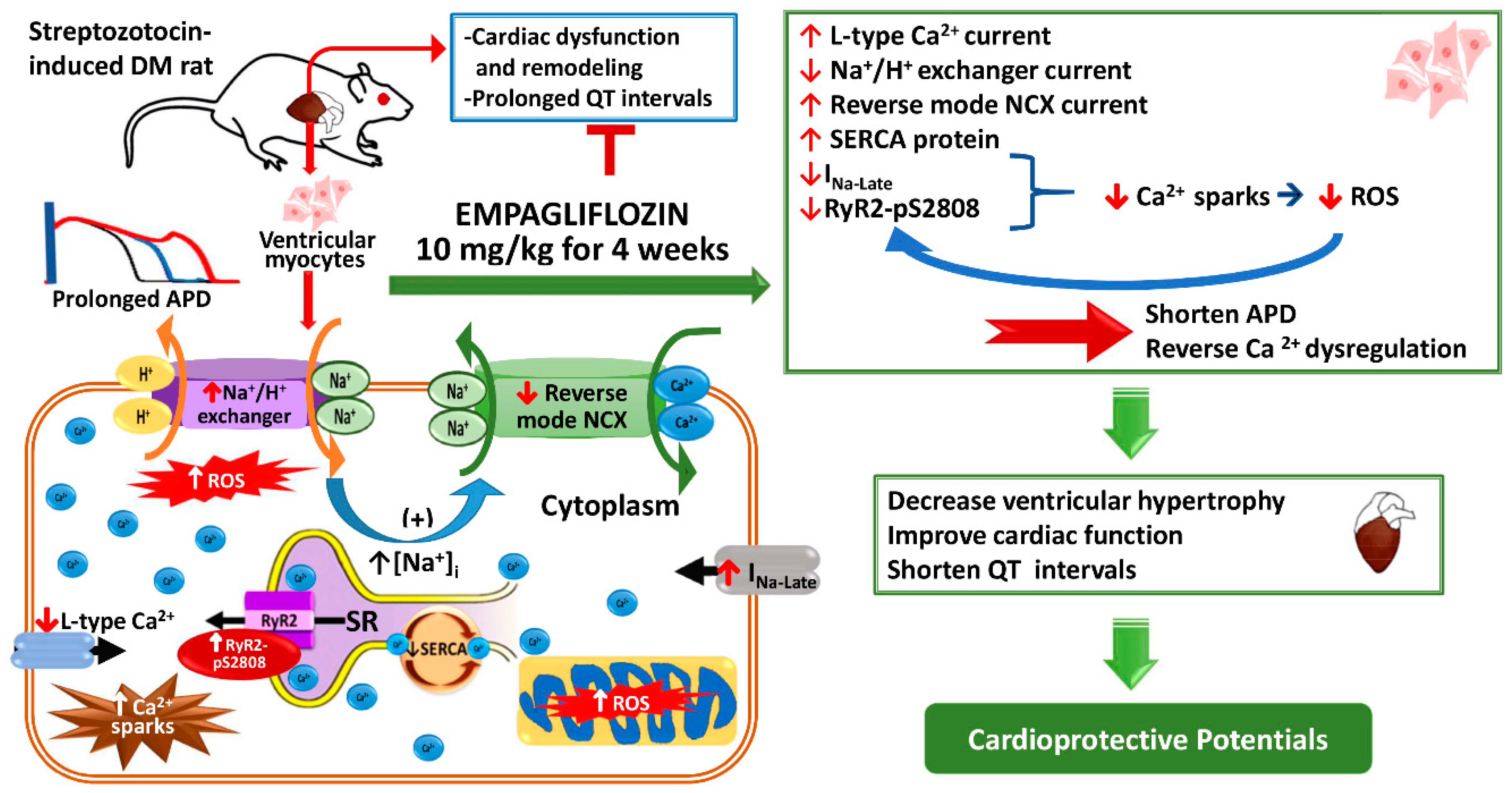
Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats

Abstract
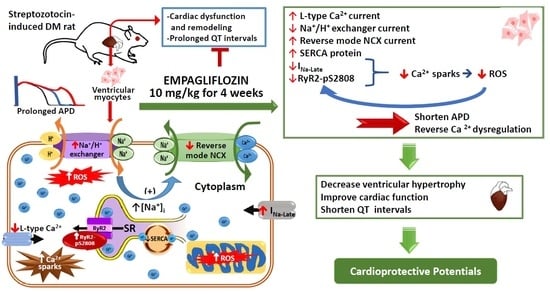
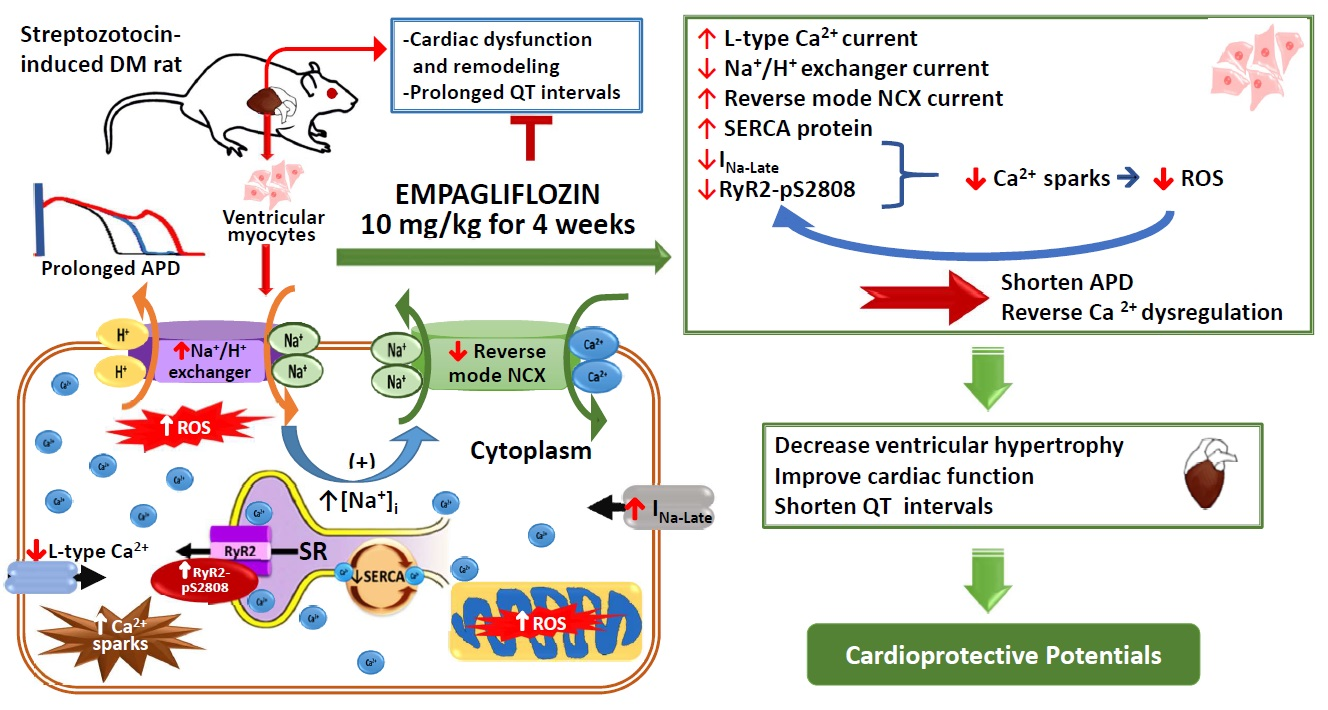
1. Introduction
2. Results
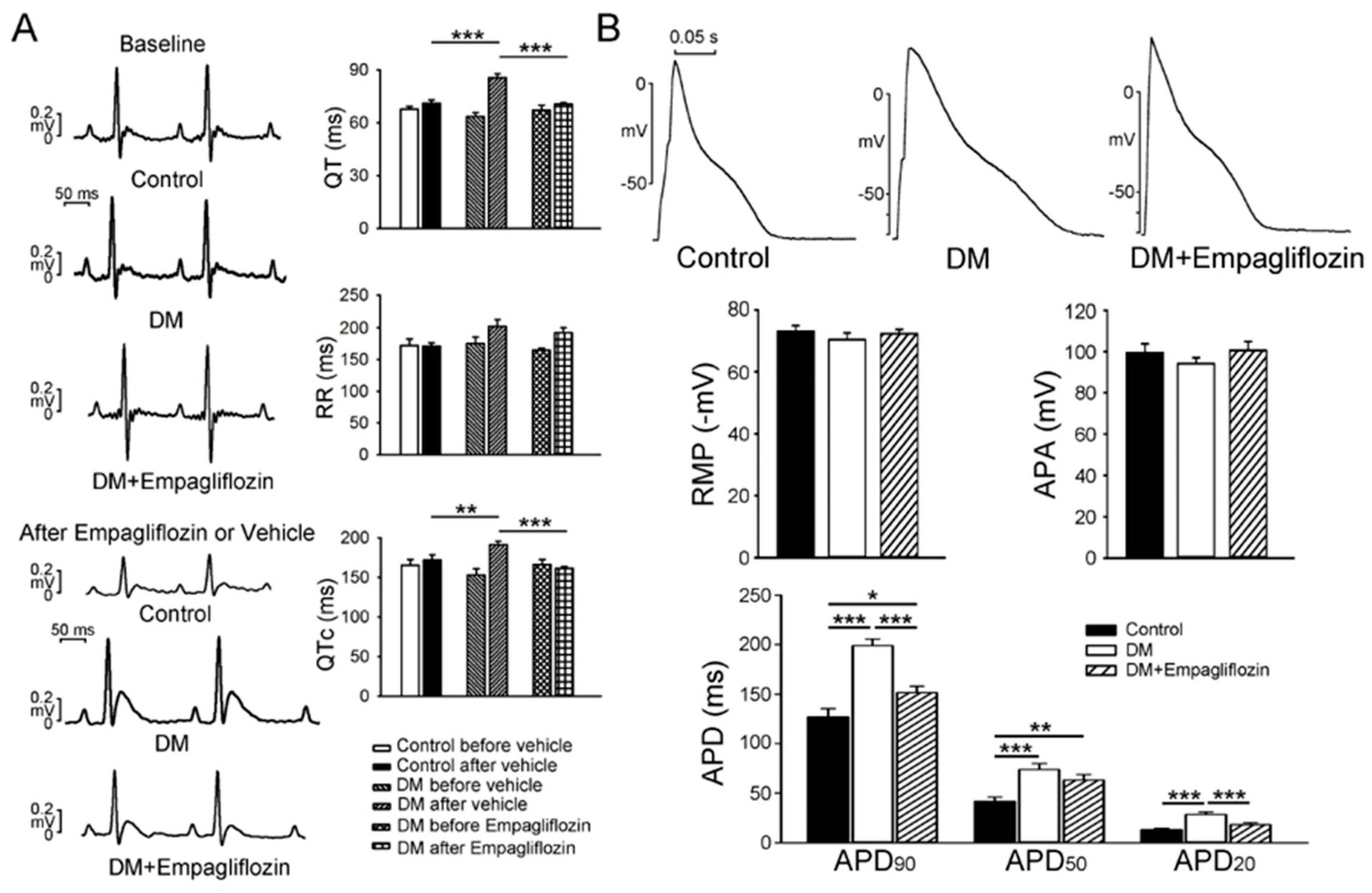
2.1. Cardiac Structure and Electrocardiography of Control, DM, and Empagliflozin-Treated DM Rats
2.2. Effects of Empagliflozin on Action Potentials (APs) in DM Rat
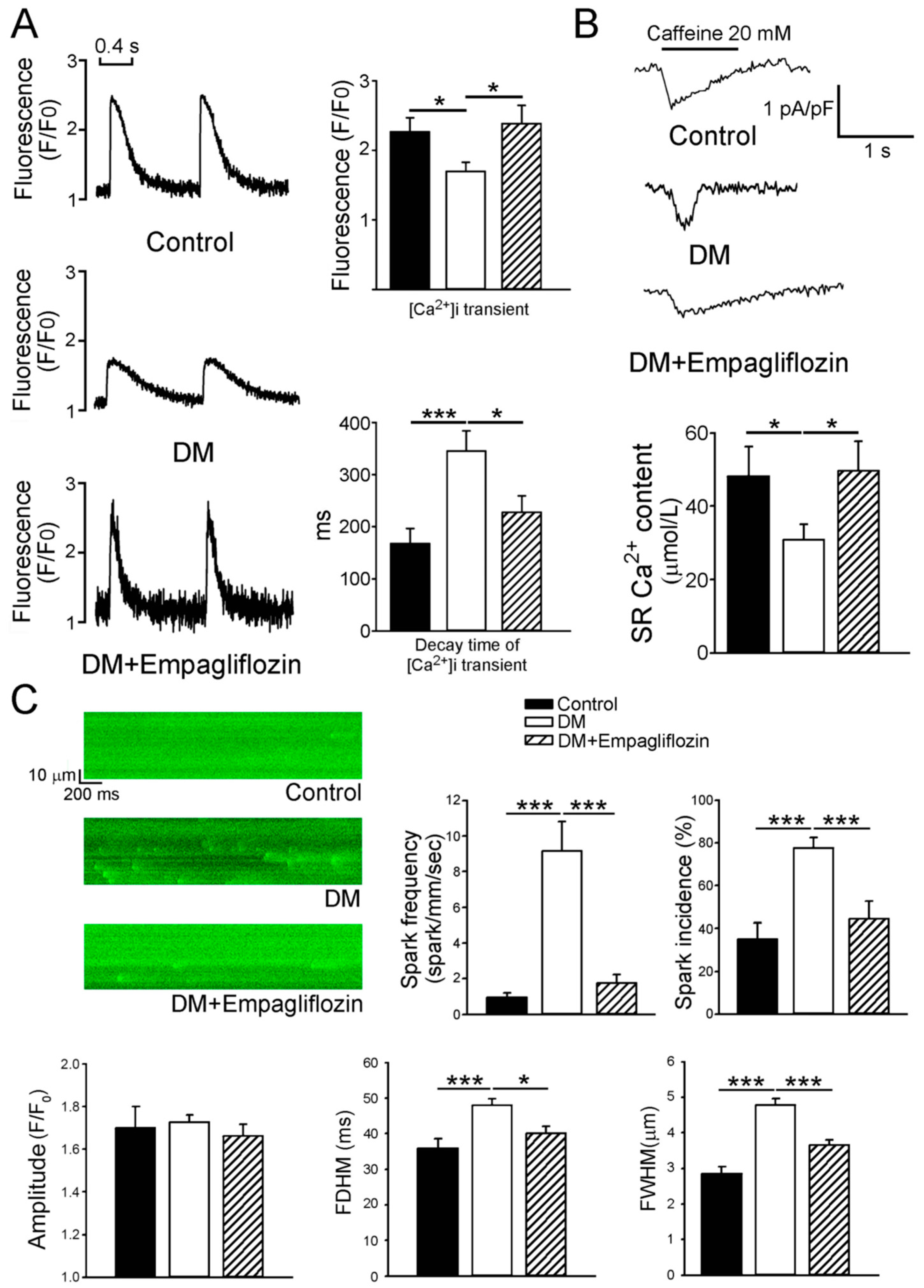
2.3. Effects of Empagliflozin on Ca2+ Stores in DM Rats
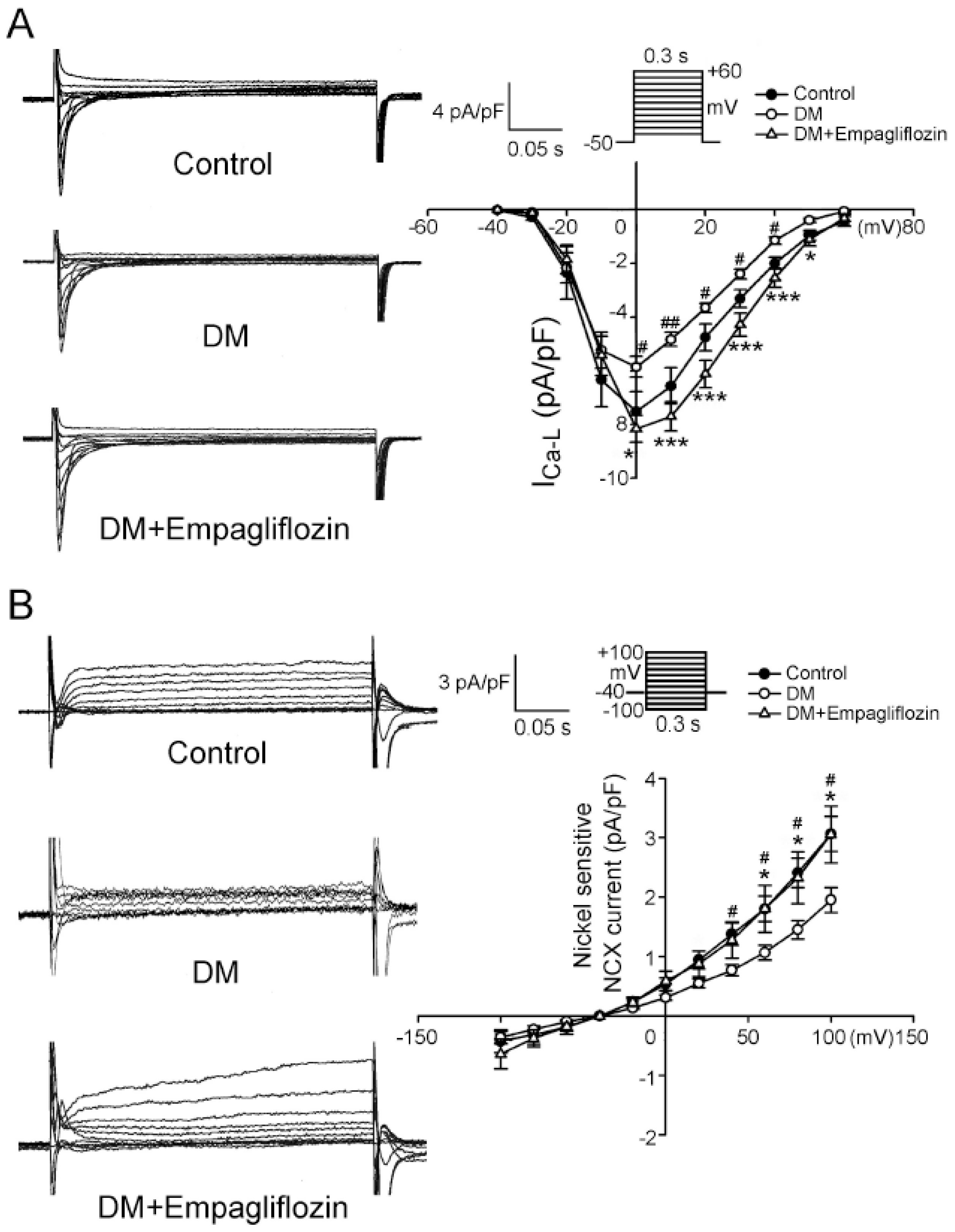
2.4. Effects of Empagliflozin on L-type Ca2+ Channel (ICa-L) Current and NCX Current in DM Rats
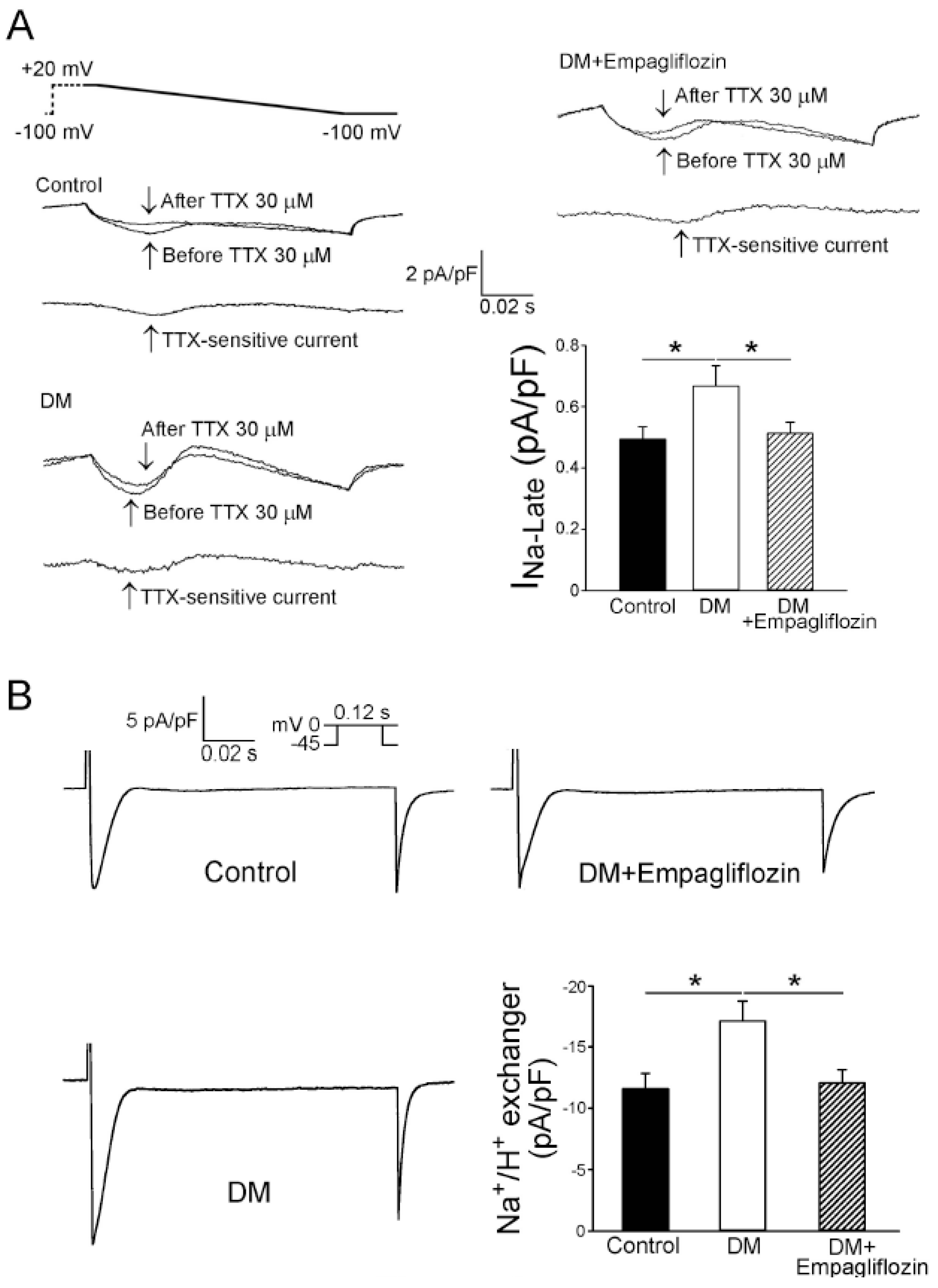
2.5. Effects of Empagliflozin on Late Na+ Channel (INa-late) Current and Na+/Hydrogen (Na+/H+) Exchanger Current in DM Rats
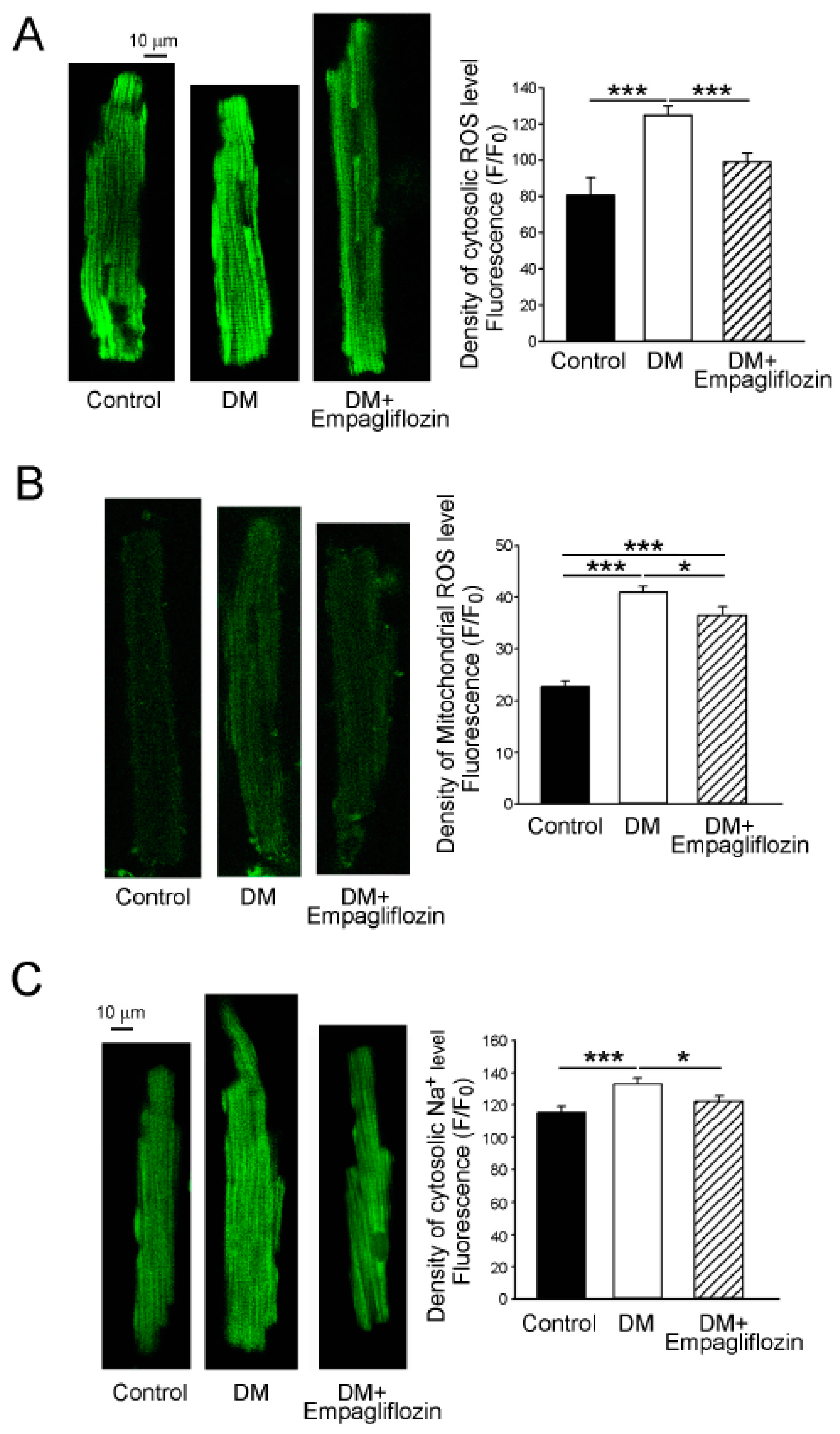
2.6. Effects of Empagliflozin on Oxidative Stress in DM Rats
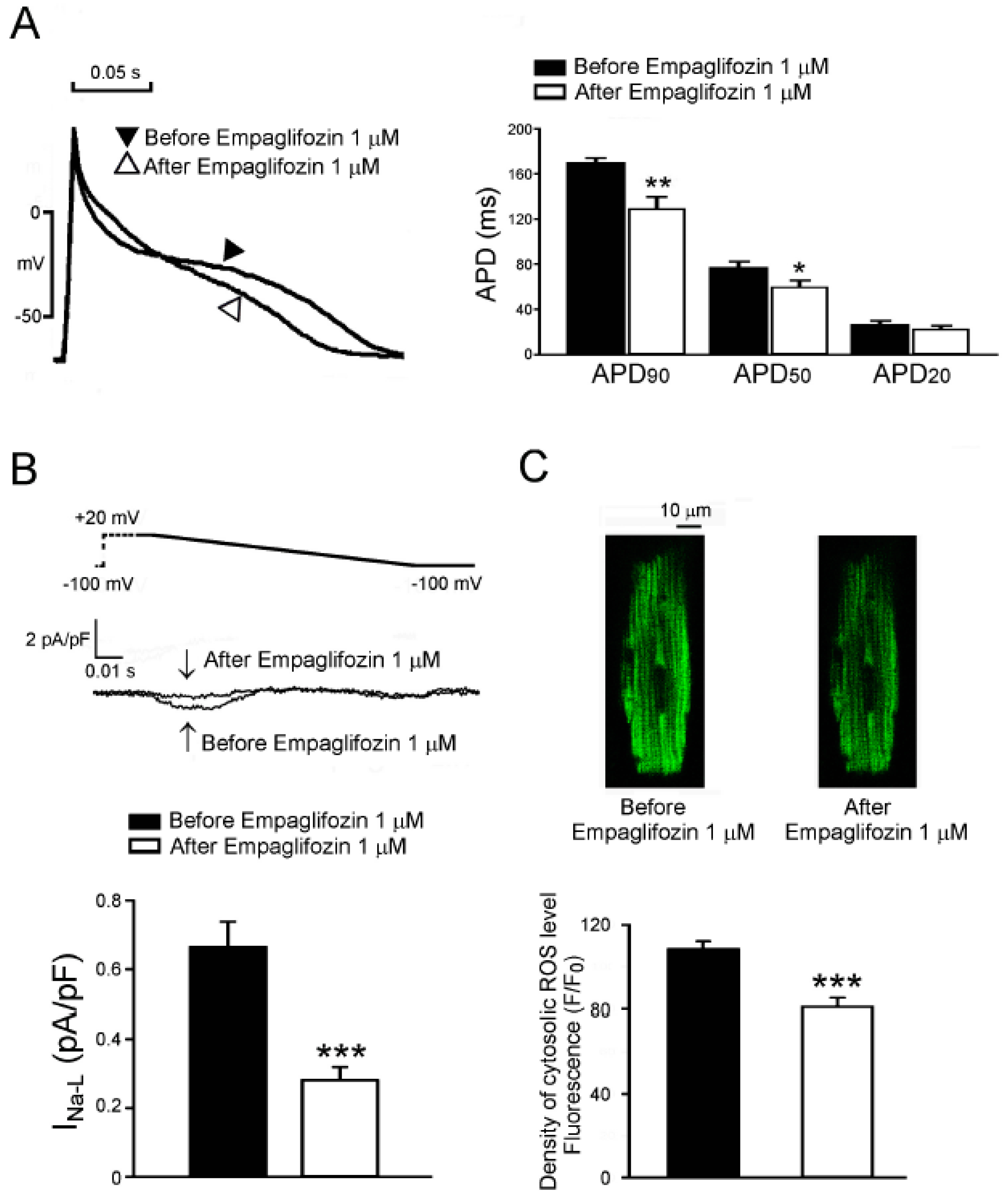
2.7. Acute Effects of Empagliflozin on DM Rat Ventricular Myocytes
