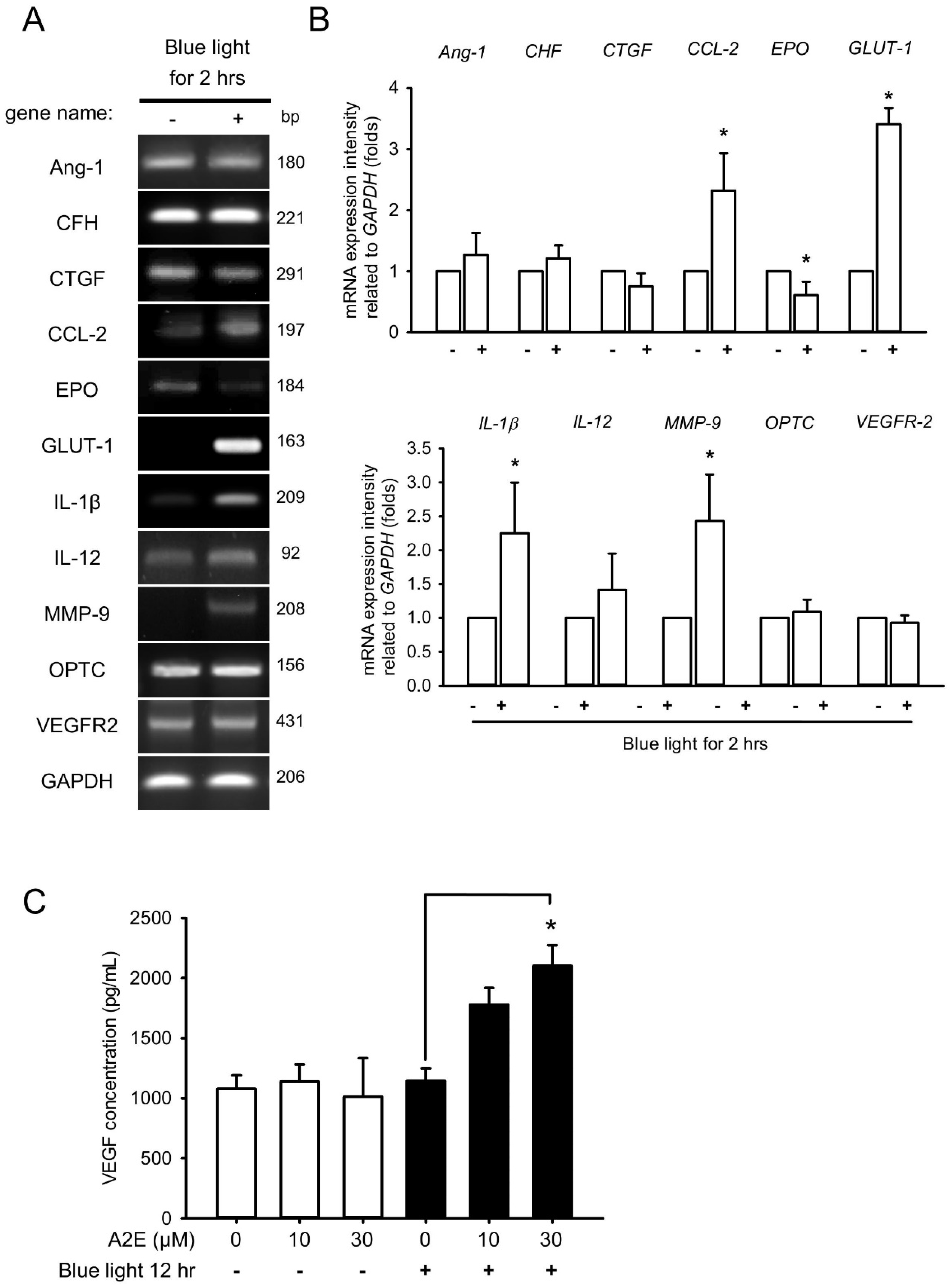
2.3. Blue Light Induces Early Inflammation and Angiogenesis in RPE Cells and Increases VEGF Levels in A2E-Laden RPE Cells
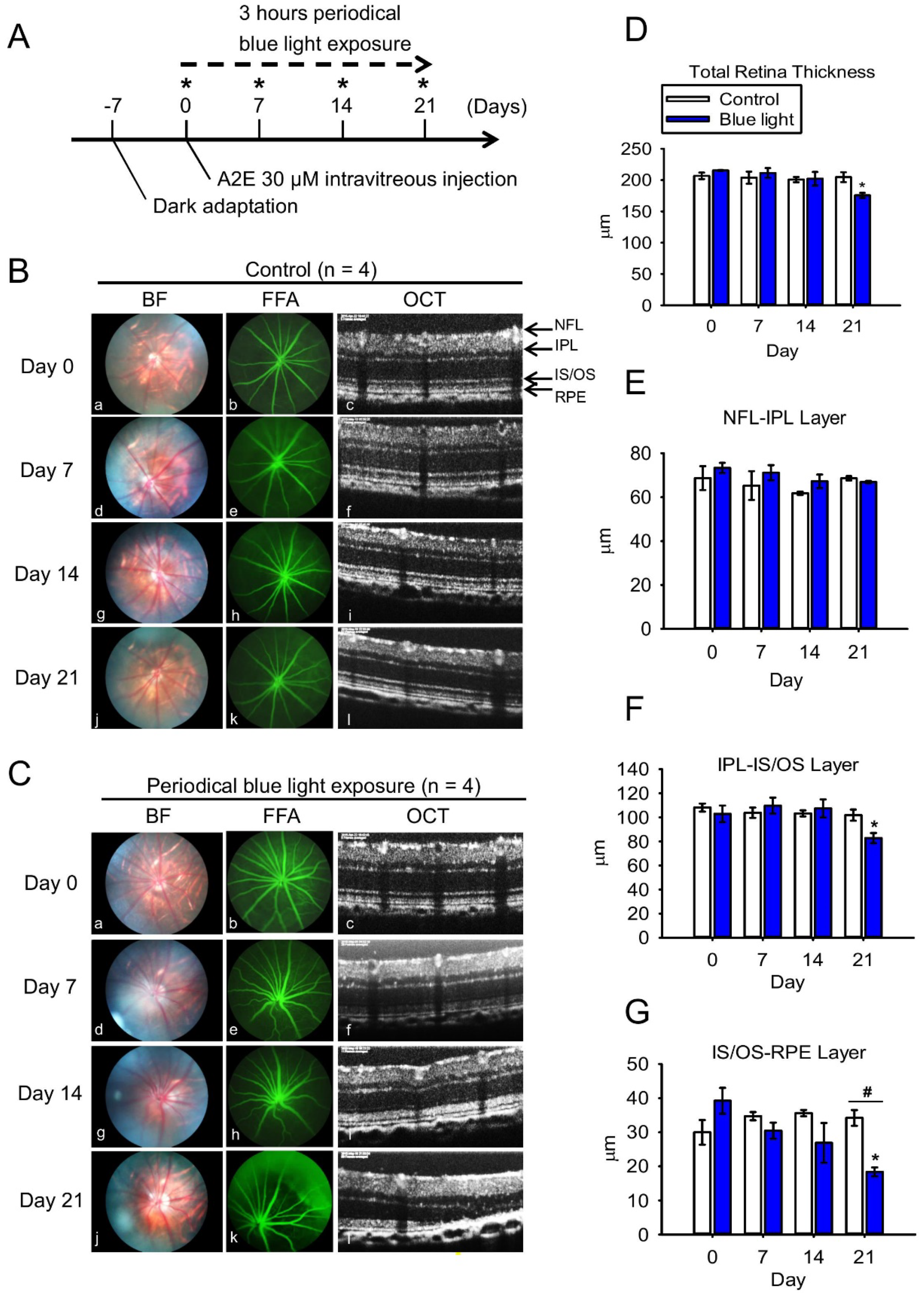
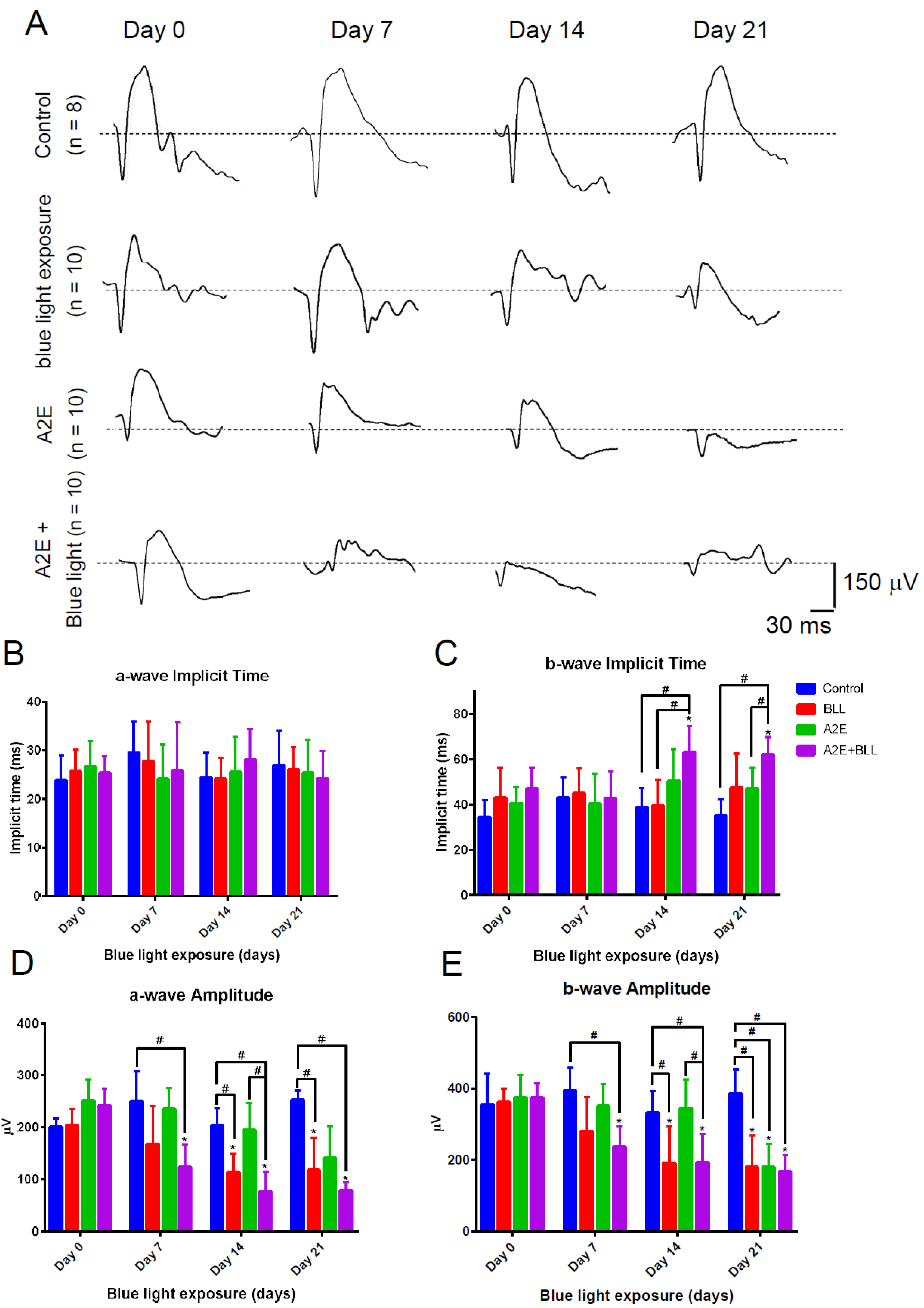
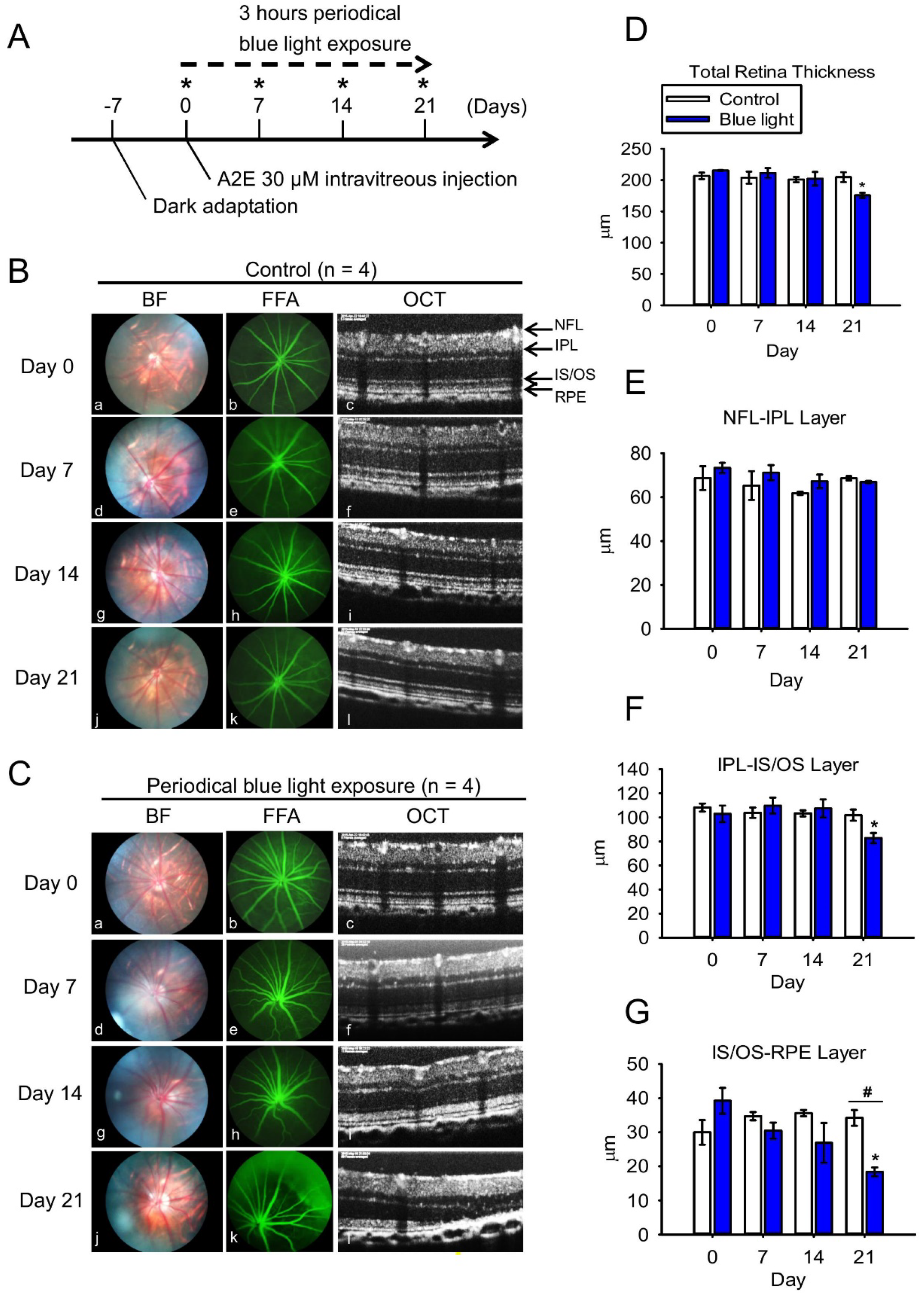
2.4. Blue Light Reduces Retinal Thickness and Damages Retinal Neuron Transduction in BN Rats
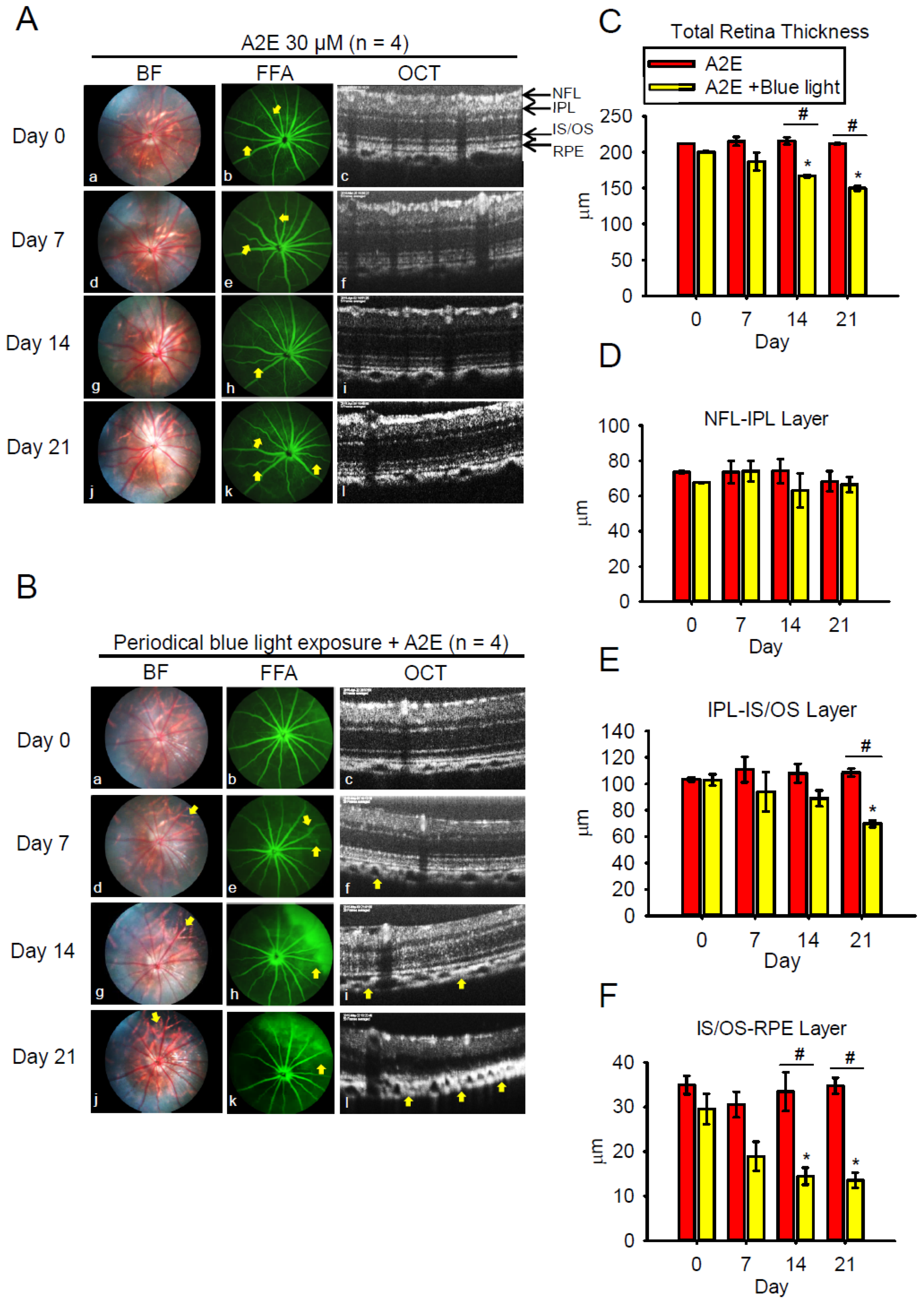
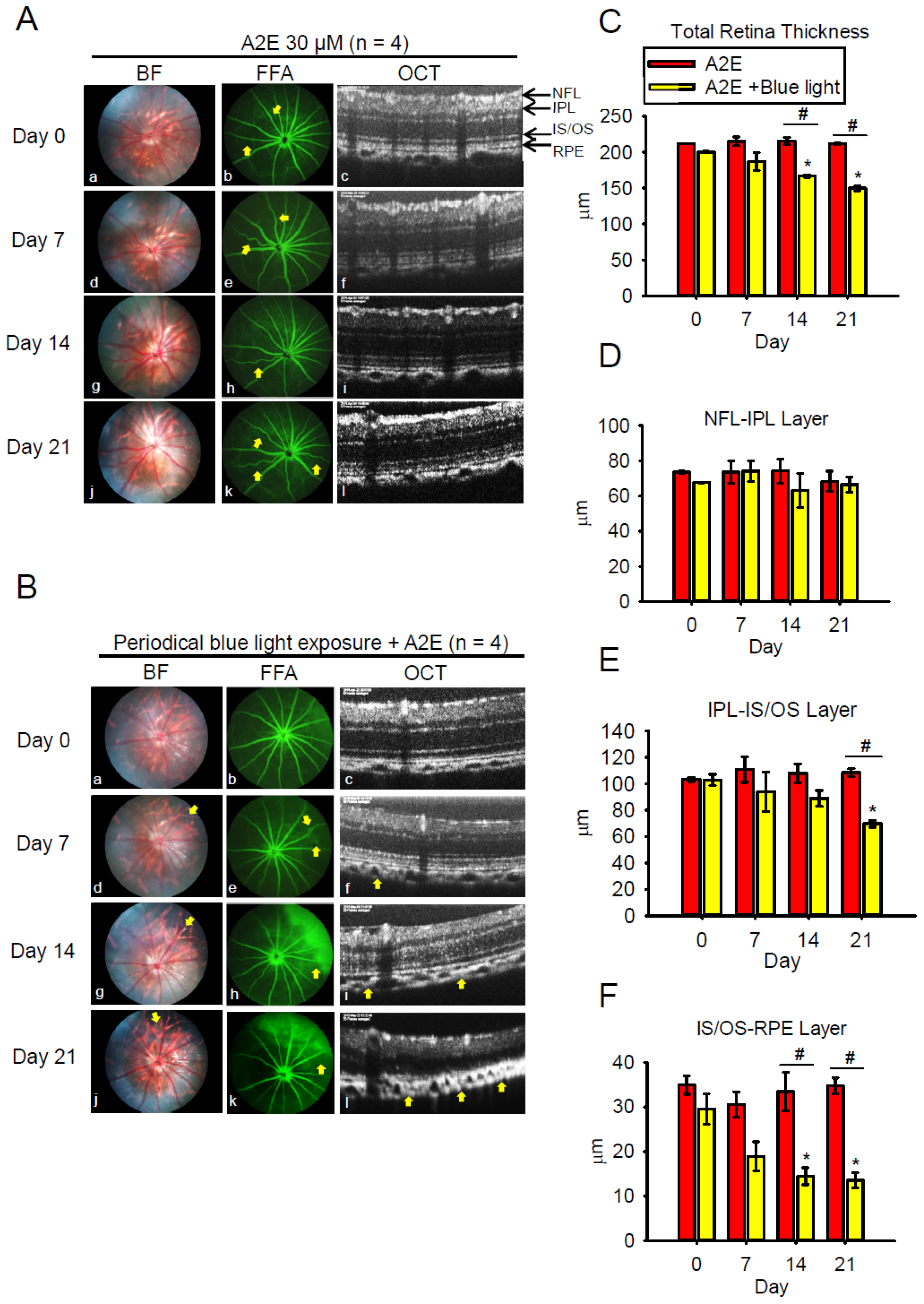
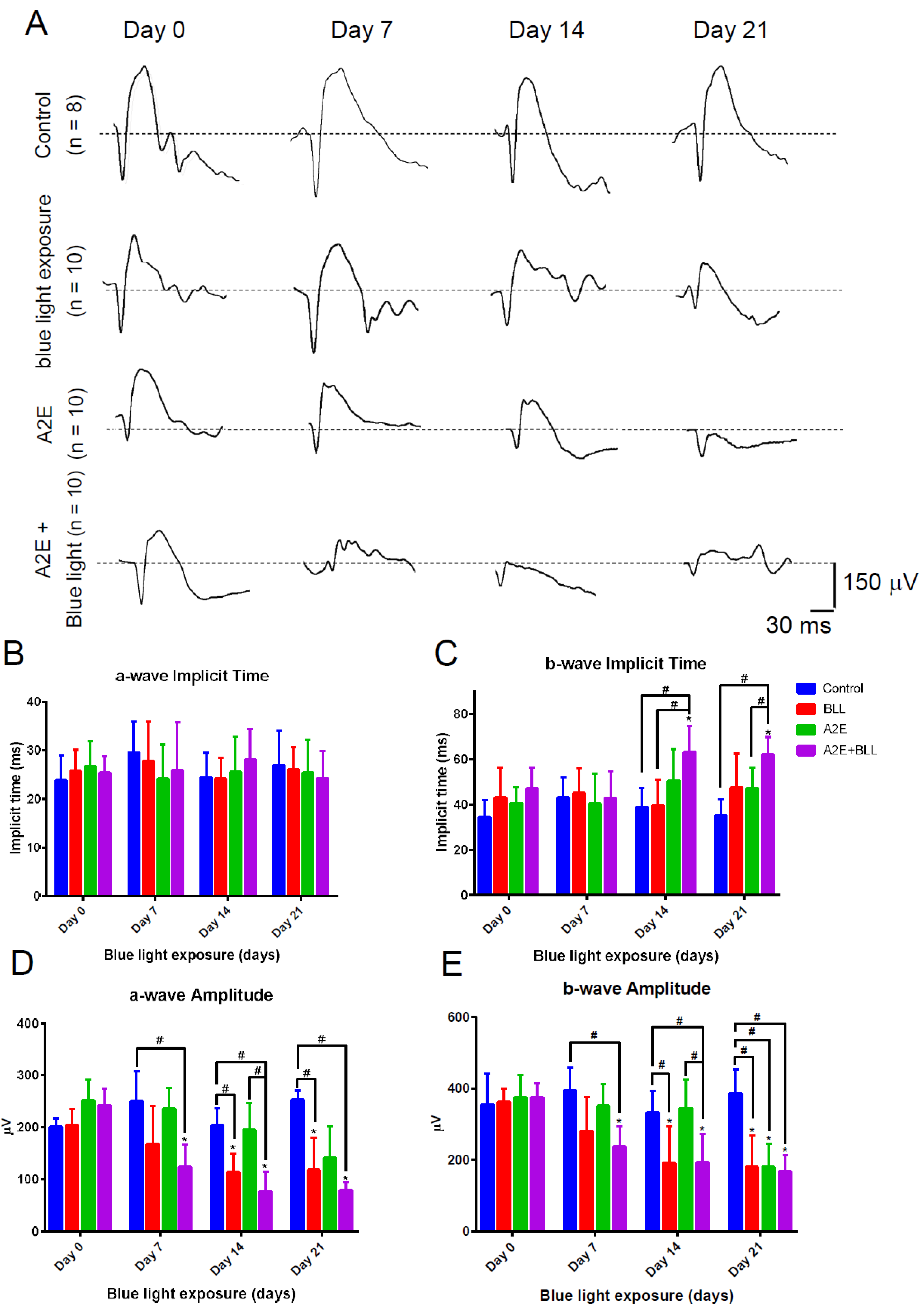
2.5. Blue Light Enhances Retinal Neuron Transduction Injury in A2E-Treated BN Rats
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Synthesis of A2E and Treatment of RPE Cells
4.3. In Vitro Exposure Protocols
4.4. Cell Viability Assay
4.5. Immunofluorescence
4.6. Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
4.7. Trans-Epithelial Electrical Resistance (TEER) Measurements in RPE Cells
4.8. VEGF Quantification
4.9. Animal Handling
4.10. In Vivo Exposure Protocols
4.11. Fundus Images and Fundus Angiography Experiments
4.12. Spectral-Domain Optical Coherence Tomography (SD-OCT)
4.13. Electroretinographic (ERG) Recording
4.14. Data and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shi, Q.; Wang, Q.; Li, J.; Zhou, X.; Fan, H.; Wang, F.; Liu, H.; Sun, X.; Sun, X. A2E Suppresses Regulatory Function of RPE Cells in Th1 Cell Differentiation Via Production of IL-1beta and Inhibition of PGE2. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7728–7738. [Google Scholar] [CrossRef]
- Stiles, M.; Moiseyev, G.P.; Budda, M.L.; Linens, A.; Brush, R.S.; Qi, H.; White, G.L.; Wolf, R.F.; Ma, J.-X.; Floyd, R.; et al. PBN (Phenyl-N-Tert-Butylnitrone)-Derivatives Are Effective in Slowing the Visual Cycle and Rhodopsin Regeneration and in Protecting the Retina from Light-Induced Damage. PLoS ONE 2015, 10, e0145305. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef]
