Recent Insight into the Role of Fibrosis in Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma

Abstract
:1. Introduction
2. Pathophysiological Mechanisms of NAFLD Progression
3. Fibrosis-Dependent Hepatocarcinogenesis
3.1. Extracellular Matrix, Hepatic Stellate Cells, and Integrins
3.2. Matrix Stiffness
3.3. Matrix Metalloproteinases and Tissue Inhibitor of Metalloproteases
3.4. Hypoxia
3.5. Hedgehog Signaling
3.6. Hepatic Progenitor Cells
3.7. Autophagy
3.8. Dysregulation of the Immune System
3.9. Crosstalk between NASH and Hepatocellular Carcinoma
4. Therapeutic Perspective
5. Conclusions and Discussion
Funding
Conflicts of Interest
Abbreviations
| NASH | Non-alcoholic steatohepatitis |
| NAFLD | Non-alcoholic fatty liver disease |
| HCC | Hepatocellular carcinoma |
| HSCs | Hepatic stellate cells |
| ECM | Extracellular matrix |
| PDGF | Platelet derived growth factor |
| TGF-β | Transforming growth factors-β |
| ROS | Reactive oxygen species |
| KCs | Kupffer cells |
| DC | Dendritic cells |
| TLR | Toll-like receptor |
| NF | Nuclear factor |
| SFA | Saturated fatty acids |
| DAMPs | Damage-endogenous-associated molecular patterns |
| PAMPs | Pathogen-associated molecular patterns |
| DR | Ductular reaction |
| HPCs | Hepatic progenitor cells |
| DEN | Diethylnitrosamine |
| LPS | Lipopolysaccharide |
| DDR | Discoidin domain receptor |
| PI3K | Phosphatidylinositol 3-kinase |
| MAPK | Mitogen-activated protein kinases |
| ERK | extracellular signal-regulated kinase |
| STAT | signal transducer and activator of transcription |
| FAK | focal adhesion kinase |
| JNK | Jun N-terminal kinase |
| VEGF | Vascular endothelial growth factor |
| HIF-1 | Hypoxia-inducible factor 1 |
| TUFT1 | Tuftelin1 |
| Hh | Hedgehog |
| HGF | Hepatocyte growth factor |
| EMT | Epithelial-mesenchymal transition |
| EGF | Epidermal growth factor |
| FGF | Fibroblast growth factor |
| EPM | Epimorphin |
| MMPs | Matrix metalloproteinases |
| TIMPs | Tissue inhibitor of metalloproteases |
| CAFs | Cancer-associated fibroblasts |
| NK | Natural killer |
| Treg | Regulatory T cells |
References
- World Health Organization. Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Liew, Z.-H.; Goh, G.B.; Hao, Y.; Chang, P.-E.; Tan, C.-K. Comparison of Hepatocellular Carcinoma in Patients with Cryptogenic Versus Hepatitis B Etiology: A Study of 1079 Cases Over 3 Decades. Dig. Dis. Sci. 2019, 64, 585–590. [Google Scholar] [CrossRef]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular carcinomas in patients with metabolic syndrome often develop without significant liver fibrosis: A pathological analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Stine, J.G.; Wentworth, B.J.; Zimmet, A.; Rinella, M.E.; Loomba, R.; Caldwell, S.H.; Argo, C.K. Systematic review with meta-analysis: Risk of hepatocellular carcinoma in non-alcoholic steatohepatitis without cirrhosis compared to other liver diseases. Aliment. Pharmacol. Ther. 2018, 48, 696–703. [Google Scholar] [CrossRef]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Prim. 2016, 2, 16018. [Google Scholar] [CrossRef]
- White, D.L.; Kanwal, F.; El-Serag, H.B. Association between nonalcoholic fatty liver disease and risk for hepatocellular cancer, based on systematic review. Clin. Gastroenterol. Hepatol. 2012, 10, 1342–1359. [Google Scholar] [CrossRef]
- Farazi, P.A.; DePinho, R.A. Hepatocellular carcinoma pathogenesis: From genes to environment. Nat. Rev. Cancer 2006, 6, 674–687. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC development is associated to peripheral insulin resistance in a mouse model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef]
- Wong, V.W.-S.; Chitturi, S.; Wong, G.L.-H.; Yu, J.; Chan, H.L.-Y.; Farrell, G.C. Pathogenesis and novel treatment options for non-alcoholic steatohepatitis. Lancet Gastroenterol. Hepatol. 2016, 1, 56–67. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The Inflammatory Microenvironment in Hepatocellular Carcinoma: A Pivotal Role for Tumor-Associated Macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef]
- Ma, C.; Kesarwala, A.H.; Eggert, T.; Medina-Echeverz, J.; Kleiner, D.E.; Jin, P.; Stroncek, D.F.; Terabe, M.; Kapoor, V.; ElGindi, M.; et al. NAFLD causes selective CD4+ T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016, 531, 253–257. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef]
- Park, E.J.; Lee, J.H.; Yu, G.-Y.; He, G.; Ali, S.R.; Holzer, R.G.; Osterreicher, C.H.; Takahashi, H.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef]
- Grohmann, M.; Wiede, F.; Dodd, G.T. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306. [Google Scholar] [CrossRef]
- Dapito, D.H.; Mencin, A.; Gwak, G.-Y. Promotion of Hepatocellular Carcinoma by the Intestinal Microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef]
- Carloni, V.; Luong, T.V.; Rombouts, K. Hepatic stellate cells and extracellular matrix in hepatocellular carcinoma: More complicated than ever. Liver Int. 2014, 34, 834–843. [Google Scholar] [CrossRef]
- Michalopoulos, G.K. Liver regeneration. J. Cell. Physiol. 2007, 213, 286–300. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Stickel, F.; Hellerbrand, C. Non-alcoholic fatty liver disease as a risk factor for hepatocellular carcinoma: Mechanisms and implications. Gut 2010, 59, 1303–1307. [Google Scholar] [CrossRef]
- Zhang, C.-Y.; Yuan, W.-G.; He, P.; Lei, J.-H.; Wang, C.-X. Liver fibrosis and hepatic stellate cells: Etiology, pathological hallmarks and therapeutic targets. World J. Gastroenterol. 2016, 22, 10512–10522. [Google Scholar] [CrossRef]
- Leitinger, B.; Hohenester, E. Mammalian collagen receptors. Matrix Biol. 2007, 26, 146–155. [Google Scholar] [CrossRef]
- Patsenker, E.; Stickel, F. Role of integrins in fibrosing liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G425–G434. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of Liver Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Howe, A.; Aplin, A.E.; Alahari, S.K.; Juliano, R.L. Integrin signaling and cell growth control. Curr. Opin. Cell. Biol. 1998, 10, 220–231. [Google Scholar] [CrossRef]
- Yang, C.; Zeisberg, M.; Lively, J.C.; Nyberg, P.; Afdhal, N.; Kalluri, R. Integrin alpha1beta1 and alpha2beta1 are the key regulators of hepatocarcinoma cell invasion across the fibrotic matrix microenvironment. Cancer Res. 2003, 63, 8312–8317. [Google Scholar]
- Wu, Y.; Qiao, X.; Qiao, S.; Yu, L. Targeting integrins in hepatocellular carcinoma. Expert Opin. Ther. Targets 2011, 15, 421–437. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, W.; Xiang, J.; Liu, P.; Ke, M.; Wang, B.; Wu, R.; Lv, Y. Collagen I promotes hepatocellular carcinoma cell proliferation by regulating integrin β1/FAK signaling pathway in nonalcoholic fatty liver. Oncotarget 2017, 8, 95586–95595. [Google Scholar] [CrossRef]
- Walsh, L.A.; Nawshad, A.; Medici, D. Discoidin domain receptor 2 is a critical regulator of epithelial-mesenchymal transition. Matrix Biol. 2011, 30, 243–247. [Google Scholar] [CrossRef]
- Zhao, H.; Bian, H.; Bu, X.; Zhang, S.; Zhang, P.; Yu, J.; Lai, X.; Li, D.; Zhu, C.; Yao, L.; et al. Targeting of Discoidin Domain Receptor 2 (DDR2) Prevents Myofibroblast Activation and Neovessel Formation During Pulmonary Fibrosis. Mol. Ther. 2016, 24, 1734–1744. [Google Scholar] [CrossRef]
- Wells, R.G. The role of matrix stiffness in regulating cell behavior. Hepatology 2008, 47, 1394–1400. [Google Scholar] [CrossRef]
- Singh, S.; Fujii, L.L.; Murad, M.H.; Wang, Z.; Asrani, S.K.; Ehman, R.L.; Kamath, P.S.; Talwalkar, J.A. Liver stiffness is associated with risk of decompensation, liver cancer, and death in patients with chronic liver diseases: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 1573–1584. [Google Scholar] [CrossRef]
- Mouw, J.K.; Yui, Y.; Damiano, L.; Bainer, R.O.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M.; et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360–367. [Google Scholar] [CrossRef]
- Ulrich, T.A.; de Juan Pardo, E.M.; Kumar, S. The mechanical rigidity of the extracellular matrix regulates the structure, motility, and proliferation of glioma cells. Cancer Res. 2009, 69, 4167–4174. [Google Scholar] [CrossRef]
- You, Y.; Zheng, Q.; Dong, Y.; Wang, Y.; Zhang, L.; Xue, T.; Xie, X.; Hu, C.; Wang, Z.; Chen, R.; et al. Higher Matrix Stiffness Upregulates Osteopontin Expression in Hepatocellular Carcinoma Cells Mediated by Integrin β1/GSK3β/β-Catenin Signaling Pathway. PLoS ONE 2015, 10, e0134243. [Google Scholar] [CrossRef]
- Schrader, J.; Gordon-Walker, T.T.; Aucott, R.L.; van Deemter, M.; Quaas, A.; Walsh, S.; Benten, D.; Forbes, S.J.; Wells, R.G.; Iredale, J.P. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011, 53, 1192–1205. [Google Scholar] [CrossRef]
- Dong, Y.; Xie, X.; Wang, Z.; Hu, C.; Zheng, Q.; Wang, Y.; Chen, R.; Xue, T.; Chen, J.; Gao, D.; et al. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin β1. Biochem. Biophys. Res. Commun. 2014, 444, 427–432. [Google Scholar] [CrossRef]
- You, Y.; Zheng, Q.; Dong, Y.; Xie, X.; Wang, Y.; Wu, S.; Zhang, L.; Wang, Y.; Xue, T.; Wang, Z.; et al. Matrix stiffness-mediated effects on stemness characteristics occurring in HCC cells. Oncotarget 2016, 7, 32221–32231. [Google Scholar] [CrossRef]
- Naim, A.; Pan, Q.; Baig, M.S. Matrix Metalloproteinases (MMPs) in Liver Diseases. J. Clin. Exp. Hepatol. 2017, 7, 367–372. [Google Scholar] [CrossRef]
- Rodríguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef]
- Murphy, G.; Docherty, A.J.P. The Matrix Metalloproteinases and Their Inhibitors. Am. J. Respir. Cell. Mol. Biol. 1992, 7, 120–125. [Google Scholar] [CrossRef]
- Wallace, M.C.; Friedman, S.L. Hepatic fibrosis and the microenvironment: Fertile soil for hepatocellular carcinoma development. Gene Expr. 2014, 16, 77–84. [Google Scholar] [CrossRef]
- Song, T.; Dou, C.; Jia, Y.; Tu, K.; Zheng, X. TIMP-1 activated carcinoma-associated fibroblasts inhibit tumor apoptosis by activating SDF1/CXCR4 signaling in hepatocellular carcinoma. Oncotarget 2015, 6, 12061–12079. [Google Scholar] [CrossRef]
- Ando, W.; Yokomori, H.; Tsutsui, N.; Yamanouchi, E.; Suzuki, Y.; Oda, M.; Inagaki, Y.; Otori, K.; Okazaki, I. Serum matrix metalloproteinase-1 level represents disease activity as opposed to fibrosis in patients with histologically proven nonalcoholic steatohepatitis. Clin. Mol. Hepatol. 2018, 24, 61–76. [Google Scholar] [CrossRef]
- O’Rourke, J.M.; Sagar, V.M.; Shah, T. Carcinogenesis on the background of liver fibrosis: Implications for the management of hepatocellular cancer. World J. Gastroenterol. 2018, 24, 4436–4447. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Yu, D.-C.; Chen, J.; Ding, Y.-T. Hypoxic and highly angiogenic non-tumor tissues surrounding hepatocellular carcinoma: The ‘niche’ of endothelial progenitor cells. Int. J. Mol. Sci. 2010, 11, 2901–2909. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen Sensing, Homeostasis, and Disease. N. Engl. J. Med. 2011, 365, 537–547. [Google Scholar] [CrossRef]
- Lichtenberger, B.M.; Tan, P.K.; Niederleithner, H.; Ferrara, N.; Petzelbauer, P.; Sibilia, M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell 2010, 140, 268–279. [Google Scholar] [CrossRef]
- Hernandez-Gea, V.; Toffanin, S.; Friedman, S.L.; Llovet, J.M. Role of the microenvironment in the pathogenesis and treatment of hepatocellular carcinoma. Gastroenterology 2013, 144, 512–527. [Google Scholar] [CrossRef]
- Leiser, Y.; Blumenfeld, A.; Haze, A.; Dafni, L.; Taylor, A.L.; Rosenfeld, E.; Fermon, E.; Gruenbaum-Cohen, Y.; Shay, B.; Deutsch, D. Localization, quantification, and characterization of tuftelin in soft tissues. Anat. Rec. 2007, 290, 449–454. [Google Scholar] [CrossRef]
- Dou, C.; Zhou, Z.; Xu, Q.; Liu, Z.; Zeng, Y.; Wang, Y.; Li, Q.; Wang, L.; Yang, W.; Liu, Q.; et al. Hypoxia-induced TUFT1 promotes the growth and metastasis of hepatocellular carcinoma by activating the Ca2+/PI3K/AKT pathway. Oncogene 2018, 38, 1239–1255. [Google Scholar] [CrossRef]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar]
- Choi, S.S.; Omenetti, A.; Syn, W.-K.; Diehl, A.M. The role of Hedgehog signaling in fibrogenic liver repair. Int. J. Biochem. Cell. Biol. 2011, 43, 238–244. [Google Scholar] [CrossRef]
- Ochoa, B.; Syn, W.-K.; Delgado, I.; Karaca, G.F.; Jung, Y.; Wang, J.; Zubiaga, A.M.; Fresnedo, O.; Omenetti, A.; Zdanowicz, M.; et al. Hedgehog signaling is critical for normal liver regeneration after partial hepatectomy in mice. Hepatology 2010, 51, 1712–1723. [Google Scholar] [CrossRef]
- Zheng, X.; Zeng, W.; Gai, X.; Xu, Q.; Li, C.; Liang, Z.; Tuo, H.; Liu, Q. Role of the Hedgehog pathway in hepatocellular carcinoma (Review). Oncol. Rep. 2013, 30, 2020–2026. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, P.; Ma, Z.; Li, M.; Teng, X.; Sun, L.; Wan, G.; Li, Y.; Guo, L.; Liu, H. Novel Interplay Between Sonic Hedgehog and Transforming Growth Factor-β1 in Human Nonalcoholic Steatohepatitis. Appl. Immunohistochem. Mol. Morphol. AIMM 2019. [Google Scholar] [CrossRef]
- Philips, G.M.; Chan, I.S.; Swiderska, M.; Schroder, V.T.; Guy, C.; Karaca, G.F.; Moylan, C.; Venkatraman, T.; Feuerlein, S.; Syn, W-K.; et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PLoS ONE 2011, 6, e23943. [Google Scholar] [CrossRef]
- Carpino, G.; Renzi, A.; Onori, P.; Gaudio, E. Role of Hepatic Progenitor Cells in Nonalcoholic Fatty Liver Disease Development: Cellular Cross-Talks and Molecular Networks. Int. J. Mol. Sci. 2013, 14, 20112–20130. [Google Scholar] [CrossRef]
- Mishra, L.; Banker, T.; Murray, J.; Byers, S.; Thenappan, A.; He, A.R.; Shetty, K.; Johnson, L.; Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar] [CrossRef]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; de Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef]
- Czaja, M.J. Function of Autophagy in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1304–1313. [Google Scholar] [CrossRef]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef]
- Thoen, L.F.R.; Guimarães, E.L.M.; Dollé, L.; Mannaerts, I.; Najimi, M.; Sokal, E.; van Grunsven, L.A. A role for autophagy during hepatic stellate cell activation. J. Hepatol. 2011, 55, 1353–1360. [Google Scholar] [CrossRef]
- Elmansi, A.; El-Karef, A.; Shishtawy, M.; Eissa, L. Hepatoprotective Effect of Curcumin on Hepatocellular Carcinoma Through Autophagic and Apoptic Pathways. Ann. Hepatol. 2017, 16, 607–618. [Google Scholar] [CrossRef]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef]
- Sun, K.; Xu, L.; Jing, Y.; Han, Z.; Chen, X.; Cai, C.; Zhao, P.; Zhao, X.; Yang, L.; Wei, L. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017, 388, 198–207. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Lanthier, N. Targeting Kupffer cells in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: Why and how? World J. Hepatol. 2015, 7, 2184–2188. [Google Scholar] [CrossRef]
- Ganz, M.; Szabo, G. Immune and inflammatory pathways in NASH. Hepatol. Int. 2013, 7 (Suppl. S2), 771–781. [Google Scholar] [CrossRef]
- Kong, L.; Zhou, Y.; Bu, H.; Lv, T.; Shi, Y.; Yang, J. Deletion of interleukin-6 in monocytes/macrophages suppresses the initiation of hepatocellular carcinoma in mice. J. Exp. Clin. Cancer Res. 2016, 35, 131. [Google Scholar] [CrossRef]
- Kubes, P.; Mehal, W.Z. Sterile Inflammation in the Liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- Koh, M.Y.; Gagea, M.; Sargis, T.; Lemos, R.; Grandjean, G.; Charbono, A.; Bekiaris, V.; Sedy, J.; Kiriakova, G.; Liu, X.; et al. A new HIF-1α/RANTES-driven pathway to hepatocellular carcinoma mediated by germline haploinsufficiency of SART1/HAF in mice. Hepatology 2016, 63, 1576–1591. [Google Scholar] [CrossRef]
- Cai, J.; Zhang, X.-J.; Li, H. The Role of Innate Immune Cells in Nonalcoholic Steatohepatitis. Hepatology 2019. [Google Scholar] [CrossRef]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Nati, M.; Haddad, D.; Birkenfeld, A.L.; Koch, C.A.; Chavakis, T.; Chatzigeorgiou, A. The role of immune cells in metabolism-related liver inflammation and development of non-alcoholic steatohepatitis (NASH). Rev. Endocr. Metab. Disord. 2016, 17, 29–39. [Google Scholar] [CrossRef]
- Tian, Z.; Chen, Y.; Gao, B. Natural killer cells in liver disease. Hepatology 2013, 57, 1654–1662. [Google Scholar] [CrossRef]
- Jeong, W.-I.; Park, O.; Suh, Y.-G.; Byun, J.-S.; Park, S.-Y.; Choi, E.; Kim, J.-K.; Ko, H.; Wang, H.; Miller, A.M.; et al. Suppression of innate immunity (natural killer cell/interferon-γ) in the advanced stages of liver fibrosis in mice. Hepatology 2011, 53, 1342–1351. [Google Scholar] [CrossRef]
- Male, V.; Stegmann, K.; Easom, N.; Maini, M. Natural Killer Cells in Liver Disease. Semin. Liver Dis. 2017, 37, 198–209. [Google Scholar] [CrossRef]
- Albertsson, P.A.; Basse, P.H.; Hokland, M.; Goldfarb, R.H.; Nagelkerke, J.F.; Nannmark, U.; Kuppen, P.J.K. NK cells and the tumour microenvironment: Implications for NK-cell function and anti-tumour activity. Trends Immunol. 2003, 24, 603–609. [Google Scholar] [CrossRef]
- Sachdeva, M.; Chawla, Y.K.; Arora, S.K. Immunology of hepatocellular carcinoma. World J. Hepatol. 2015, 7, 2080–2090. [Google Scholar] [CrossRef]
- Hassin, D.; Garber, O.G.; Meiraz, A.; Schiffenbauer, Y.S.; Berke, G. Cytotoxic T lymphocyte perforin and Fas ligand working in concert even when Fas ligand lytic action is still not detectable. Immunology 2011, 133, 190–196. [Google Scholar] [CrossRef]
- Lucifora, J.; Xia, Y.; Reisinger, F.; Zhang, K.; Stadler, D.; Cheng, X.; Sprinzl, M.F.; Koppensteiner, H.; Makowska, Z.; Volz, T.; et al. Specific and Nonhepatotoxic Degradation of Nuclear Hepatitis B Virus cccDNA. Science 2014, 343, 1221–1228. [Google Scholar] [CrossRef]
