Bioreactor-Controlled Physoxia Regulates TGF-β Signaling to Alter Extracellular Matrix Synthesis by Human Chondrocytes



Abstract
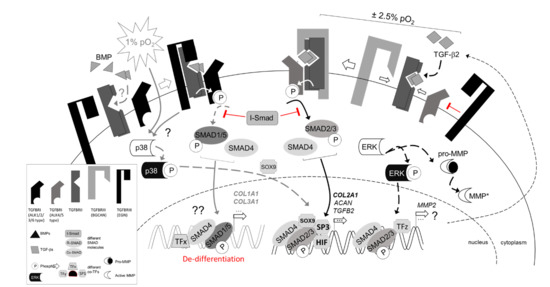
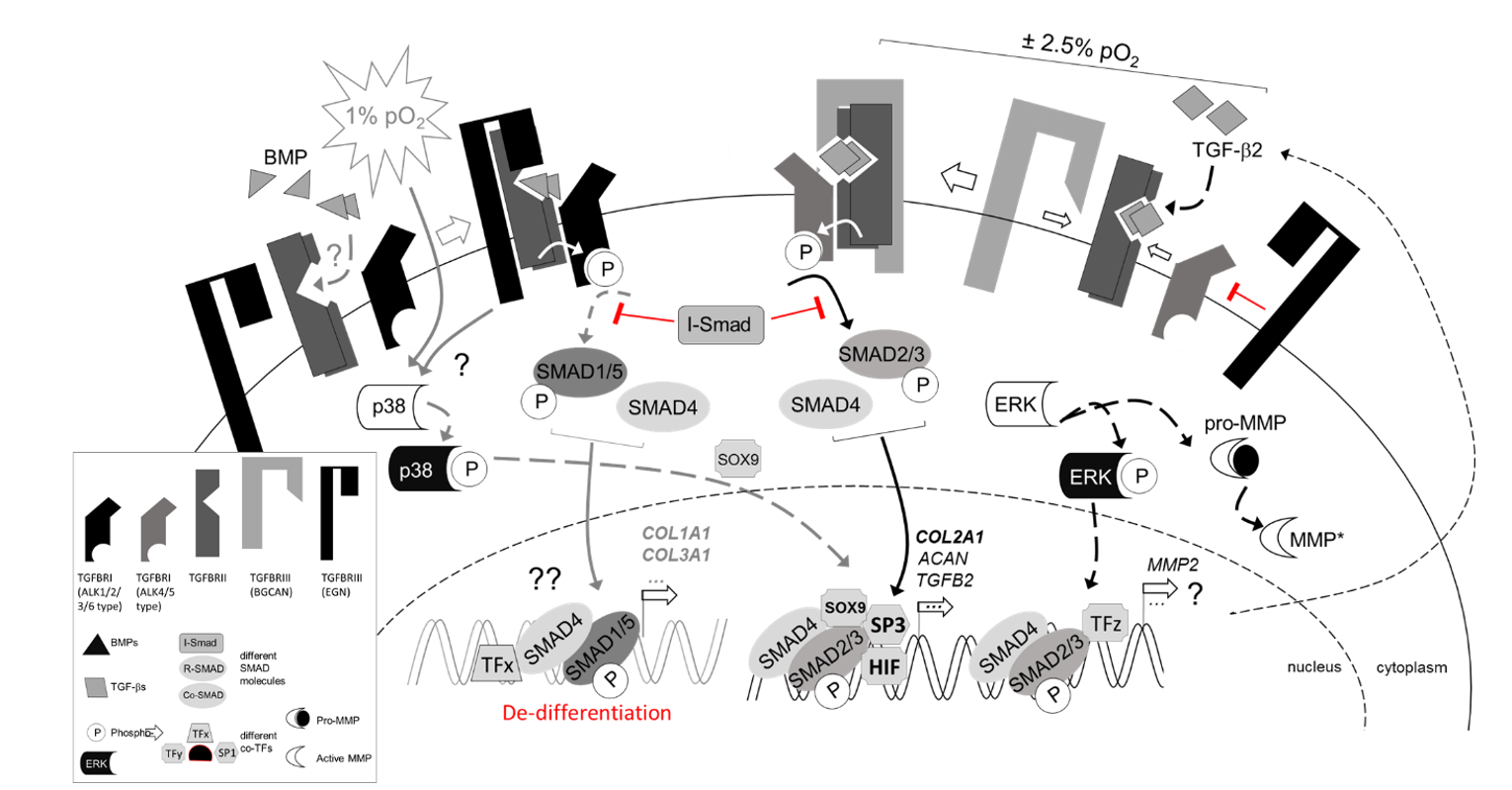{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Articular Cartilage and Hypoxia
1.2. TGF-β Signaling in Articular Cartilage
1.3. Hypoxia and Chondrocyte Metabolism
2. Results
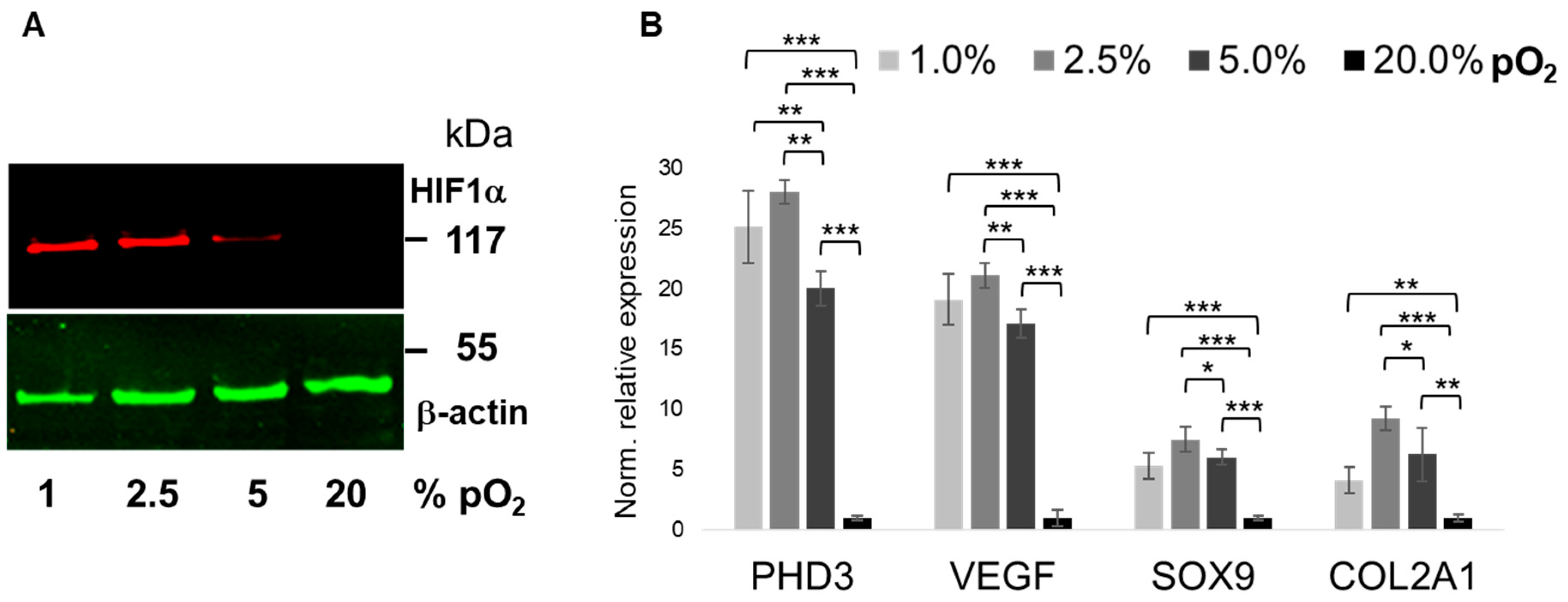
2.1. Bioreactor Validation
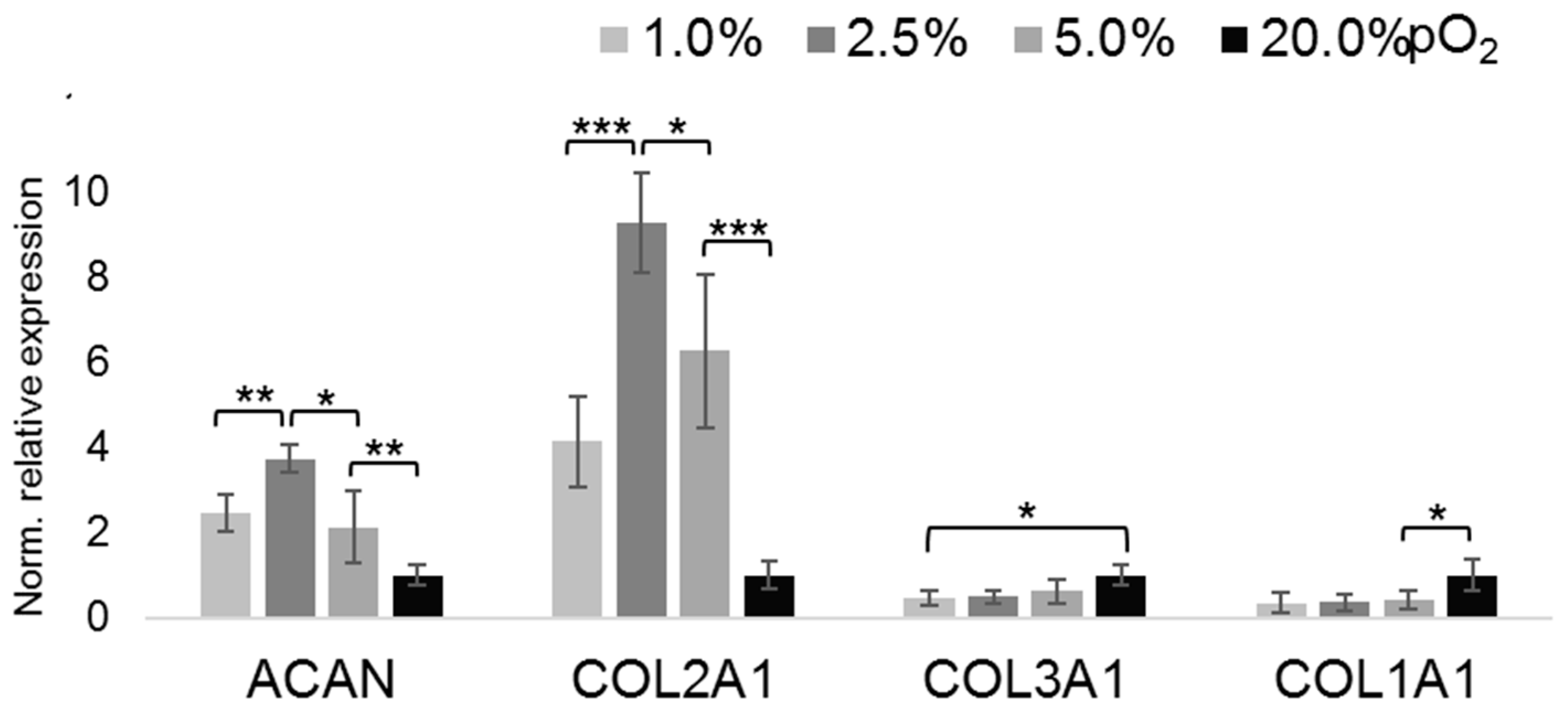
2.2. Oxygenation-Dependent ECM Marker Expression
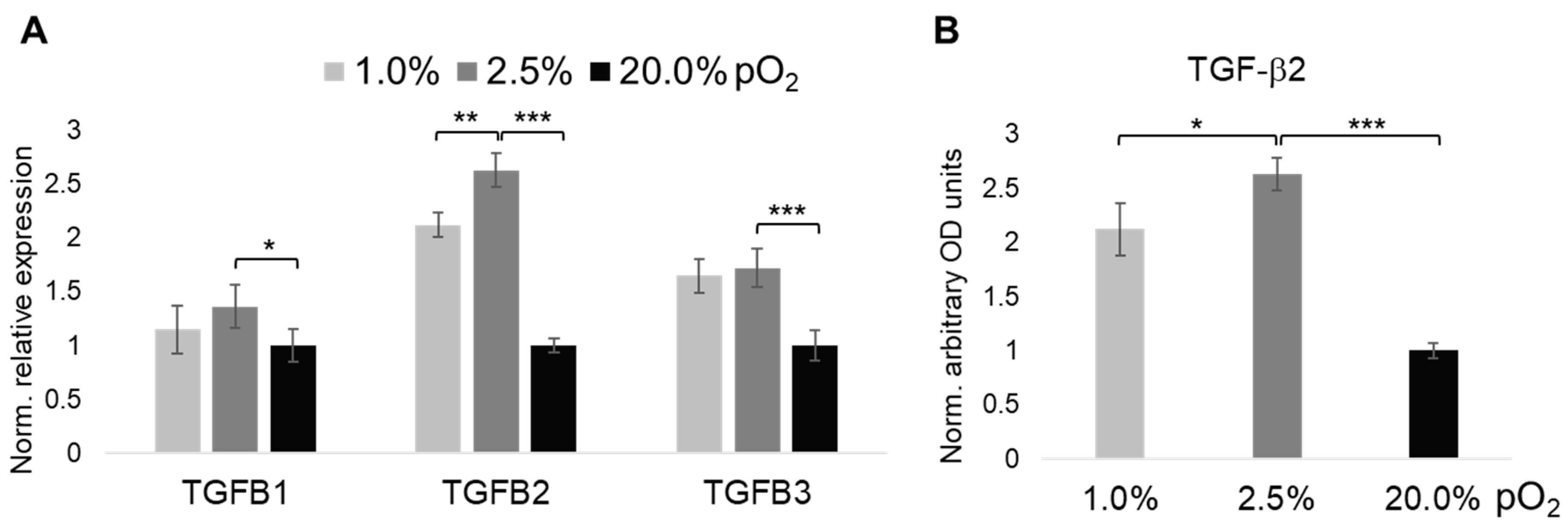
2.3. Oxygenation-Dependent Expression of TGF-β Ligands
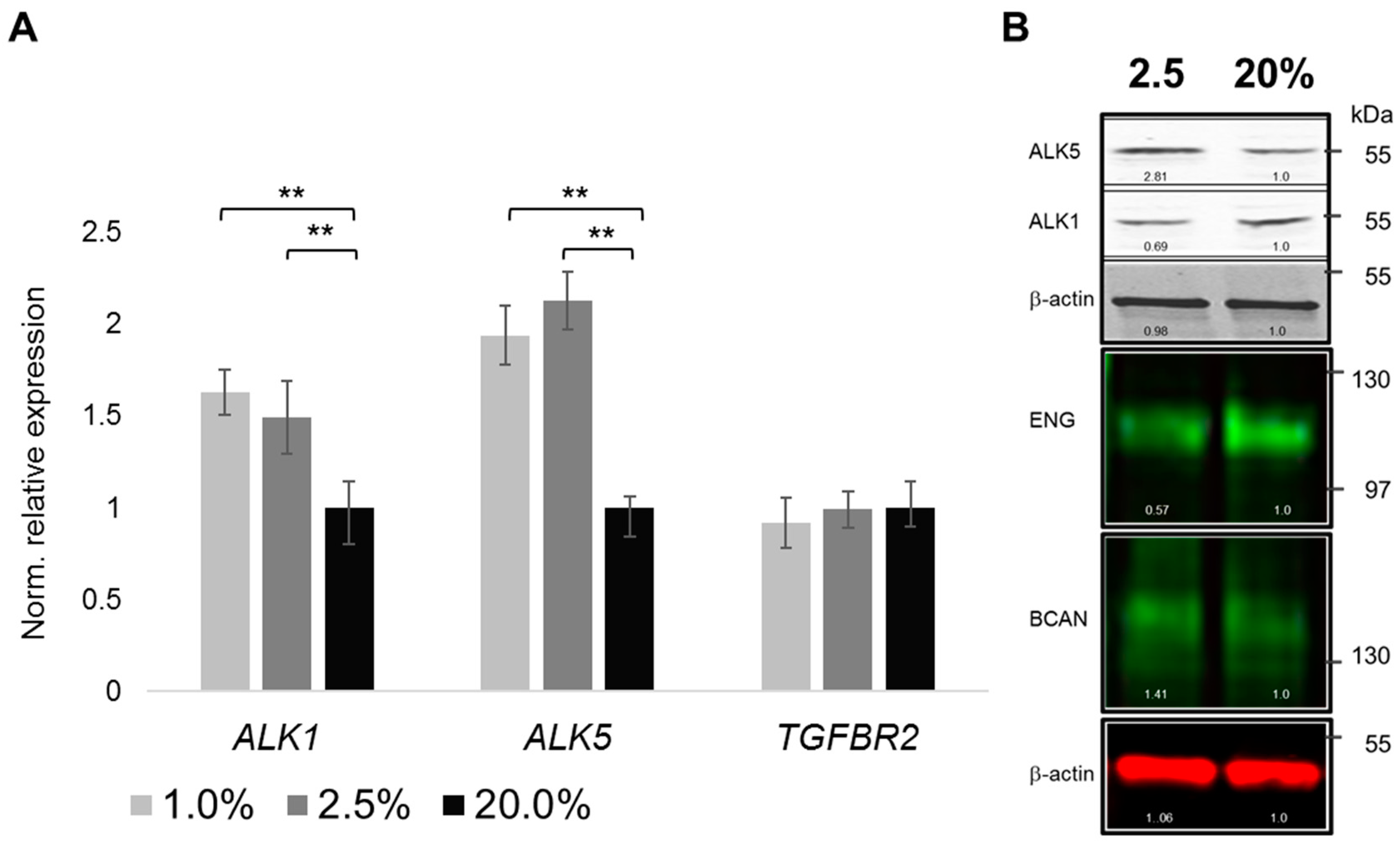
2.4. Oxygenation-Dependent Regulation of TGF-β Receptors
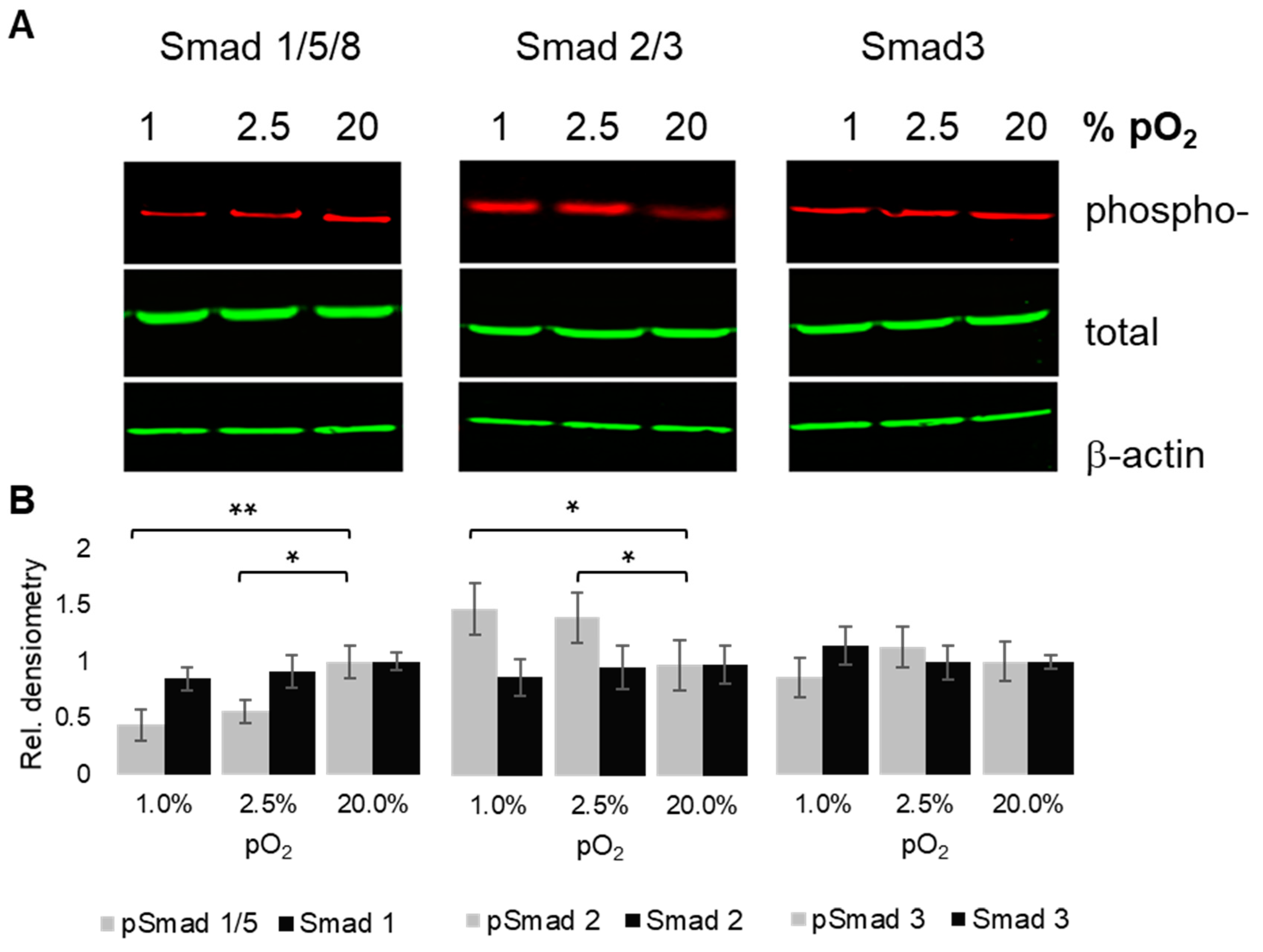
2.5. Oxygenation-Dependent Regulation of R-Smad Phosphorylation
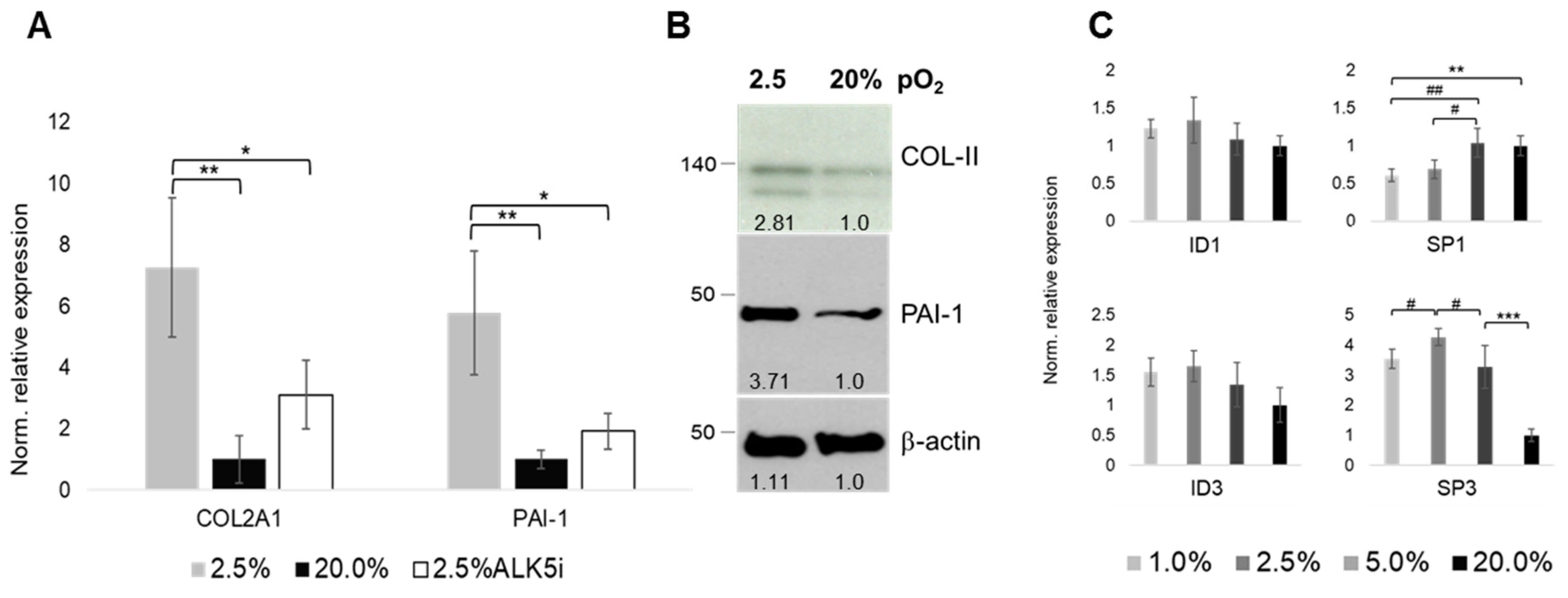
2.6. Oxygen- and ALK-Dependency of The Articular Chondrocyte Phenotype
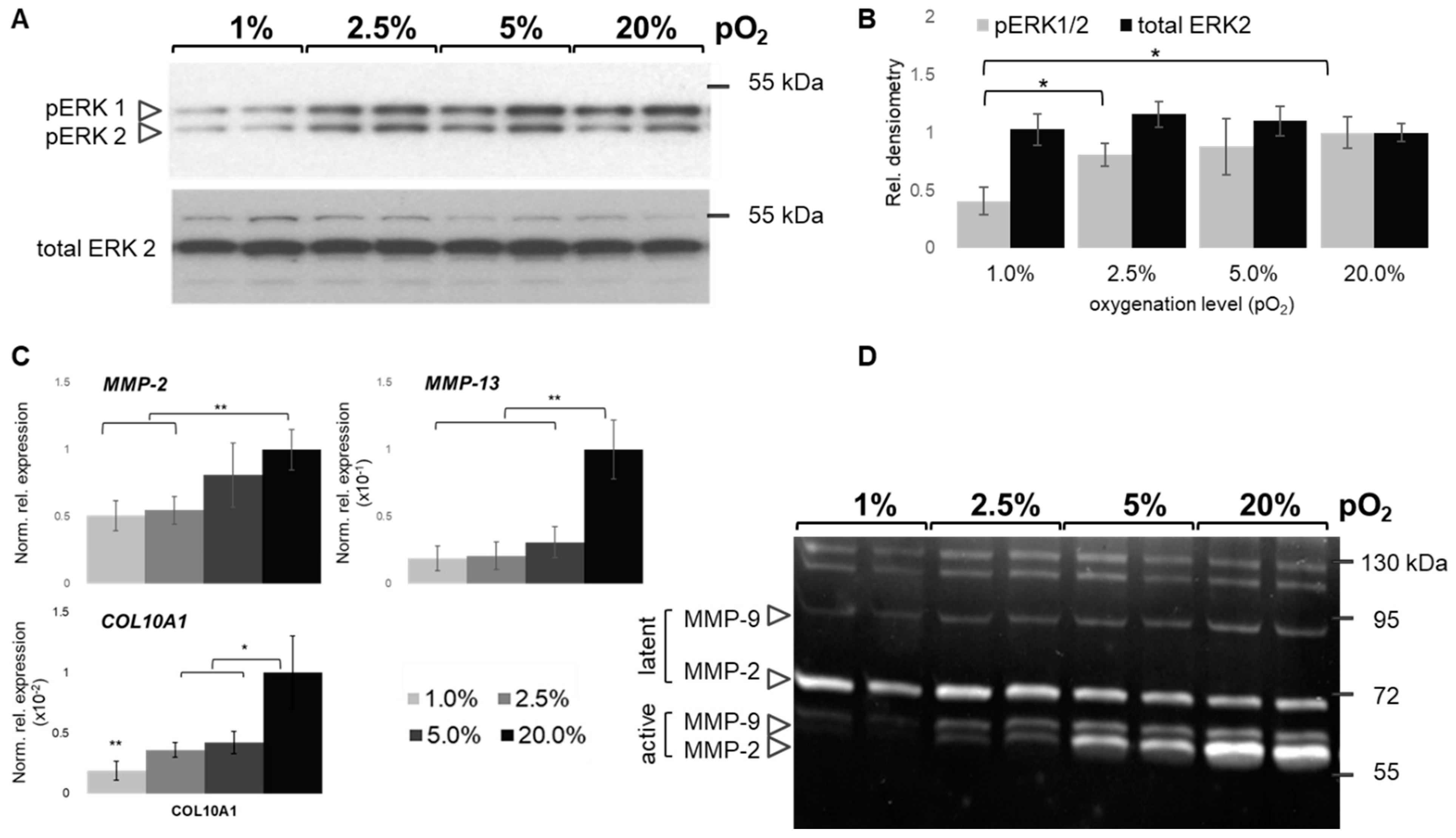
2.7. Oxygen-Dependent MAPK and MMP Activation
3. Discussion
3.1. Physoxia-Preserved Chondrocyte Phenotype
3.2. Regulation of Collagen and MMP Expression
3.3. Regulation of TGFβ Signaling Balance under Physoxia
4. Materials and Methods
4.1. Chondrocyte Expansion Culture
4.2. Bioreactor Culture System and ALK5 Inhibition
4.3. RNA Isolation and RT-qPCR Experiments
4.4. Protein Isolation, Immunoblotting, and ELISA
4.5. ELISA and Zymography
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| ECM | Extracellular Matrix |
| TBSF | TGF-β superfamily |
| TF | Transcription factor |
References
- Silver, I.A. Measurement of pH and ionic composition of pericellular sites. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1975, 271, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Fermor, B.; Christensen, S.E.; Youn, I.; Cernanec, J.M.; Davies, C.M.; Weinberg, J.B. Oxygen, nitric oxide and articular cartilage. Eur. Cell Mater. 2007, 13, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Timur, U.T.; Edip, S.; Haak, E.; Wruck, C.; Weinans, H.; Jahr, H. TGF-β2 is involved in the preservation of the chondrocyte phenotype under hypoxic conditions. Ann. Anat. 2015, 198, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Grimshaw, M.J.; Mason, R.M. Bovine articular chondrocyte function in vitro depends upon oxygen tension. Osteoarthr. Cartil. 2000, 8, 386–392. [Google Scholar] [CrossRef]
- Grimshaw, M.J.; Mason, R.M. Modulation of bovine articular chondrocyte gene expression in vitro by oxygen tension. Osteoarthr. Cartil. 2001, 9, 357–364. [Google Scholar] [CrossRef]
- Ströbel, S.; Loparic, M.; Wendt, D.; Schenk, A.D.; Candrian, C.; Lindberg, R.L.; Moldovan, F.; Barbero, A.; Martin, I. Anabolic and catabolic responses of human articular chondrocytes to varying oxygen percentages. Arthritis Res. 2010, 12, R34. [Google Scholar] [CrossRef] [PubMed]
- Coyle, C.H.; Izzo, N.J.; Chu, C.R. Sustained hypoxia enhances chondrocyte matrix synthesis. J. Orthop. Res. 2009, 27, 793–799. [Google Scholar] [CrossRef]
- Shi, Y.; Ma, J.; Zhang, X.; Li, H.; Jiang, L.; Qin, J. Hypoxia combined with spheroid culture improves cartilage specific function in chondrocytes. Integr. Biol. 2015, 7, 289–297. [Google Scholar] [CrossRef]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Blancher, C.; Moore, J.W.; Talks, K.L.; Houlbrook, S.; Harris, A.L. Relationship of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression to vascular endothelial growth factor induction and hypoxia survival in human breast cancer cell lines. Cancer Res. 2000, 60, 7106–7113. [Google Scholar]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014, 9, 47–71. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J. How cells read TGF-beta signals. Nat. Rev. Mol. Cell. Biol. 2000, 1, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Massagué, J.; Gomis, R.R. The logic of TGFbeta signaling. Febs Lett. 2006, 580, 2811–2820. [Google Scholar] [CrossRef]
- Shi, Y.; Massagué, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural determinants of Smad function in TGF-β signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef]
- Finnson, K.W.; Parker, W.L.; ten Dijke, P.; Thorikay, M.; Philip, A. ALK1 opposes ALK5/Smad3 signaling and expression of extracellular matrix components in human chondrocytes. J. Bone Min. Res. 2008, 23, 896–906. [Google Scholar] [CrossRef]
- Bush, J.R.; Beier, F. TGF-β and osteoarthritis—The good and the bad. Nat. Med. 2013, 19, 667–669. [Google Scholar] [CrossRef]
- Blaney Davidson, E.N.; Remst, D.F.; Vitters, E.L.; van Beuningen, H.M.; Blom, A.B.; Goumans, M.J.; van den Berg, W.B.; van der Kraan, P.M. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 2009, 182, 7937–7945. [Google Scholar] [CrossRef]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. A role for age-related changes in TGFbeta signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res. 2010, 12, 201. [Google Scholar] [CrossRef]
- Gilbert, R.W.D.; Vickaryous, M.K.; Viloria-Petit, A.M. Signalling by Transforming Growth Factor Beta Isoforms in Wound Healing and Tissue Regeneration. J. Dev. Biol. 2016, 4, 21. [Google Scholar] [CrossRef]
- Barry, F.; Boynton, R.E.; Liu, B.; Murphy, J.M. Chondrogenic differentiation of mesenchymal stem cells from bone marrow: Differentiation-dependent gene expression of matrix components. Exp. Cell. Res. 2001, 268, 189–200. [Google Scholar] [CrossRef]
- Song, J.J.; Aswad, R.; Kanaan, R.A.; Rico, M.C.; Owen, T.A.; Barbe, M.F.; Safadi, F.F.; Popoff, S.N. Connective tissue growth factor (CTGF) acts as a downstream mediator of TGF-beta1 to induce mesenchymal cell condensation. J. Cell Physiol. 2007, 210, 398–410. [Google Scholar] [CrossRef]
- Tuli, R.; Tuli, S.; Nandi, S.; Huang, X.; Manner, P.A.; Hozack, W.J.; Danielson, K.G.; Hall, D.J.; Tuan, R.S. Transforming growth factor-beta-mediated chondrogenesis of human mesenchymal progenitor cells involves N-cadherin and mitogen-activated protein kinase and Wnt signaling cross-talk. J. Biol. Chem. 2003, 278, 41227–41236. [Google Scholar] [CrossRef]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical Smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef]
- Zhang, M.; Yan, Y.; Lim, Y.B.; Tang, D.; Xie, R.; Chen, A.; Tai, P.; Harris, S.E.; Xing, L.; Qin, Y.X.; et al. BMP-2 modulates beta-catenin signaling through stimulation of Lrp5 expression and inhibition of beta-TrCP expression in osteoblasts. J. Cell Biochem. 2009, 108, 896–905. [Google Scholar] [CrossRef]
- Van der Kraan, P.M. Age-related alterations in TGF beta signaling as a causal factor of cartilage degeneration in osteoarthritis. Biomed. Mater. Eng. 2014, 24, 75–80. [Google Scholar]
- van der Kraan, P.M. The changing role of TGFβ in healthy, ageing and osteoarthritic joints. Nat. Rev. Rheumatol. 2017, 13, 155–163. [Google Scholar] [CrossRef]
- van der Kraan, P.M.; van den Berg, W.B. Osteoarthritis in the context of ageing and evolution. Loss of chondrocyte differentiation block during ageing. Ageing Res. Rev. 2008, 7, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Lafont, J.E.; Poujade, F.A.; Pasdeloup, M.; Neyret, P.; Mallein-Gerin, F. Hypoxia potentiates the BMP-2 driven COL2A1 stimulation in human articular chondrocytes via p38 MAPK. Osteoarthr. Cartil. 2016, 24, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Hanna, C.; Hubchak, S.C.; Liang, X.; Rozen-Zvi, B.; Schumacker, P.T.; Hayashida, T.; Schnaper, H.W. Hypoxia-inducible factor-2α and TGF-β signaling interact to promote normoxic glomerular fibrogenesis. Am. J. Physiol. Ren. Physiol. 2013, 305, F1323–F1331. [Google Scholar] [CrossRef]
- Van der Windt, A.E.; Haak, E.; Kops, N.; Verhaar, J.A.; Weinans, H.; Jahr, H. Inhibiting calcineurin activity under physiologic tonicity elevates anabolic but suppresses catabolic chondrocyte markers. Arthritis Rheum. 2012, 64, 1929–1939. [Google Scholar] [CrossRef]
- Van der Windt, A.E.; Haak, E.; Das, R.H.; Kops, N.; Welting, T.J.; Caron, M.M.; van Til, N.P.; Verhaar, J.A.; Weinans, H.; Jahr, H. Physiological tonicity improves human chondrogenic marker expression through nuclear factor of activated T-cells 5 in vitro. Arthritis Res. 2010, 12, R100. [Google Scholar] [CrossRef]
- Das, R.; Jahr, H.; van Osch, G.J.; Farrell, E. The role of hypoxia in bone marrow-derived mesenchymal stem cells: Considerations for regenerative medicine approaches. Tissue Eng. Part B Rev. 2010, 16, 159–168. [Google Scholar] [CrossRef]
- Das, R.H.; van Osch, G.J.; Kreukniet, M.; Oostra, J.; Weinans, H.; Jahr, H. Effects of individual control of pH and hypoxia in chondrocyte culture. J. Orthop. Res. 2010, 28, 537–545. [Google Scholar] [CrossRef]