2.8. Effects of Empagliflozin on Ca2+ Regulatory Proteins in DM Rats
3. Discussion
Study Limitation
4. Methods
4.1. Ethical Approval
4.2. Induction of DM, Treatment and Tissue Harvesting
4.3. Echocardiography and Electrocardiography Measurements
4.4. Measurement of [Ca2+]i, SR Ca2+ Contents, and Ca2+ Spark Imaging
4.5. AP and Ionic Currents
4.6. Measurement of Cell Size and Intracellular ROS and Na+
4.7. Western Blot Analysis
4.8. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Stamler, J.; Vaccaro, O.; Neaton, J.D.; Wentworth, D. Diabetes, other risk factors, and 12-yr cardiovascular mortality for men screened in the Multiple Risk Factor Intervention Trial. Diabetes Care 1993, 16, 434–444. [Google Scholar] [CrossRef]
- Morrish, N.J.; Wang, S.L.; Stevens, L.K.; Fuller, J.H.; Keen, H. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia 2001, 44 (Suppl. 2), S14–S21. [Google Scholar] [CrossRef]
- Scognamiglio, R.; Avogaro, A.; Negut, C.; Piccolotto, R.; Vigili de Kreutzenberg, S.; Tiengo, A. Early myocardial dysfunction in the diabetic heart: Current research and clinical applications. Am. J. Cardiol. 2004, 93, 17A–20A. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.Y.; Prins, J.B.; Marwick, T.H. Diabetic cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Endocr. Rev. 2004, 25, 543–567. [Google Scholar] [CrossRef] [PubMed]
- Pfister, M.; Whaley, J.M.; Zhang, L.; List, J.F. Inhibition of SGLT2: A novel strategy for treatment of type 2 diabetes mellitus. Clin. Pharmacol. Ther. 2011, 89, 621–625. [Google Scholar] [CrossRef]
- Henry, R.R.; Thakkar, P.; Tong, C.; Polidori, D.; Alba, M. Efficacy and Safety of Canagliflozin, a Sodium-Glucose Cotransporter 2 Inhibitor, as Add-on to Insulin in Patients with Type 1 Diabetes. Diabetes Care 2015, 38, 2258–2265. [Google Scholar] [CrossRef]
- Van den Mooter, L.; Caerels, S.; Mathieu, C. Efficacy of dapagliflozin as an adjunct therapy in patients with inadequately controlled type 1 diabetes mellitus. Expert Opin. Pharmacother. 2018, 19, 617–622. [Google Scholar] [CrossRef]