- Endig, J.; Buitrago-Molina, L.E.; Marhenke, S.; Marhenke, S.; Reisinger, F.; Saborowski, A.; Schütt, J.; Limbourg, F.; Könecke, C.; Schreder, A.; et al. Dual Role of the Adaptive Immune System in Liver Injury and Hepatocellular Carcinoma Development. Cancer Cell 2016, 30, 308–323. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.-J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4(+) T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, Z.; Zhou, L.; Qi, Z.; Xing, S.; Lv, J.; Shi, J.; Fu, B.; Liu, Z.; Zhang, J.-Y.; et al. Impairment of CD4+ cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2013, 58, 139–149. [Google Scholar] [CrossRef]
- Shen, Y.; Wei, Y.; Wang, Z.; Jing, Y.; He, H.; Yuan, J.; Li, R.; Zhao, Q.; Wei, L.; Yang, T.; et al. TGF-β regulates hepatocellular carcinoma progression by inducing Treg cell polarization. Cell. Physiol. Biochem. 2015, 35, 1623–1632. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased regulatory T cells correlate with CD8 T-cell impairment and poor survival in hepatocellular carcinoma patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- McPherson, S.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Serum immunoglobulin levels predict fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 60, 1055–1062. [Google Scholar] [CrossRef]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Wang, H.; Chen, L. Tumor microenviroment and hepatocellular carcinoma metastasis. J. Gastroenterol. Hepatol. 2013, 28, 43–48. [Google Scholar] [CrossRef]
- Kocabayoglu, P.; Friedman, S.L. Cellular basis of hepatic fibrosis and its role in inflammation and cancer. Front. Biosci. (Schol. Ed.) 2013, 5, 217–230. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Miao, H.Q.; Medalion, B.; Danagher, P.; Ron, D. Involvement of heparan sulfate and related molecules in sequestration and growth promoting activity of fibroblast growth factor. Cancer Metastasis Rev. 1996, 15, 177–186. [Google Scholar] [CrossRef]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.T.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; Odell, M.M.; Bauer, R.L.; Ren, H.-P.; Haugen, H.S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef]
- Kelly, J.D.; Haldeman, B.A.; Grant, F.J.; Murray, M.J.; Seifert, R.A.; Bowen-Pope, D.F.; Cooper, J.A.; Kazlauskas, A. Platelet-derived growth factor (PDGF) stimulates PDGF receptor subunit dimerization and intersubunit trans-phosphorylation. J. Biol. Chem. 1991, 266, 8987–8992. [Google Scholar]
- Wright, J.H.; Johnson, M.M.; Shimizu-Albergine, M. Paracrine activation of hepatic stellate cells in platelet-derived growth factor C transgenic mice: Evidence for stromal induction of hepatocellular carcinoma. Int. J. Cancer 2014, 134, 778–788. [Google Scholar] [CrossRef]
- Jia, Y.-L.; Shi, L.; Zhou, J.-N.; Fu, C.-J.; Chen, L.; Yuan, H.-F.; Wang, Y.-F.; Yan, X.-L.; Xu, Y.-C.; Zeng, Q.; et al. Epimorphin promotes human hepatocellular carcinoma invasion and metastasis through activation of focal adhesion kinase/extracellular signal-regulated kinase/matrix metalloproteinase-9 axis. Hepatology 2011, 54, 1808–1818. [Google Scholar] [CrossRef]
- Imai, Y.; Yoshida, O.; Watanabe, T.; Yukimoto, A.; Koizumi, Y.; Ikeda, Y.; Tokumoto, Y.; Hirooka, M.; Abe, M.; Hiasa, Y. Stimulated hepatic stellate cell promotes progression of hepatocellular carcinoma due to protein kinase R activation. PLoS ONE 2019, 14, e0212589. [Google Scholar] [CrossRef]
- Kubo, N.; Araki, K.; Kuwano, H.; Shirabe, K. Cancer-associated fibroblasts in hepatocellular carcinoma. World J. Gastroenterol. 2016, 22, 6841–6850. [Google Scholar] [CrossRef]
- Cesselli, D.; Beltrami, A.P.; Poz, A.; Marzinotto, S.; Comisso, E.; Bergamin, N.; Bourkoula, E.; Pucer, A.; Puppato, E.; Toffoletto, B.; et al. Role of tumor associated fibroblasts in human liver regeneration, cirrhosis, and cancer. Int. J. Hepatol. 2011, 2011, 120925. [Google Scholar] [CrossRef]
- Neaud, V.; Faouzi, S.; Guirouilh, J.; Le Bail, B.; Balabaud, C.; Bioulac-Sage, P.; Rosenbaum, J. Human hepatic myofibroblasts increase invasiveness of hepatocellular carcinoma cells: Evidence for a role of hepatocyte growth factor. Hepatology 1997, 26, 1458–1466. [Google Scholar] [CrossRef]
- Feng, T.; Yu, H.; Xia, Q.; Ma, Y.; Yin, H.; Shen, Y.; Liu, X. Cross-talk mechanism between endothelial cells and hepatocellular carcinoma cells via growth factors and integrin pathway promotes tumor angiogenesis and cell migration. Oncotarget 2017, 8, 69577–69593. [Google Scholar] [CrossRef]
- Kareva, I.; Abou-Slaybi, A.; Dodd, O.; Dashevsky, O.; Klement, G.L. Normal Wound Healing and Tumor Angiogenesis as a Game of Competitive Inhibition. PLoS ONE 2016, 11, e0166655. [Google Scholar] [CrossRef]
- Welti, J.; Loges, S.; Dimmeler, S.; Carmeliet, P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J. Clin. Investig. 2013, 123, 3190–3200. [Google Scholar] [CrossRef]
- Makarova-Rusher, O.V.; Medina-Echeverz, J.; Duffy, A.G.; Greten, T.F. The yin and yang of evasion and immune activation in HCC. J. Hepatol. 2015, 62, 1420–1429. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Fabregat, I.; Fernando, J.; Mainez, J.; Sancho, P. TGF-beta signaling in cancer treatment. Curr. Pharm. Des. 2014, 20, 2934–2947. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, T.; Tang, W.; Deng, B.; Chen, Y.; Zhu, J.; Shen, X. Hepatocellular Carcinoma Cells Induce Regulatory T Cells and Lead to Poor Prognosis via Production of Transforming Growth Factor-β1. Cell. Physiol. Biochem. 2016, 38, 306–318. [Google Scholar] [CrossRef]
- Calon, A.; Tauriello, D.V.F.; Batlle, E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin. Cancer Biol. 2014, 25, 15–22. [Google Scholar] [CrossRef]
- Fransvea, E.; Angelotti, U.; Antonaci, S.; Giannelli, G. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008, 47, 1557–1566. [Google Scholar] [CrossRef]
- Mazzocca, A.; Fransvea, E.; Dituri, F.; Lupo, L.; Antonaci, S.; Giannelli, G. Down-regulation of connective tissue growth factor by inhibition of transforming growth factor beta blocks the tumor-stroma cross-talk and tumor progression in hepatocellular carcinoma. Hepatology 2010, 51, 523–534. [Google Scholar] [CrossRef]
- Luangmonkong, T.; Suriguga, S.; Bigaeva, E.; Boersema, M.; Oosterhuis, D.; de Jong, K.P.; Schuppan, D.; Mutsaers, H.A.M.; Olinga, P. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br. J. Pharmacol. 2017, 174, 3107–3117. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for chemokines in liver disease. Gastroenterology 2014, 147, 577–594. [Google Scholar] [CrossRef]
- Puengel, T.; Krenkel, O.; Mossanen, J.; Longerich, T.; Lefebvre, E.; Trautwein, C.; Tacke, F. The Dual Ccr2/Ccr5 Antagonist Cenicriviroc Ameliorates Steatohepatitis and Fibrosis in Vivo by Inhibiting the Infiltration of Inflammatory Monocytes into Injured Liver. J. Hepatol. 2016, 64, S160. [Google Scholar] [CrossRef]
- Friedman, S.L.; Ratziu, V.; Harrison, S.; Abdelmalek, M.F.; Aithal, G.P.; Caballeria, J.; Francque, S.; Farrell, G.; Kowdley, K.V.; Craxi, A.; et al. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018, 67, 1754–1767. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Stefano, J.T.; Pereira, I.V.A.; Torres, M.M.; Bida, P.M.; Coelho, A.M.M.; Xerfan, M.P.; Cogliati, B.; Barbeiro, D.F.; Mazo, D.F.C.; Kubrusly, M.S.; et al. Sorafenib prevents liver fibrosis in a non-alcoholic steatohepatitis (NASH) rodent model. Braz. J. Med. Biol. Res. 2015, 48, 408–414. [Google Scholar] [CrossRef]
- Jiménez Calvente, C.; Sehgal, A.; Popov, Y.; Kim, Y.O.; Zevallos, V.; Sahin, U.; Diken, M.; Schuppan, D. Specific hepatic delivery of procollagen α1(I) small interfering RNA in lipid-like nanoparticles resolves liver fibrosis. Hepatology 2015, 62, 1285–1297. [Google Scholar] [CrossRef]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68–69, 435–451. [Google Scholar] [CrossRef]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H.; Pellicoro, A.; Raschperger, E.; Betsholtz, C.; Ruminski, P.G.; et al. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef]
- Becker, L.R.; Aakhus, A.E.; Reich, H.C.; Lee, P.K. A Novel Alternate Dosing of Vismodegib for Treatment of Patients with Advanced Basal Cell Carcinomas. JAMA Dermatol. 2017, 153, 321–322. [Google Scholar] [CrossRef]
- Whiteside, T.L.; Demaria, S.; Rodriguez-Ruiz, M.E.; Zarour, H.M.; Melero, I. Emerging Opportunities and Challenges in Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 1845–1855. [Google Scholar] [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.-Y.; Choo, S.-P.; Trojan, J.; Welling, T.H.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Machado, M.V.; Michelotti, G.A.; Xie, G.; de Almeida, T.P.; Boursier, J.; Bohnic, B.; Guy, C.D.; Diehl, A.M.; Diehl, A.M. Mouse Models of Diet-Induced Nonalcoholic Steatohepatitis Reproduce the Heterogeneity of the Human Disease. PLoS ONE 2015, 10, e0127991. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.-I.; Hoshida, Y.; et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef]
- Alkreathy, H.M.; Khan, R.A.; Khan, M.R.; Sahreen, S. CCl4 induced genotoxicity and DNA oxidative damages in rats: Hepatoprotective effect of Sonchus arvensis. BMC Complement. Altern. Med. 2014, 14, 452. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Paschetta, E.; Gambino, R. Thiazolidinediones and Advanced Liver Fibrosis in Nonalcoholic Steatohepatitis: A Meta-analysis. JAMA Intern. Med. 2017, 177, 633–640. [Google Scholar] [CrossRef]
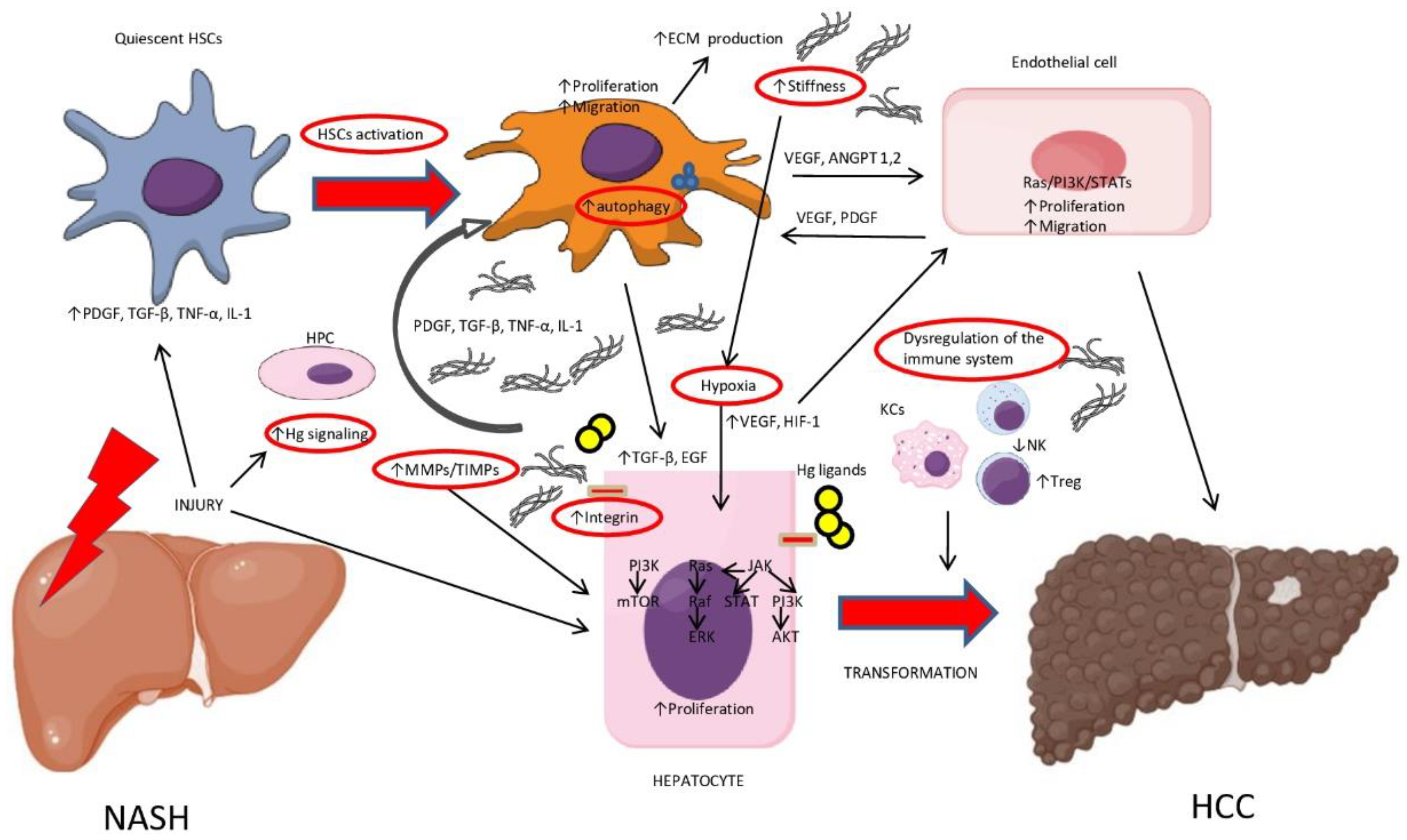

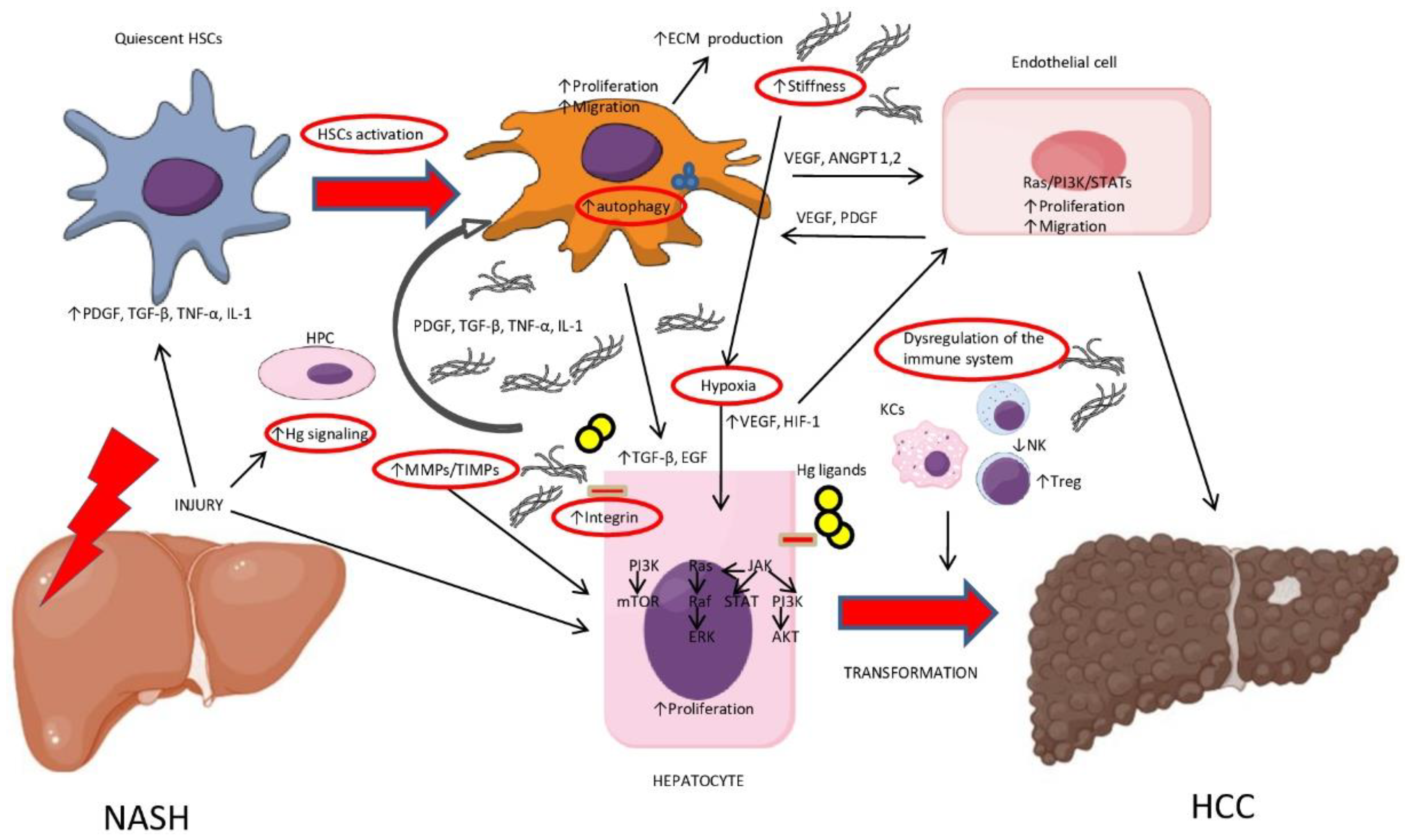{kind=link}
| Factor | Mechanism | Biological Effects |
|---|---|---|
| Inflammation | ↑ PDGF, TGF-β, TNF-α, IL-1 and chemokines | HSCs and KCs activation |
| Activated HSCs |
|
|
| ↑ Stiffness |
|
|
| Integrins |
|
|
| MMPs/TIMPs imbalance |
|
|
| Hypoxia |
|
|
| Hedgehog (Hg) |
|
|
| HPCs and ductular reaction (DR) |
|
|
| Autophagy |
|
|
| Kupffer Cells (KCs) |
|
|
| ↓ NK cells |
|
|
| NKT cells |
|
|
| ↑ Treg cells |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sircana, A.; Paschetta, E.; Saba, F.; Molinaro, F.; Musso, G. Recent Insight into the Role of Fibrosis in Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma. Int. J. Mol. Sci. 2019, 20, 1745. https://doi.org/10.3390/ijms20071745
Sircana A, Paschetta E, Saba F, Molinaro F, Musso G. Recent Insight into the Role of Fibrosis in Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma. International Journal of Molecular Sciences. 2019; 20(7):1745. https://doi.org/10.3390/ijms20071745
Chicago/Turabian StyleSircana, Antonio, Elena Paschetta, Francesca Saba, Federica Molinaro, and Giovanni Musso. 2019. "Recent Insight into the Role of Fibrosis in Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma" International Journal of Molecular Sciences 20, no. 7: 1745. https://doi.org/10.3390/ijms20071745
APA StyleSircana, A., Paschetta, E., Saba, F., Molinaro, F., & Musso, G. (2019). Recent Insight into the Role of Fibrosis in Nonalcoholic Steatohepatitis-Related Hepatocellular Carcinoma. International Journal of Molecular Sciences, 20(7), 1745. https://doi.org/10.3390/ijms20071745