- Tan Timur, U.; Caron, M.; van den Akker, G.; van der Windt, A.; Visser, J.; van Rhijn, L.; Weinans, H.; Welting, T.; Emans, P.; Jahr, H. Increased TGF-β and BMP Levels and Improved Chondrocyte-Specific Marker Expression In Vitro under Cartilage-Specific Physiological Osmolarity. Int. J. Mol. Sci. 2019, 20, 795. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Wang, X.; Zhao, Y.; Gao, F. Expression of MMPs is dependent on the activity of mitogen-activated protein kinase in chondrosarcoma. Mol. Med. Rep. 2017, 15, 915–921. [Google Scholar] [CrossRef]
- Markway, B.D.; Cho, H.; Johnstone, B. Hypoxia promotes redifferentiation and suppresses markers of hypertrophy and degeneration in both healthy and osteoarthritic chondrocytes. Arthritis Res. 2013, 15, R92. [Google Scholar] [CrossRef]
- Lafont, J.E.; Talma, S.; Murphy, C.L. Hypoxia-inducible factor 2alpha is essential for hypoxic induction of the human articular chondrocyte phenotype. Arthritis Rheum. 2007, 56, 3297–3306. [Google Scholar] [CrossRef]
- Duval, E.; Leclercq, S.; Elissalde, J.M.; Demoor, M.; Galéra, P.; Boumédiene, K. Hypoxia-inducible factor 1alpha inhibits the fibroblast-like markers type I and type III collagen during hypoxia-induced chondrocyte redifferentiation: Hypoxia not only induces type II collagen and aggrecan, but it also inhibits type I and type III collagen in the hypoxia-inducible factor 1alpha-dependent redifferentiation of chondrocytes. Arthritis Rheum. 2009, 60, 3038–3048. [Google Scholar]
- Huang, X.; Zhong, L.; Post, J.N.; Karperien, M. Co-treatment of TGF-β3 and BMP7 is superior in stimulating chondrocyte redifferentiation in both hypoxia and normoxia compared to single treatments. Sci. Rep. 2018, 8, 10251. [Google Scholar] [CrossRef]
- Murphy, C.L.; Thoms, B.L.; Vaghjiani, R.J.; Lafont, J.E. Hypoxia. HIF-mediated articular chondrocyte function: Prospects for cartilage repair. Arthritis Res. 2009, 11, 213. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, R.; Sakai, A.; Nakamura, T.; Kunugita, N.; Norimura, T.; Suzuki, K. Effects of transforming growth factor beta s and basic fibroblast growth factor on articular chondrocytes obtained from immobilised rabbit knees. Ann. Rheum. Dis. 1996, 55, 181–186. [Google Scholar] [CrossRef]
- Roberts, A.B.; Sporn, M.B. Differential expression of the TGF-beta isoforms in embryogenesis suggests specific roles in developing and adult tissues. Mol. Reprod. Dev. 1992, 32, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Deng, L.; Wang, P.; Cheng, C.; Xu, K. Post-translational modification of CREB-1 decreases collagen I expression by inhibiting the TGF-β1 signaling pathway in rat hepatic stellate cells. Mol. Med. Rep. 2016, 14, 5751–5759. [Google Scholar] [CrossRef] [PubMed]
- Akman, H.O.; Zhang, H.; Siddiqui, M.A.; Solomon, W.; Smith, E.L.; Batuman, O.A. Response to hypoxia involves transforming growth factor-beta2 and Smad proteins in human endothelial cells. Blood 2001, 98, 3324–3331. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Maeno, T.; Nomura, M.; Aoyagi-Ikeda, K.; Matsui, H.; Hara, K.; Tanaka, T.; Iso, T.; Suga, T.; Kurabayashi, M. Hypoxia-inducible factor-1α mediates TGF-β-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am. J. Physiol. Lung. Cell Mol. Physiol. 2011, 300, L740–L752. [Google Scholar] [CrossRef] [PubMed]
- Finnson, K.W.; Parker, W.L.; Chi, Y.; Hoemann, C.D.; Goldring, M.B.; Antoniou, J.; Philip, A. Endoglin differentially regulates TGF-β-induced Smad2/3 and Smad1/5 signalling and its expression correlates with extracellular matrix production and cellular differentiation state in human chondrocytes. Osteoarthr. Cartil. 2010, 18, 1518–1527. [Google Scholar] [CrossRef] [PubMed]
- Cheifetz, S.; Bellón, T.; Calés, C.; Vera, S.; Bernabeu, C.; Massagué, J.; Letarte, M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J. Biol. Chem. 1992, 267, 19027–19030. [Google Scholar]
- Zhu, Y.; Sun, Y.; Xie, L.; Jin, K.; Sheibani, N.; Greenberg, D.A. Hypoxic induction of endoglin via mitogen-activated protein kinases in mouse brain microvascular endothelial cells. Stroke 2003, 34, 2483–2488. [Google Scholar] [CrossRef]
- Parker, W.L.; Goldring, M.B.; Philip, A. Endoglin is expressed on human chondrocytes and forms a heteromeric complex with betaglycan in a ligand and type II TGFbeta receptor independent manner. J. Bone Min. Res. 2003, 18, 289–302. [Google Scholar] [CrossRef]
- Sánchez-Elsner, T.; Botella, L.M.; Velasco, B.; Langa, C.; Bernabéu, C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. J. Biol. Chem. 2002, 277, 43799–43808. [Google Scholar] [CrossRef]
- Martin, G.; Andriamanalijaona, R.; Grässel, S.; Dreier, R.; Mathy-Hartert, M.; Bogdanowicz, P.; Boumédiene, K.; Henrotin, Y.; Bruckner, P.; Pujol, J.P. Effect of hypoxia and reoxygenation on gene expression and response to interleukin-1 in cultured articular chondrocytes. Arthritis Rheum. 2004, 50, 3549–3560. [Google Scholar] [CrossRef] [PubMed]
- Denis, J.F.; Sader, F.; Gatien, S.; Villiard, É.; Philip, A.; Roy, S. Activation of Smad2 but not Smad3 is required to mediate TGF-β signaling during axolotl limb regeneration. Development 2016, 143, 3481–3490. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Law, B.K.; Chytil, A.M.; Brown, K.A.; Aakre, M.E.; Moses, H.L. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia 2004, 6, 603–610. [Google Scholar] [CrossRef]
- Principe, D.R.; Diaz, A.M.; Torres, C.; Mangan, R.J.; DeCant, B.; McKinney, R.; Tsao, M.S.; Lowy, A.; Munshi, H.G.; Jung, B.; et al. TGFβ engages MEK/ERK to differentially regulate benign and malignant pancreas cell function. Oncogene 2017, 36, 4336–4348. [Google Scholar] [CrossRef] [PubMed]
- Appleton, C.T.; Usmani, S.E.; Mort, J.S.; Beier, F. Rho/ROCK and MEK/ERK activation by transforming growth factor-alpha induces articular cartilage degradation. Lab. Investig. 2010, 90, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Rigueur, D.; Lyons, K.M. TGFβ signaling in cartilage development and maintenance. Birth Defects Res. C Embryo Today 2014, 102, 37–51. [Google Scholar] [CrossRef]
- Shu, B.; Zhang, M.; Xie, R.; Wang, M.; Jin, H.; Hou, W.; Tang, D.; Harris, S.E.; Mishina, Y.; O’Keefe, R.J.; et al. BMP2, but not BMP4, is crucial for chondrocyte proliferation and maturation during endochondral bone development. J. Cell Sci. 2011, 124, 3428–3440. [Google Scholar] [CrossRef] [PubMed]
- Caron, M.M.; Emans, P.J.; Cremers, A.; Surtel, D.A.; Coolsen, M.M.; van Rhijn, L.W.; Welting, T.J. Hypertrophic differentiation during chondrogenic differentiation of progenitor cells is stimulated by BMP-2 but suppressed by BMP-7. Osteoarthr. Cartil. 2013, 21, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Coricor, G.; Serra, R. TGF-β regulates phosphorylation and stabilization of Sox9 protein in chondrocytes through p38 and Smad dependent mechanisms. Sci. Rep. 2016, 6, 38616. [Google Scholar] [CrossRef]
- Jenkins, G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [CrossRef]
- Bernabeu, C.; Lopez-Novoa, J.M.; Quintanilla, M. The emerging role of TGF-beta superfamily coreceptors in cancer. Biochim. Biophys. Acta 2009, 1792, 954–973. [Google Scholar] [CrossRef]
- Velasco, S.; Alvarez-Muñoz, P.; Pericacho, M.; Dijke, P.T.; Bernabéu, C.; López-Novoa, J.M.; Rodríguez-Barbero, A. L- and S-endoglin differentially modulate TGFbeta1 signaling mediated by ALK1 and ALK5 in L6E9 myoblasts. J. Cell Sci. 2008, 121, 913–919. [Google Scholar] [CrossRef]
- Asp, J.; Thornemo, M.; Inerot, S.; Lindahl, A. The helix-loop-helix transcription factors Id1 and Id3 have a functional role in control of cell division in human normal and neoplastic chondrocytes. Febs Lett. 1998, 438, 85–90. [Google Scholar] [CrossRef]
- Dehne, T.; Schenk, R.; Perka, C.; Morawietz, L.; Pruss, A.; Sittinger, M.; Kaps, C.; Ringe, J. Gene expression profiling of primary human articular chondrocytes in high-density micromasses reveals patterns of recovery, maintenance, re- and dedifferentiation. Gene 2010, 462, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Wiercinska, E.; Wickert, L.; Denecke, B.; Said, H.M.; Hamzavi, J.; Gressner, A.M.; Thorikay, M.; ten Dijke, P.; Mertens, P.R.; Breitkopf, K.; et al. Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology 2006, 43, 1032–1041. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; He, S.; Sun, J.M.; Davie, J.R. Gene regulation by Sp1 and Sp3. Biochem. Cell Biol. 2004, 82, 460–471. [Google Scholar] [CrossRef]
- Magee, C.; Nurminskaya, M.; Faverman, L.; Galera, P.; Linsenmayer, T.F. SP3/SP1 transcription activity regulates specific expression of collagen type X in hypertrophic chondrocytes. J. Biol. Chem. 2005, 280, 25331–25338. [Google Scholar] [CrossRef]
- Chadjichristos, C.; Ghayor, C.; Herrouin, J.F.; Ala-Kokko, L.; Suske, G.; Pujol, J.P.; Galéra, P. Down-regulation of human type II collagen gene expression by transforming growth factor-beta 1 (TGF-beta 1) in articular chondrocytes involves SP3/SP1 ratio. J. Biol. Chem. 2002, 277, 43903–43917. [Google Scholar] [CrossRef]
- Ghayor, C.; Chadjichristos, C.; Herrouin, J.F.; Ala-Kokko, L.; Suske, G.; Pujol, J.P.; Galera, P. Sp3 represses the Sp1-mediated transactivation of the human COL2A1 gene in primary and de-differentiated chondrocytes. J. Biol. Chem. 2001, 276, 36881–36895. [Google Scholar] [CrossRef]
- Duval, E.; Bouyoucef, M.; Leclercq, S.; Baugé, C.; Boumédiene, K. Hypoxia inducible factor 1 alpha down-regulates type i collagen through Sp3 transcription factor in human chondrocytes. Iubmb. Life 2016, 68, 756–763. [Google Scholar] [CrossRef]
- Yang, X.; Chen, L.; Xu, X.; Li, C.; Huang, C.; Deng, C.X. TGF-beta/Smad3 signals repress chondrocyte hypertrophic differentiation and are required for maintaining articular cartilage. J. Cell Biol. 2001, 153, 35–46. [Google Scholar] [CrossRef]
- Van Caam, A.; Madej, W.; Garcia de Vinuesa, A.; Goumans, M.J.; Ten Dijke, P.; Blaney Davidson, E.; van der Kraan, P. TGFβ1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. 2017, 19, 112. [Google Scholar] [CrossRef]
- Tchetina, E.V.; Markova, G.A. Regulation of energy metabolism in the growth plate and osteoarthritic chondrocytes. Rheumatol. Int. 2018, 38, 1963–1974. [Google Scholar] [CrossRef]
- Matsumoto, H.; Silverton, S.F.; Debolt, K.; Shapiro, I.M. Superoxide dismutase and catalase activities in the growth cartilage: Relationship between oxidoreductase activity and chondrocyte maturation. J. Bone Min. Res. 1991, 6, 569–574. [Google Scholar] [CrossRef]
- Schalkwijk, J.; van den Berg, W.B.; van de Putte, L.B.; Joosten, L.A. Hydrogen peroxide suppresses the proteoglycan synthesis of intact articular cartilage. J. Rheumatol. 1985, 12, 205–210. [Google Scholar]
- Das, R.H.; Jahr, H.; Verhaar, J.A.; van der Linden, J.C.; van Osch, G.J.; Weinans, H. In vitro expansion affects the response of chondrocytes to mechanical stimulation. Osteoarthr. Cartil. 2008, 16, 385–391. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef]
- van der Windt, A.E.; Jahr, H.; Farrell, E.; Verhaar, J.A.; Weinans, H.; van Osch, G.J. Calcineurin inhibitors promote chondrogenic marker expression of dedifferentiated human adult chondrocytes via stimulation of endogenous TGFbeta1 production. Tissue Eng. Part A 2010, 16, 1–10. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Uitterlinden, E.J.; Jahr, H.; Koevoet, J.L.; Jenniskens, Y.M.; Bierma-Zeinstra, S.M.; Degroot, J.; Verhaar, J.A.; Weinans, H.; van Osch, G.J. Glucosamine decreases expression of anabolic and catabolic genes in human osteoarthritic cartilage explants. Osteoarthr. Cartil. 2006, 14, 250–257. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mueller, M.R.; Folz, R.J. Transcription factors sp1 and sp3 regulate expression of human extracellular superoxide dismutase in lung fibroblasts. Am. J. Respir Cell Mol. Biol. 2008, 39, 243–251. [Google Scholar] [CrossRef]
- Jahr, H.; van Driel, M.; van Osch, G.J.; Weinans, H.; van Leeuwen, J.P. Identification of acid-sensing ion channels in bone. Biochem. Biophys. Res. Commun. 2005, 337, 349–354. [Google Scholar] [CrossRef]
- Caron, M.M.; van der Windt, A.E.; Emans, P.J.; van Rhijn, L.W.; Jahr, H.; Welting, T.J. Osmolarity determines the in vitro chondrogenic differentiation capacity of progenitor cells via nuclear factor of activated T-cells 5. Bone 2013, 53, 94–102. [Google Scholar] [CrossRef]
- Caron, M.M.; Emans, P.J.; Coolsen, M.M.; Voss, L.; Surtel, D.A.; Cremers, A.; van Rhijn, L.W.; Welting, T.J. Redifferentiation of dedifferentiated human articular chondrocytes: Comparison of 2D and 3D cultures. Osteoarthr. Cartil. 2012, 20, 1170–1178. [Google Scholar] [CrossRef]
- Siebelt, M.; Jahr, H.; Groen, H.C.; Sandker, M.; Waarsing, J.H.; Kops, N.; Müller, C.; van Eden, W.; de Jong, M.; Weinans, H. Hsp90 inhibition protects against biomechanically induced osteoarthritis in rats. Arthritis Rheum. 2013, 65, 2102–2112. [Google Scholar] [CrossRef]
- Farrell, E.; van der Jagt, O.P.; Koevoet, W.; Kops, N.; van Manen, C.J.; Hellingman, C.A.; Jahr, H.; O’Brien, F.J.; Verhaar, J.A.; Weinans, H.; et al. Chondrogenic priming of human bone marrow stromal cells: A better route to bone repair? Tissue Eng. Part C Methods 2009, 15, 285–295. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahr, H.; Gunes, S.; Kuhn, A.-R.; Nebelung, S.; Pufe, T. Bioreactor-Controlled Physoxia Regulates TGF-β Signaling to Alter Extracellular Matrix Synthesis by Human Chondrocytes. Int. J. Mol. Sci. 2019, 20, 1715. https://doi.org/10.3390/ijms20071715
Jahr H, Gunes S, Kuhn A-R, Nebelung S, Pufe T. Bioreactor-Controlled Physoxia Regulates TGF-β Signaling to Alter Extracellular Matrix Synthesis by Human Chondrocytes. International Journal of Molecular Sciences. 2019; 20(7):1715. https://doi.org/10.3390/ijms20071715
Chicago/Turabian StyleJahr, Holger, Seval Gunes, Annika-Ricarda Kuhn, Sven Nebelung, and Thomas Pufe. 2019. "Bioreactor-Controlled Physoxia Regulates TGF-β Signaling to Alter Extracellular Matrix Synthesis by Human Chondrocytes" International Journal of Molecular Sciences 20, no. 7: 1715. https://doi.org/10.3390/ijms20071715
APA StyleJahr, H., Gunes, S., Kuhn, A.-R., Nebelung, S., & Pufe, T. (2019). Bioreactor-Controlled Physoxia Regulates TGF-β Signaling to Alter Extracellular Matrix Synthesis by Human Chondrocytes. International Journal of Molecular Sciences, 20(7), 1715. https://doi.org/10.3390/ijms20071715