- Nauck, M.A. Update on developments with SGLT2 inhibitors in the management of type 2 diabetes. Drug Des. Dev. Ther. 2014, 8, 1335–1380. [Google Scholar] [CrossRef]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef]
- Fein, F.S.; Aronson, R.S.; Nordin, C.; Miller-Green, B.; Sonnenblick, E.H. Altered myocardial response to ouabain in diabetic rats: Mechanics and electrophysiology. J. Mol. Cell. Cardiol. 1983, 15, 769–784. [Google Scholar] [CrossRef]
- Lambert, R.; Srodulski, S.; Peng, X.; Margulies, K.B.; Despa, F.; Despa, S. Intracellular Na+ Concentration ([Na+]i) Is Elevated in Diabetic Hearts Due to Enhanced Na+-Glucose Cotransport. J. Am. Heart Assoc. 2015, 4, e002183. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef]
- Lee, T.I.; Chen, Y.C.; Kao, Y.H.; Hsiao, F.C.; Lin, Y.K.; Chen, Y.J. Rosiglitazone induces arrhythmogenesis in diabetic hypertensive rats with calcium handling alteration. Int. J. Cardiol. 2013, 165, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Huang, J.H.; Hsu, M.I.; Chen, Y.J. The dipeptidyl peptidase-4 inhibitor-sitagliptin modulates calcium dysregulation, inflammation, and PPARs in hypertensive cardiomyocytes. Int. J. Cardiol. 2013, 168, 5390–5395. [Google Scholar] [CrossRef]
- Huang, J.H.; Chen, Y.C.; Lee, T.I.; Kao, Y.H.; Chazo, T.F.; Chen, S.A.; Chen, Y.J. Glucagon-like peptide-1 regulates calcium homeostasis and electrophysiological activities of HL-1 cardiomyocytes. Peptides 2016, 78, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Trost, S.U.; Belke, D.D.; Bluhm, W.F.; Meyer, M.; Swanson, E.; Dillmann, W.H. Overexpression of the sarcoplasmic reticulum Ca2+-ATPase improves myocardial contractility in diabetic cardiomyopathy. Diabetes 2002, 51, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Matsuda, N.; Kimura, J.; Ishitani, T.; Tamada, A.; Gando, S.; Kemmotsu, O.; Kanno, M. Diminished function and expression of the cardiac Na+-Ca2+ exchanger in diabetic rats: Implication in Ca2+ overload. J. Physiol. 2000, 527 Pt 1, 85–94. [Google Scholar] [CrossRef]
- Teshima, Y.; Takahashi, N.; Saikawa, T.; Hara, M.; Yasunaga, S.; Hidaka, S.; Sakata, T. Diminished expression of sarcoplasmic reticulum Ca2+-ATPase and ranodine sensitive Ca2+Channel mRNA in streptozotocin-induced diabetic rat heart. J. Mol. Cell. Cardiol. 2000, 32, 655–664. [Google Scholar] [CrossRef]
- Vetter, R.; Rehfeld, U.; Reissfelder, C.; Weiss, W.; Wagner, K.D.; Gunther, J.; Hammes, A.; Tschope, C.; Dillmann, W.; Paul, M. Transgenic overexpression of the sarcoplasmic reticulum Ca2+ATPase improves reticular Ca2+ handling in normal and diabetic rat hearts. FASEB J. 2002, 16, 1657–1659. [Google Scholar] [CrossRef]
- Fein, F.S.; Zola, B.E.; Malhotra, A.; Cho, S.; Factor, S.M.; Scheuer, J.; Sonnenblick, E.H. Hypertensive-diabetic cardiomyopathy in rats. Am. J. Physiol. 1990, 258, H793–H805. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.K.; Sisson, J.C.; Vinik, A.I. QT interval prolongation and sudden cardiac death in diabetic autonomic neuropathy. J. Clin. Endocrinol. Metab. 1987, 64, 751–754. [Google Scholar] [CrossRef]
- Magyar, J.; Rusznak, Z.; Szentesi, P.; Szucs, G.; Kovacs, L. Action potentials and potassium currents in rat ventricular muscle during experimental diabetes. J. Mol. Cell. Cardiol. 1992, 24, 841–853. [Google Scholar] [CrossRef]
- Habibi, J.; Aroor, A.R.; Sowers, J.R.; Jia, G.; Hayden, M.R.; Garro, M.; Barron, B.; Mayoux, E.; Rector, R.S.; Whaley-Connell, A.; et al. Sodium glucose transporter 2 (SGLT2) inhibition with empagliflozin improves cardiac diastolic function in a female rodent model of diabetes. Cardiovasc. Diabetol. 2017, 16, 9. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Ungvari, Z.; Nanasi, P.P.; Kecskemeti, V. Electrophysiological changes in rat ventricular and atrial myocardium at different stages of experimental diabetes. Acta Physiol. Scand. 1999, 166, 7–13. [Google Scholar] [CrossRef]
- Despa, S. Myocyte [Na+]i Dysregulation in Heart Failure and Diabetic Cardiomyopathy. Front. Physiol. 2018, 9, 1303. [Google Scholar] [CrossRef]
- Choi, K.M.; Zhong, Y.; Hoit, B.D.; Grupp, I.L.; Hahn, H.; Dilly, K.W.; Guatimosim, S.; Lederer, W.J.; Matlib, M.A. Defective intracellular Ca2+ signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1398–H1408. [Google Scholar] [CrossRef]
- Kotsanas, G.; Delbridge, L.M.; Wendt, I.R. Stimulus interval-dependent differences in Ca2+ transients and contractile responses of diabetic rat cardiomyocytes. Cardiovasc. Res. 2000, 46, 450–462. [Google Scholar] [CrossRef]
- Asghar, O.; Al-Sunni, A.; Khavandi, K.; Khavandi, A.; Withers, S.; Greenstein, A.; Heagerty, A.M.; Malik, R.A. Diabetic cardiomyopathy. Clin. Sci. 2009, 116, 741–760. [Google Scholar] [CrossRef]
- Hammoudi, N.; Jeong, D.; Singh, R.; Farhat, A.; Komajda, M.; Mayoux, E.; Hajjar, R.; Lebeche, D. Empagliflozin Improves Left Ventricular Diastolic Dysfunction in a Genetic Model of Type 2 Diabetes. Cardiovasc. Drugs Ther. 2017, 31, 233–246. [Google Scholar] [CrossRef]
- