Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes



Abstract
1. Introduction
2. Results
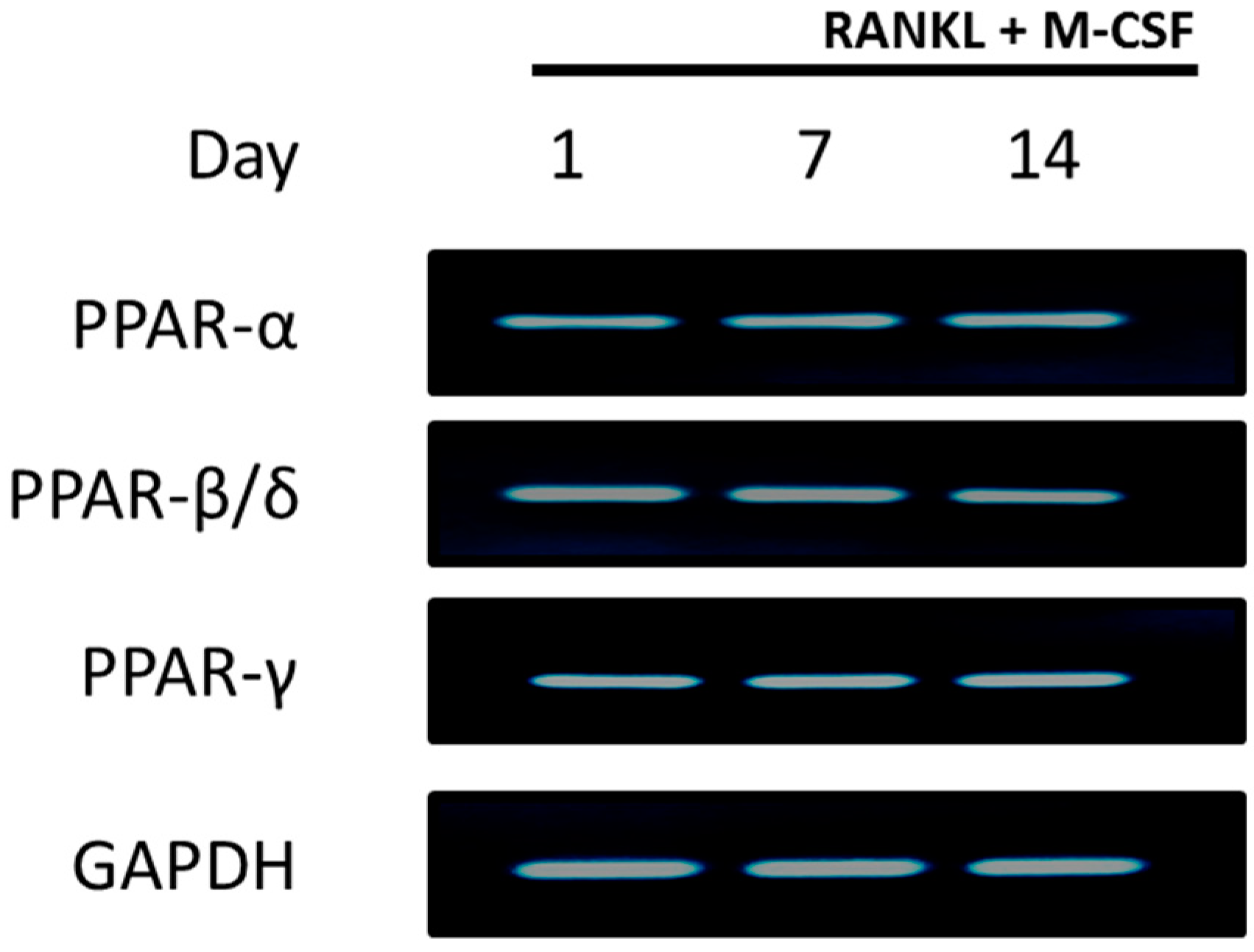
2.1. PPARs are Expressed in CD14+ Monocytes
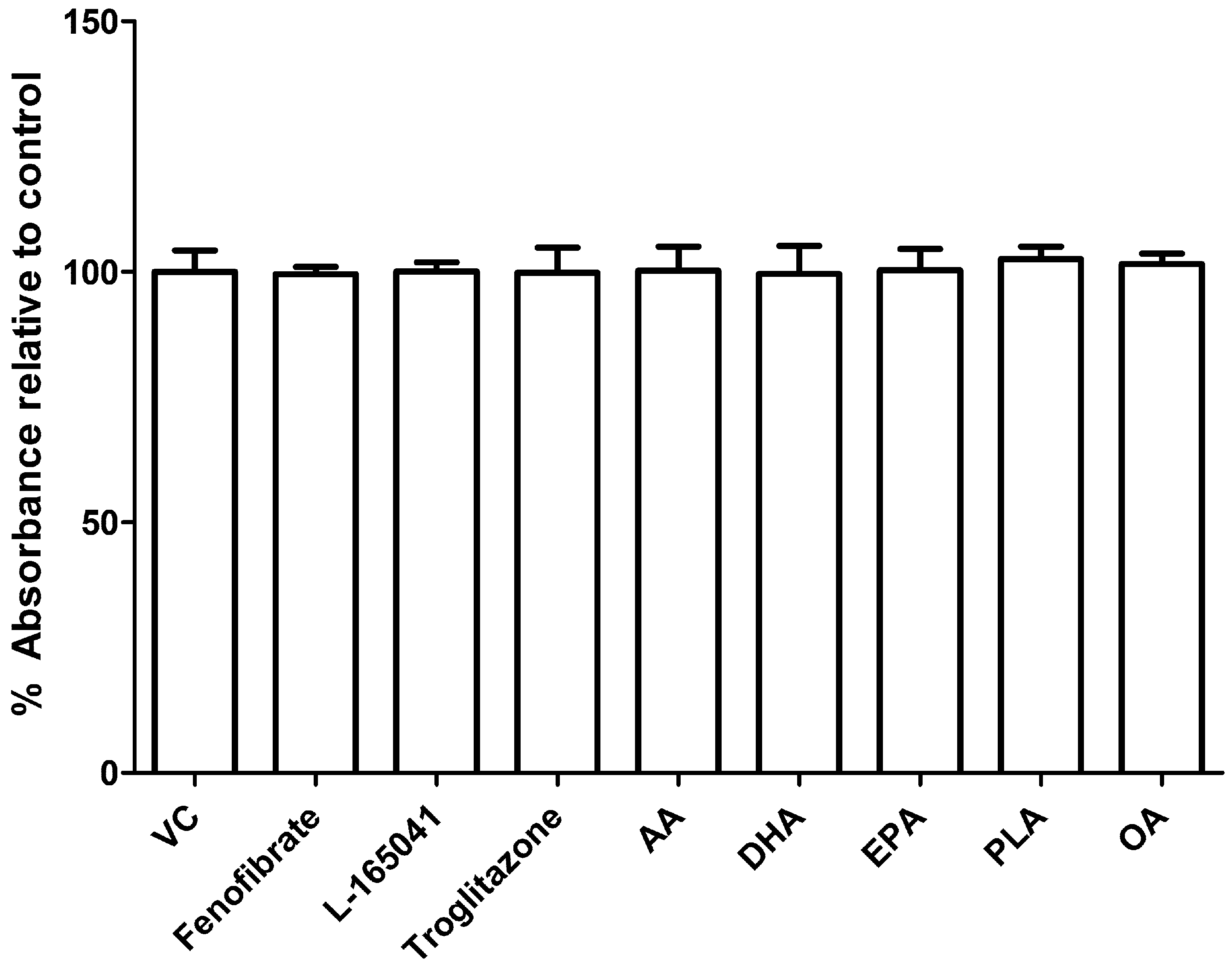
2.2. UFAs and PPAR Agonists do not Affect Cell Viability in Human CD14+ Monocytes
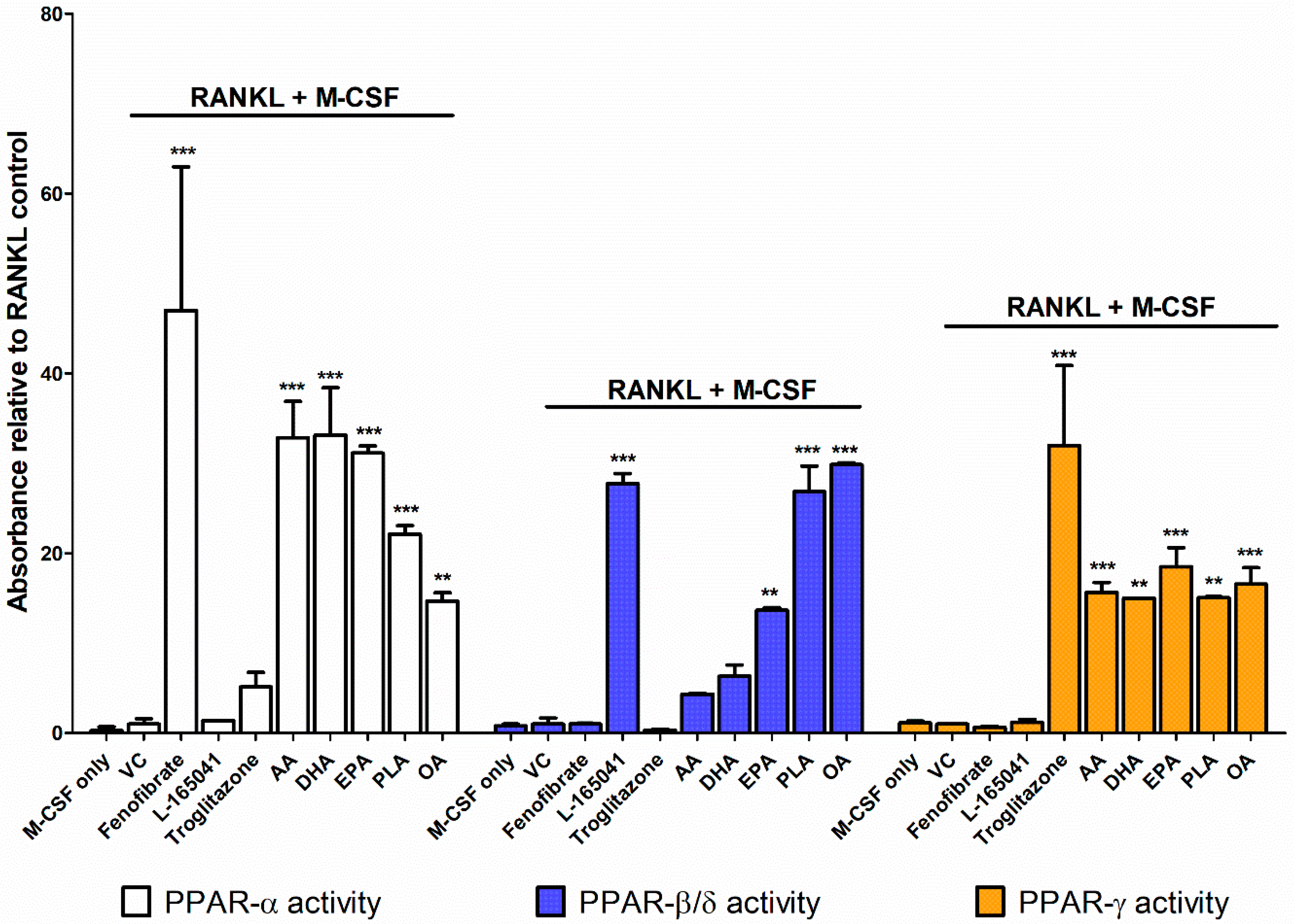
2.3. PPARs are Differentially Activated by Unsaturated Fatty Acids
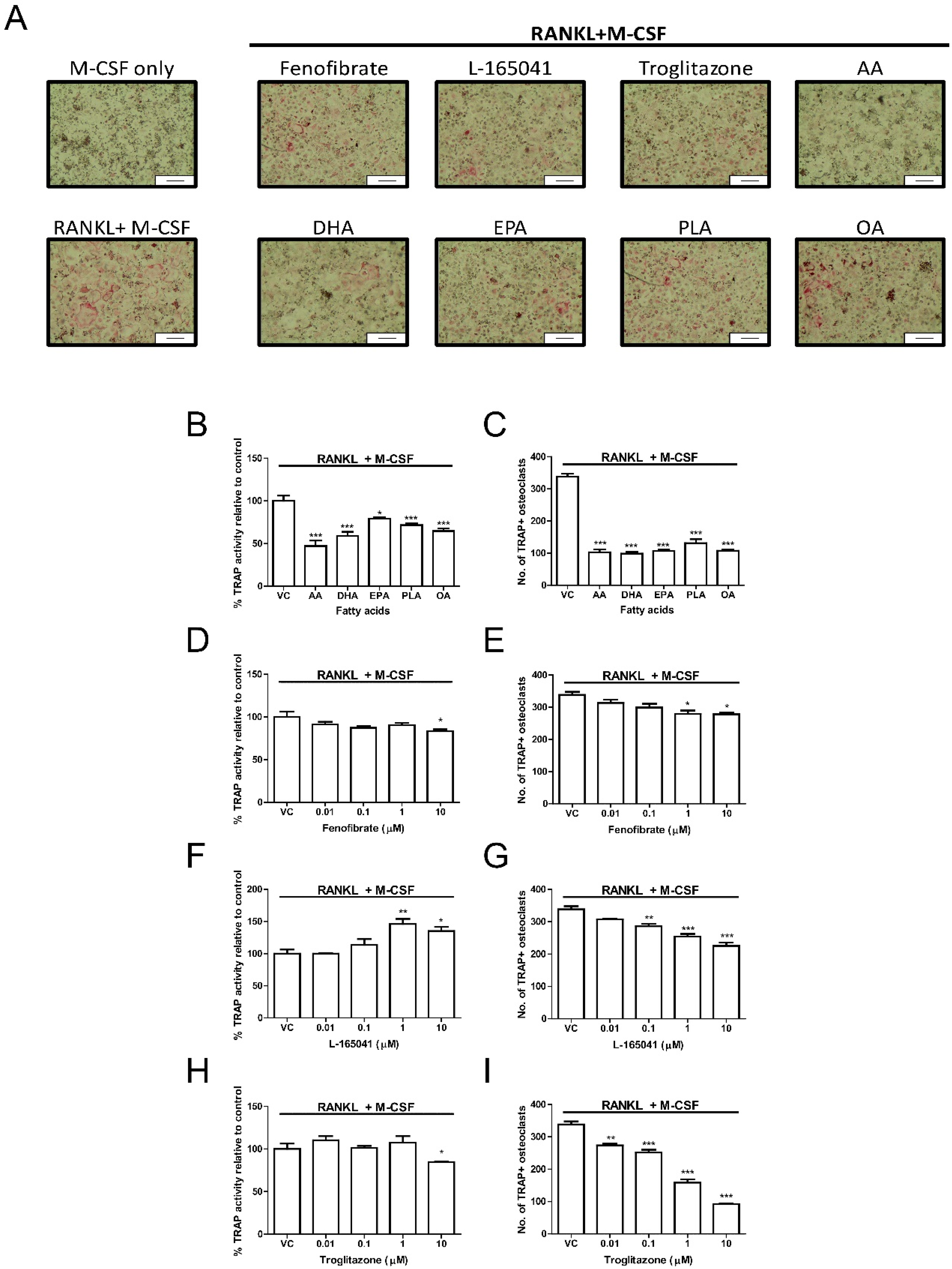
2.4. PPAR Agonists Modulate Osteoclast Formation
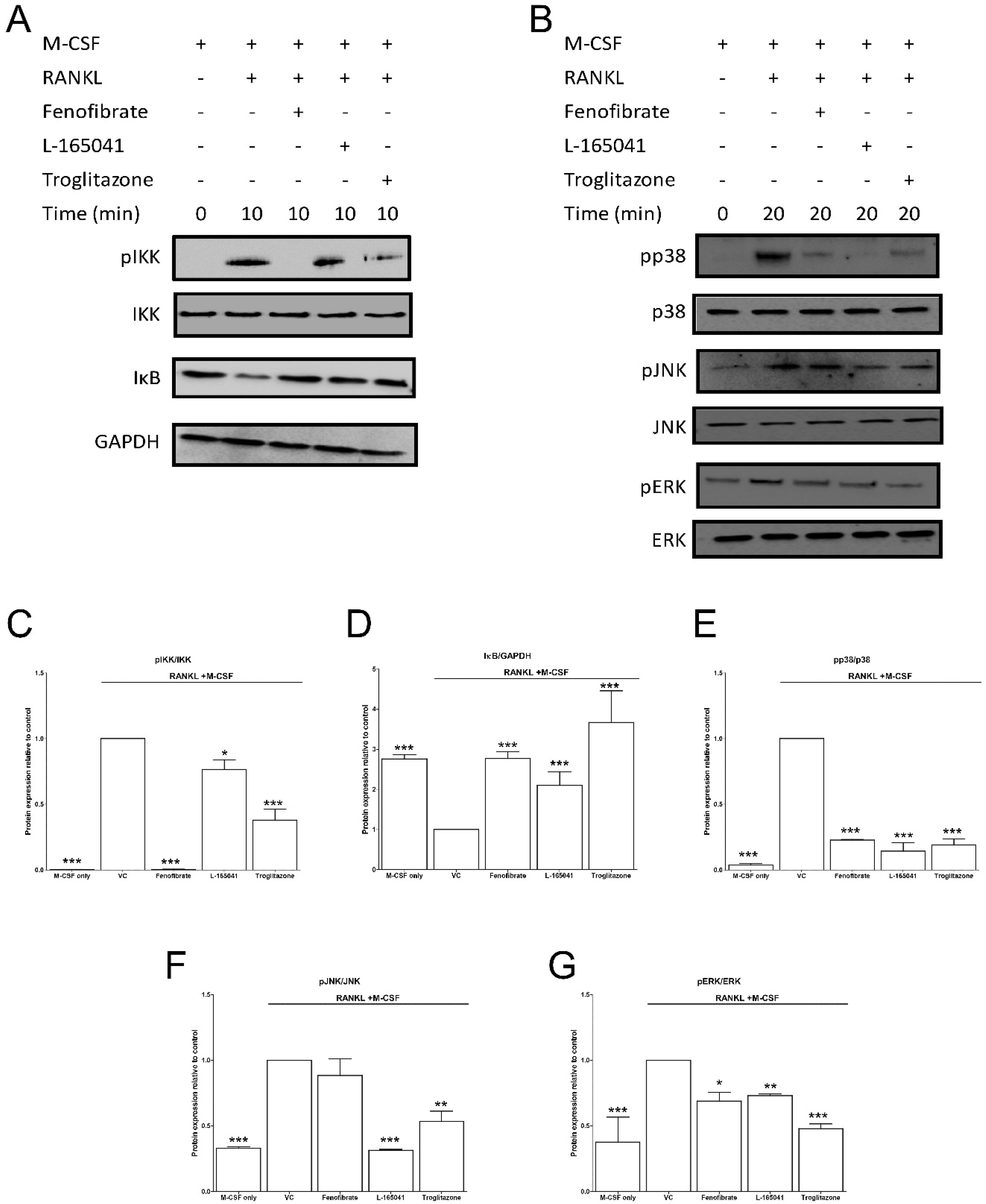
2.5. PPAR Agonists Modulate RANKL Signalling in CD14+ Monocytes
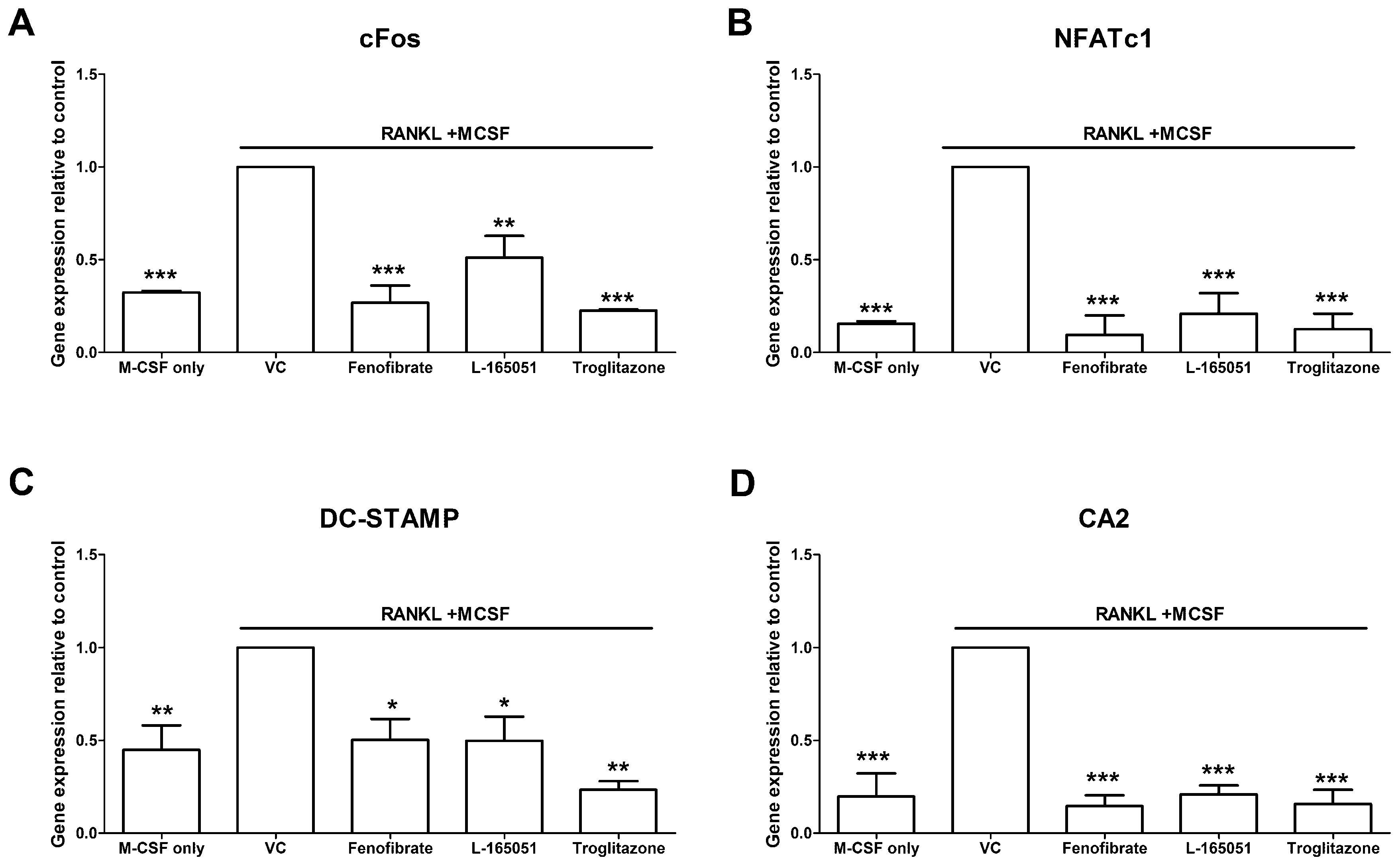
2.6. PPAR Agonists Modulate Osteoclast Specific Gene Expression
3. Discussion
4. Materials and Methods
4.1. Reagents and Materials
4.2. Preparation of Fatty Acids and Agonists
4.3. Ethics Statement
4.4. CD14+ Monocyte Isolation
4.5. PPAR Expression
4.6. Resazurin Assay
4.7. PPAR Activation Assay
4.7.1. Nuclear Fractionation
4.7.2. PPAR Assay
4.8. Differentiation of CD14+ Monocytes
4.8.1. TRAP Activity
4.8.2. TRAP Stain
4.9. Western Blot
4.10. Quantitative PCR
4.11. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AA | Arachidonic acid |
| CA2 | Carbonic anhydrase 2 |
| DC-STAMP | Dendritic cell-specific transmembrane protein |
| DHA | Docosahexaenoic acid |
| EPA | Eicosapentaenoic acid |
| ERK | Extracellular signal–regulated kinase |
| IκB | Inhibitor of κB |
| IKK | Inhibitor of κB kinase |
| JNK | c-Jun N-terminal kinase |
| MAPK | Mitogen activated protein kinases |
| M-CSF | Macrophage colony stimulating factor |
| MUFA | Monounsaturated fatty acid |
| NF-κB | Nuclear factor κB |
| NFATc1 | Nuclear factor of activated T-cells, cytoplasmic 1 |
| OA | Oleic acid |
| PBMC | Peripheral blood mononuclear cells |
| PLA | Palmitoleic acid |
| PPAR | Peroxisome proliferator activated receptor |
| PUFA | Polyunsaturated fatty acid |
| RANK | Receptor activator of nuclear factor κB |
| RANKL | Receptor activator of nuclear factor κB ligand |
| TRAP | Tartrate resistant acid phosphatase |
| UFA | Unsaturated fatty acid |
References
- Vaananen, H.K.; Zhao, H.; Mulari, M.; Halleen, J.M. The cell biology of osteoclast function. J. Cell Sci. 2000, 113 Pt 3, 377–381. [Google Scholar]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. Soc. Nephrol. 2008, 3 (Suppl. 3), S131–S139. [Google Scholar] [CrossRef]
- Poole, K.E.; Compston, J.E. Osteoporosis and its management. BMJ 2006, 333, 1251–1256. [Google Scholar] [CrossRef]
- Bar-Shavit, Z. The osteoclast: A multinucleated, hematopoietic-origin, bone-resorbing osteoimmune cell. J. Cell. Biochem. 2007, 102, 1130–1139. [Google Scholar] [CrossRef]
- Boyce, B.F. Advances in the regulation of osteoclasts and osteoclast functions. J. Dent. Res. 2013, 92, 860–867. [Google Scholar] [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef]
- Aeschlimann, D.; Evans, B.A. The vital osteoclast: How is it regulated? Cell Death Differ. 2004, 11 (Suppl. 1), S5–S7. [Google Scholar] [CrossRef] [PubMed]
- Issemann, I.; Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990, 347, 645–650. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, M.; Baldessin, L.; Boccia, D.; Racagni, G.; Mitro, N. Non-insulin anti-diabetic drugs: An update on pharmacological interactions. Pharmacol. Res. 2017, 115, 14–24. [Google Scholar] [CrossRef]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and Safety of Pemafibrate Versus Fenofibrate in Patients with High Triglyceride and Low HDL Cholesterol Levels: A Multicenter, Placebo-Controlled, Double-Blind, Randomized Trial. J. Atheroscler. Thromb. 2018, 25, 521–538. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Yamashita, S.; Yokote, K.; Araki, E.; Suganami, H.; Ishibashi, S.; Group, K.S. Efficacy and safety of K-877, a novel selective peroxisome proliferator-activated receptor alpha modulator (SPPARMalpha), in combination with statin treatment: Two randomised, double-blind, placebo-controlled clinical trials in patients with dyslipidaemia. Atherosclerosis 2017, 261, 144–152. [Google Scholar] [CrossRef]
- Jani, R.H.; Pai, V.; Jha, P.; Jariwala, G.; Mukhopadhyay, S.; Bhansali, A.; Joshi, S. A multicenter, prospective, randomized, double-blind study to evaluate the safety and efficacy of Saroglitazar 2 and 4 mg compared with placebo in type 2 diabetes mellitus patients having hypertriglyceridemia not controlled with atorvastatin therapy (PRESS VI). Diabetes Technol. Ther. 2014, 16, 63–71. [Google Scholar]
- Cariou, B.; Zair, Y.; Staels, B.; Bruckert, E. Effects of the new dual PPAR alpha/delta agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011, 34, 2008–2014. [Google Scholar] [CrossRef]
- Botta, M.; Audano, M.; Sahebkar, A.; Sirtori, C.R.; Mitro, N.; Ruscica, M. PPAR Agonists and Metabolic Syndrome: An Established Role? Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Hu, J.P.; Yu, S.; Li, B.K.; Cui, Y.; Ren, L.; Zhang, L.D. Astragaloside IV, a Natural PPARgamma Agonist, Reduces Abeta Production in Alzheimer’s Disease Through Inhibition of BACE1. Mol. Neurobiol 2017, 54, 2939–2949. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Qiao, P.F.; Zhao, F.L.; Yan, Y. PPAR-alpha agonist regulates amyloid-beta generation via inhibiting BACE-1 activity in human neuroblastoma SH-SY5Y cells transfected with APPswe gene. Mol. Cell. Biochem. 2015, 408, 37–46. [Google Scholar] [CrossRef]
- Gou, Q.; Gong, X.; Jin, J.; Shi, J.; Hou, Y. Peroxisome proliferator-activated receptors (PPARs) are potential drug targets for cancer therapy. Oncotarget 2017, 8, 60704–60709. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Y.; Xu, Q.; Shi, H.; Shi, J.; Hou, Y. PPARdelta promotes tumor progression via activation of Glut1 and SLC1-A5 transcription. Carcinogenesis 2017, 38, 748–755. [Google Scholar] [CrossRef]
- Baker, B.G.; Ball, G.R.; Rakha, E.A.; Nolan, C.C.; Caldas, C.; Ellis, I.O.; Green, A.R. Lack of expression of the proteins GMPR2 and PPARalpha are associated with the basal phenotype and patient outcome in breast cancer. Breast Cancer Res. Treat. 2013, 137, 127–137. [Google Scholar] [CrossRef]
- Heudobler, D.; Rechenmacher, M.; Luke, F.; Vogelhuber, M.; Pukrop, T.; Herr, W.; Ghibelli, L.; Gerner, C.; Reichle, A. Peroxisome Proliferator-Activated Receptors (PPAR)gamma Agonists as Master Modulators of Tumor Tissue. Int J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Poulsen, R.C.; Moughan, P.J.; Kruger, M.C. Long-chain polyunsaturated fatty acids and the regulation of bone metabolism. Exp. Biol. Med. 2007, 232, 1275–1288. [Google Scholar] [CrossRef]
- Grey, A. Skeletal consequences of thiazolidinedione therapy. Osteoporos Int. 2008, 19, 129–137. [Google Scholar] [CrossRef]
- Cho, E.S.; Kim, M.K.; Son, Y.O.; Lee, K.S.; Park, S.M.; Lee, J.C. The effects of rosiglitazone on osteoblastic differentiation, osteoclast formation and bone resorption. Mol. Cells 2012, 33, 173–181. [Google Scholar] [CrossRef]
- Wan, Y.; Chong, L.W.; Evans, R.M. PPAR-gamma regulates osteoclastogenesis in mice. Nat. Med. 2007, 13, 1496–1503. [Google Scholar] [CrossRef]
- Chan, B.Y.; Gartland, A.; Wilson, P.J.; Buckley, K.A.; Dillon, J.P.; Fraser, W.D.; Gallagher, J.A. PPAR agonists modulate human osteoclast formation and activity in vitro. Bone 2007, 40, 149–159. [Google Scholar] [CrossRef]
- Kruger, M.C.; Coetzee, M.; Haag, M.; Weiler, H. Long-chain polyunsaturated fatty acids: Selected mechanisms of action on bone. Prog. Lipid Res. 2010, 49, 438–449. [Google Scholar] [CrossRef]
- Sauma, L.; Stenkula, K.G.; Kjolhede, P.; Stralfors, P.; Soderstrom, M.; Nystrom, F.H. PPAR-gamma response element activity in intact primary human adipocytes: Effects of fatty acids. Nutrition 2006, 22, 60–68. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Sundseth, S.S.; Jones, S.A.; Brown, P.J.; Wisely, G.B.; Koble, C.S.; Devchand, P.; Wahli, W.; Willson, T.M.; Lenhard, J.M.; et al. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. USA 1997, 94, 4318–4323. [Google Scholar] [CrossRef]
- Keller, H.; Dreyer, C.; Medin, J.; Mahfoudi, A.; Ozato, K.; Wahli, W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid X receptor heterodimers. Proc. Natl. Acad. Sci. USA 1993, 90, 2160–2164. [Google Scholar] [CrossRef]
- Pawar, A.; Jump, D.B. Unsaturated fatty acid regulation of peroxisome proliferator-activated receptor alpha activity in rat primary hepatocytes. J. Biol. Chem. 2003, 278, 35931–35939. [Google Scholar] [CrossRef]
- Grygiel-Górniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications-a review. J. Nutr. 2014, 13, e17. [Google Scholar] [CrossRef]
- Sorensen, M.G.; Henriksen, K.; Schaller, S.; Henriksen, D.B.; Nielsen, F.C.; Dziegiel, M.H.; Karsdal, M.A. Characterization of osteoclasts derived from CD14+ monocytes isolated from peripheral blood. J. Bone Miner. Metab. 2007, 25, 36–45. [Google Scholar] [CrossRef]
- Aljada, A.; Ghanim, H.; Friedman, J.; Garg, R.; Mohanty, P.; Dandona, P. Troglitazone reduces the expression of PPARgamma while stimulating that of PPARalpha in mononuclear cells in obese subjects. J. Clin. Endocrinol. Metab. 2001, 86, 3130–3133. [Google Scholar]
- Ravnskjaer, K.; Frigerio, F.; Boergesen, M.; Nielsen, T.; Maechler, P.; Mandrup, S. PPARdelta is a fatty acid sensor that enhances mitochondrial oxidation in insulin-secreting cells and protects against fatty acid-induced dysfunction. J. Lipid Res. 2010, 51, 1370–1379. [Google Scholar] [CrossRef]
- Lorenzo, J.A.; Choi, Y.; Horowitz, M.; Takayanagi, H. (Eds.) Osteoimmunology; Academic Press: London, UK, 2011. [Google Scholar]
- Zou, W.; Rohatgi, N.; Chen, T.H.; Schilling, J.; Abu-Amer, Y.; Teitelbaum, S.L. PPAR-gamma regulates pharmacological but not physiological or pathological osteoclast formation. Nat. Med. 2016, 22, 1203–1205. [Google Scholar] [CrossRef]
- Okazaki, R.; Toriumi, M.; Fukumoto, S.; Miyamoto, M.; Fujita, T.; Tanaka, K.; Takeuchi, Y. Thiazolidinediones inhibit osteoclast-like cell formation and bone resorption in vitro. Endocrinology 1999, 140, 5060–5065. [Google Scholar] [CrossRef]
- Young, P.W.; Buckle, D.R.; Cantello, B.C.; Chapman, H.; Clapham, J.C.; Coyle, P.J.; Haigh, D.; Hindley, R.M.; Holder, J.C.; Kallender, H.; et al. Identification of high-affinity binding sites for the insulin sensitizer rosiglitazone (BRL-49653) in rodent and human adipocytes using a radioiodinated ligand for peroxisomal proliferator-activated receptor gamma. J. Pharmacol. Exp. Ther. 1998, 284, 751–759. [Google Scholar]
- Patel, J.J.; Butters, O.R.; Arnett, T.R. PPAR agonists stimulate adipogenesis at the expense of osteoblast differentiation while inhibiting osteoclast formation and activity. Cell Biochem. Funct. 2014, 32, 368–377. [Google Scholar] [CrossRef]
- Hauner, H. The mode of action of thiazolidinediones. Diabetes Metab. Res. Rev. 2002, 18 (Suppl. 2), S10–S15. [Google Scholar] [CrossRef]
- Takano, M.; Otsuka, F.; Matsumoto, Y.; Inagaki, K.; Takeda, M.; Nakamura, E.; Tsukamoto, N.; Miyoshi, T.; Sada, K.E.; Makino, H. Peroxisome proliferator-activated receptor activity is involved in the osteoblastic differentiation regulated by bone morphogenetic proteins and tumor necrosis factor-alpha. Mol. Cell. Endocrinol. 2012, 348, 224–232. [Google Scholar] [CrossRef]
- Stunes, A.K.; Westbroek, I.; Gustafsson, B.I.; Fossmark, R.; Waarsing, J.H.; Eriksen, E.F.; Petzold, C.; Reseland, J.E.; Syversen, U. The peroxisome proliferator-activated receptor (PPAR) alpha agonist fenofibrate maintains bone mass, while the PPAR gamma agonist pioglitazone exaggerates bone loss, in ovariectomized rats. BMC Endocr. Disord. 2011, 11, 11. [Google Scholar] [CrossRef]
- Brun, R.P.; Tontonoz, P.; Forman, B.M.; Ellis, R.; Chen, J.; Evans, R.M.; Spiegelman, B.M. Differential activation of adipogenesis by multiple PPAR isoforms. Genes Dev. 1996, 10, 974–984. [Google Scholar] [CrossRef]
- Kasonga, A.E.; Deepak, V.; Kruger, M.C.; Coetzee, M. Arachidonic acid and docosahexaenoic acid suppress osteoclast formation and activity in human CD14+ monocytes, in vitro. PLoS ONE 2015, 10, e0125145. [Google Scholar] [CrossRef]
- van Heerden, B.; Kasonga, A.; Kruger, M.C.; Coetzee, M. Palmitoleic acid inhibits RANKL-induced osteoclastogenesis and bone resorption by suppressing NF-kappaB and MAPK signalling pathways. Nutrients 2017, 9, 441. [Google Scholar] [CrossRef]
- Drosatos-Tampakaki, Z.; Drosatos, K.; Siegelin, Y.; Gong, S.; Khan, S.; Van Dyke, T.; Goldberg, I.J.; Schulze, P.C.; Schulze-Spate, U. Palmitic acid and DGAT1 deficiency enhance osteoclastogenesis, while oleic acid-induced triglyceride formation prevents it. J. Bone Miner. Res. 2014, 29, 1183–1195. [Google Scholar] [CrossRef]
- Agrawal, A.; Gallagher, J.A.; Gartland, A. Human osteoclast culture and phenotypic characterization. Methods Mol. Biol. 2012, 806, 357–375. [Google Scholar]
- Deepak, V.; Kasonga, A.; Kruger, M.C.; Coetzee, M. Inhibitory effects of eugenol on RANKL-induced osteoclast formation via attenuation of NF-kappaB and MAPK pathways. Connect. Tissue Res. 2014, 56, 195–203. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (5′–3′) |
|---|---|---|
| GAPDH | CATGTTGCAACCGGGAAGG | CGCCCAATACGACCAAATCA |
| PPAR-α | TCATCAAGAAGACGGAGTCG | CGGTTACCTACAGCTCAGAC |
| PPAR-β/δ | GCCCTTTGTGATCCACGACA | GGATGCTCTTGGCGAACTCAG |
| PPAR-γ | ATGACAGCGACTTGGCAATA | GCAACTGGAAGAAGGGAAAT |
| Gene | Forward Primer Sequence (5′–3′) | Reverse Primer Sequence (5′–3′) |
|---|---|---|
| GAPDH | CATGTTGCAACCGGGAAGG | CGCCCAATACGACCAAATCA |
| cFos | CCCATCGCAGACCAGAGC | ATCTTGCAGGCAGGTCGGT |
| NFATc1 | GTGGAGAAGCAGAGCAC | ACGCTGGTACTGGCTTC |
| DC-STAMP | ATGACTTGCAACCTAAGGGCAAAG | GTCTGGTTCCAAGAAACAAGGTCAT |
| CA2 | GAGTTTGATGACTCTCAGGACAA | CATATTTGGTGTTCCAGTGAACCA |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasonga, A.; Kruger, M.C.; Coetzee, M. Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes. Int. J. Mol. Sci. 2019, 20, 1798. https://doi.org/10.3390/ijms20071798
Kasonga A, Kruger MC, Coetzee M. Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes. International Journal of Molecular Sciences. 2019; 20(7):1798. https://doi.org/10.3390/ijms20071798
Chicago/Turabian StyleKasonga, Abe, Marlena C. Kruger, and Magdalena Coetzee. 2019. "Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes" International Journal of Molecular Sciences 20, no. 7: 1798. https://doi.org/10.3390/ijms20071798
APA StyleKasonga, A., Kruger, M. C., & Coetzee, M. (2019). Activation of PPARs Modulates Signalling Pathways and Expression of Regulatory Genes in Osteoclasts Derived from Human CD14+ Monocytes. International Journal of Molecular Sciences, 20(7), 1798. https://doi.org/10.3390/ijms20071798