- Hanus, J.; Zhao, F.; Wang, S. Current therapeutic developments in atrophic age-related macular degeneration. Br. J. Ophthalmol. 2016, 100, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar] [CrossRef]
- Liang, F.Q.; Godley, B.F. Oxidative stress-induced mitochondrial DNA damage in human retinal pigment epithelial cells: A possible mechanism for RPE aging and age-related macular degeneration. Exp. Eye Res. 2003, 76, 397–403. [Google Scholar] [CrossRef]
- Bian, Q.; Gao, S.; Zhou, J.; Qin, J.; Taylor, A.; Johnson, E.J.; Tang, G.; Sparrow, J.R.; Gierhart, D.; Shang, F. Lutein and zeaxanthin supplementation reduces photooxidative damage and modulates the expression of inflammation-related genes in retinal pigment epithelial cells. Free Radic. Biol. Med. 2012, 53, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shabat, S.; Itagaki, Y.; Jockusch, S.; Sparrow, J.R.; Turro, N.J.; Nakanishi, K. Formation of a nonaoxirane from A2E, a lipofuscin fluorophore related to macular degeneration, and evidence of singlet oxygen involvement. Angew. Chem. 2002, 41, 814–817. [Google Scholar] [CrossRef]
- Jang, Y.P.; Matsuda, H.; Itagaki, Y.; Nakanishi, K.; Sparrow, J.R. Characterization of peroxy-A2E and furan-A2E photooxidation products and detection in human and mouse retinal pigment epithelial cell lipofuscin. J. Biol. Chem. 2005, 280, 39732–39739. [Google Scholar] [CrossRef]
- Dillon, J.; Zheng, L.; Merriam, J.C.; Gaillard, E.R. Transmission spectra of light to the mammalian retina. Photochem. Photobiol. 2000, 71, 225–229. [Google Scholar] [CrossRef]
- Thao, M.T.; Renfus, D.J.; Dillon, J.; Gaillard, E.R. A2E-mediated photochemical modification to fibronectin and its implications to age-related changes in Bruch’s membrane. Photochem. Photobiol. 2014, 90, 329–334. [Google Scholar] [CrossRef]
- Lu, B.; Zhang, P.; Zhou, M.; Wang, W.; Gu, Q.; Feng, J.; Luo, X.; Sun, X.; Wang, F.; Sun, X. Involvement of XBP1s in Blue Light-Induced A2E-Containing Retinal Pigment Epithelium Cell Death. Ophthalmic Res. 2017, 57, 252–262. [Google Scholar] [CrossRef]
- Kernt, M.; Neubauer, A.S.; Liegl, R.; Eibl, K.H.; Alge, C.S.; Lackerbauer, C.A.; Ulbig, M.W.; Kampik, A. Cytoprotective effects of a blue light-filtering intraocular lens on human retinal pigment epithelium by reducing phototoxic effects on vascular endothelial growth factor-alpha, Bax, and Bcl-2 expression. J. Cataract Refract. Surg. 2009, 35, 354–362. [Google Scholar] [CrossRef]
- Wang, L.; Cai, S.; Wu, Z.; Gong, X.; Lyu, J.; Su, G.; Wang, L. The correlation between the concentrations of VEGF and PEDF and Ca2+-PKC signaling pathways in human retinal pigment epithelial cells cultured in vitro after exposuring to blue light. [Zhonghua yan ke za zhi] Chin. J. Ophthalmol. 2015, 51, 839–843. [Google Scholar]
- Cachafeiro, M.; Bemelmans, A.-P.; Samardzija, M.; Afanasieva, T.; Pournaras, J.-A.; Grimm, C.; Kostic, C.; Philippe, S.; Wenzel, A.; Arsenijevic, Y.; et al. Hyperactivation of retina by light in mice leads to photoreceptor cell death mediated by VEGF and retinal pigment epithelium permeability. Cell Death Dis. 2013, 4, e781. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Obert, E.; Strauss, R.; Brandon, C.; Grek, C.; Ghatnekar, G.; Gourdie, R.; Rohrer, B. Targeting the tight junction protein, zonula occludens-1, with the connexin43 mimetic peptide, alphaCT1, reduces VEGF-dependent RPE pathophysiology. J. Mol. Med. 2017, 95, 535–552. [Google Scholar] [CrossRef] [PubMed]
- Ablonczy, Z.; Higbee, D.; Anderson, D.M.; Dahrouj, M.; Grey, A.C.; Gutierrez, D.; Koutalos, Y.; Schey, K.L.; Hanneken, A.; Crouch, R.K. Lack of Correlation Between the Spatial Distribution of A2E and Lipofuscin Fluorescence in the Human Retinal Pigment Epithelium. Investig. Opthalmol. Vis. Sci. 2013, 54, 5535–5542. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Zhao, J.; Kim, H.J.; Sparrow, J.R. Photodegradation of retinal bisretinoids in mouse models and implications for macular degeneration. Proc. Natl. Acad. Sci. USA 2016, 113, 6904–6909. [Google Scholar] [CrossRef] [PubMed]
- Algvere, P.V.; Kvanta, A.; Seregard, S. Drusen maculopathy: A risk factor for visual deterioration. Acta Ophthalmol. 2016, 94, 427–433. [Google Scholar] [CrossRef]
- Crabb, J.W. The proteomics of drusen. Cold Spring Harb. Perspect. Med. 2014, 4, a017194. [Google Scholar] [CrossRef]
- Anderson, O.A.; Finkelstein, A.; Shima, D.T. A2E induces IL-1ss production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS ONE 2013, 8, e67263. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Parish, C.A.; Hashimoto, M.; Nakanishi, K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Investig. Ophthalmol. Vis. Sci. 1999, 40, 2988–2995. [Google Scholar]
- Poliakov, E.; Strunnikova, N.V.; Jiang, J.-K.; Martinez, B.; Parikh, T.; Lakkaraju, A.; Thomas, C.; Brooks, B.P.; Redmond, T.M. Multiple A2E treatments lead to melanization of rod outer segment–challenged ARPE-19 cells. Mol. Vis. 2014, 20, 285–300. [Google Scholar]
- O’Hagan, J.B.; Khazova, M.; Price, L.L. Low-energy light bulbs, computers, tablets and the blue light hazard. Eye 2016, 30, 230–233. [Google Scholar] [CrossRef]
- Tosini, G.; Ferguson, I.; Tsubota, K. Effects of blue light on the circadian system and eye physiology. Mol. Vis. 2016, 22, 61–72. [Google Scholar]
- Tokarz, P.; Kaarniranta, K.; Blasiak, J. Role of antioxidant enzymes and small molecular weight antioxidants in the pathogenesis of age-related macular degeneration (AMD). Biogerontology 2013, 14, 461–482. [Google Scholar] [CrossRef]
- Luthra, S.; Fardin, B.; Dong, J.; Hertzog, D.; Kamjoo, S.; Gebremariam, S.; Butani, V.; Narayanan, R.; Mungcal, J.K.; Kuppermann, B.D.; et al. Activation of Caspase-8 and Caspase-12 Pathways by 7-Ketocholesterol in Human Retinal Pigment Epithelial Cells. Investig. Opthalmol. Vis. Sci. 2006, 47, 5569–5575. [Google Scholar] [CrossRef]
- Lin, C.H.; Wu, M.R.; Li, C.H.; Cheng, H.W.; Huang, S.H.; Tsai, C.H.; Lin, F.L.; der Ho, J.; Kang, J.J.; Hsiao, G.; et al. Periodic exposure to smartphone-mimic low-luminance blue light induces retina damage through Bcl-2/BAX-dependent apoptosis. Toxicol. Sci. 2017, 157, 196–210. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Cai, B. Blue light-induced apoptosis of A2E-containing RPE: Involvement of caspase-3 and protection by Bcl-2. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1356–1362. [Google Scholar]
- Sparrow, J.R.; Zhou, J.; Ben-Shabat, S.; Vollmer, H.; Itagaki, Y.; Nakanishi, K. Involvement of oxidative mechanisms in blue-light-induced damage to A2E-laden RPE. Investig. Ophthalmol. Vis. Sci. 2002, 43, 1222–1227. [Google Scholar]
- London, A.; Benhar, I.; Schwartz, M. The retina as a window to the brain-from eye research to CNS disorders. Nat. Rev. Neurol. 2013, 9, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Hageman, G.S.; Luthert, P.J.; Victor Chong, N.H.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef]
- Neumann, E.; Schaefer-Ridder, M.; Wang, Y.; Hofschneider, P.H. Gene transfer into mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982, 1, 841–845. [Google Scholar] [CrossRef] [PubMed]
- De, S.; Sakmar, T.P. Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol. 2002, 120, 147–157. [Google Scholar] [CrossRef]
- Liu, J.; Itagaki, Y.; Ben-Shabat, S.; Nakanishi, K.; Sparrow, J.R. The biosynthesis of A2E, a fluorophore of aging retina, involves the formation of the precursor, A2-PE, in the photoreceptor outer segment membrane. J. Biol. Chem. 2000, 275, 29354–29360. [Google Scholar] [CrossRef] [PubMed]
- Bavik, C.; Henry, S.H.; Zhang, Y.; Mitts, K.; McGinn, T.; Budzynski, E.; Pashko, A.; Lieu, K.L.; Zhong, S.; Blumberg, B.; et al. Visual Cycle Modulation as an Approach toward Preservation of Retinal Integrity. PLoS ONE 2015, 10, e0124940. [Google Scholar] [CrossRef]
- Jin, H.L.; Lee, S.C.; Kwon, Y.S.; Choung, S.Y.; Jeong, K.W. A novel fluorescence-based assay for measuring A2E removal from human retinal pigment epithelial cells to screen for age-related macular degeneration inhibitors. J. Pharm. Biomed. Anal. 2016, 117, 560–567. [Google Scholar] [CrossRef]
- Narimatsu, T.; Ozawa, Y.; Miyake, S.; Kubota, S.; Hirasawa, M.; Nagai, N.; Shimmura, S.; Tsubota, K. Disruption of Cell-Cell Junctions and Induction of Pathological Cytokines in the Retinal Pigment Epithelium of Light-Exposed Mice. Investig. Opthalmol. Vis. Sci. 2013, 54, 4555–4562. [Google Scholar] [CrossRef]
- Salcedo, R.; Ponce, M.L.; A Young, H.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Oppenheim, J.J.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar]
- Rutar, M.; Natoli, R.; Provis, J.M. Small interfering RNA-mediated suppression of Ccl2 in Muller cells attenuates microglial recruitment and photoreceptor death following retinal degeneration. J. Neuroinflamm. 2012, 9, 221. [Google Scholar] [CrossRef]
- Nakaigawa, N.; Kondo, K.; Ueno, D.; Namura, K.; Makiyama, K.; Kobayashi, K.; Shioi, K.; Ikeda, I.; Kishida, T.; Kaneta, T.; et al. The acceleration of glucose accumulation in renal cell carcinoma assessed by FDG PET/CT demonstrated acquisition of resistance to tyrosine kinase inhibitor therapy. BMC Cancer 2017, 17, 39. [Google Scholar] [CrossRef]
- Tochio, T.; Tanaka, H.; Nakata, S. Glucose transporter member 1 is involved in UVB-induced epidermal hyperplasia by enhancing proliferation in epidermal keratinocytes. Int. J. Dermatol. 2013, 52, 300–308. [Google Scholar] [CrossRef]
- Weinkauf, B.; Rukwied, R.; Quiding, H.; Dahllund, L.; Johansson, P.; Schmelz, M. Local gene expression changes after UV-irradiation of human skin. PLoS ONE 2012, 7, e39411. [Google Scholar] [CrossRef]
- Iriyama, A.; Inoue, Y.; Takahashi, H.; Tamaki, Y.; Jang, W.D.; Yanagi, Y. A2E, a component of lipofuscin, is pro-angiogenic in vivo. J. Cell. Physiol. 2009, 220, 469–475. [Google Scholar] [CrossRef]
- Maeda, A.; Golczak, M.; Maeda, T.; Palczewski, K. Limited roles of Rdh8, Rdh12, and Abca4 in all-trans-retinal clearance in mouse retina. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5435–5443. [Google Scholar] [CrossRef]
- Parish, C.A.; Hashimoto, M.; Nakanishi, K.; Dillon, J.; Sparrow, J. Isolation and one-step preparation of A2E and iso-A2E, fluorophores from human retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 1998, 95, 14609–14613. [Google Scholar] [CrossRef]
- Moiseyev, G.; Nikolaeva, O.; Chen, Y.; Farjo, K.; Takahashi, Y.; Ma, J.X. Inhibition of the visual cycle by A2E through direct interaction with RPE65 and implications in Stargardt disease. Proc. Natl. Acad. Sci. USA 2010, 107, 17551–17556. [Google Scholar] [CrossRef]
- Liao, P.-L.; Li, C.-H.; Huang, S.-H.; Tsai, C.-H.; Wu, M.-R.; Ho, J.-D.; Lin, C.-H.; Hsiao, G.; Cheng, H.-W. Long-term Fluorometholone Topical Use Induces Ganglion Cell Damage in Rats Analyzed With Optical Coherence Tomography. Toxicol. Sci. 2015, 147, 317–325. [Google Scholar]
- Lin, C.; Li, C.; Liao, P.; Tse, L.; Huang, W.; Cheng, H.; Cheng, Y. Silibinin inhibits VEGF secretion and age-related macular degeneration in a hypoxia-dependent manner through the PI-3 kinase/Akt/mTOR pathway. Br. J. Pharmacol. 2013, 168, 920–931. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, C.-H.; Wu, M.-R.; Huang, W.-J.; Chow, D.S.-L.; Hsiao, G.; Cheng, Y.-W. Low-Luminance Blue Light-Enhanced Phototoxicity in A2E-Laden RPE Cell Cultures and Rats. Int. J. Mol. Sci. 2019, 20, 1799. https://doi.org/10.3390/ijms20071799
Lin C-H, Wu M-R, Huang W-J, Chow DS-L, Hsiao G, Cheng Y-W. Low-Luminance Blue Light-Enhanced Phototoxicity in A2E-Laden RPE Cell Cultures and Rats. International Journal of Molecular Sciences. 2019; 20(7):1799. https://doi.org/10.3390/ijms20071799
Chicago/Turabian StyleLin, Cheng-Hui, Man-Ru Wu, Wei-Jan Huang, Diana Shu-Lian Chow, George Hsiao, and Yu-Wen Cheng. 2019. "Low-Luminance Blue Light-Enhanced Phototoxicity in A2E-Laden RPE Cell Cultures and Rats" International Journal of Molecular Sciences 20, no. 7: 1799. https://doi.org/10.3390/ijms20071799
APA StyleLin, C.-H., Wu, M.-R., Huang, W.-J., Chow, D. S.-L., Hsiao, G., & Cheng, Y.-W. (2019). Low-Luminance Blue Light-Enhanced Phototoxicity in A2E-Laden RPE Cell Cultures and Rats. International Journal of Molecular Sciences, 20(7), 1799. https://doi.org/10.3390/ijms20071799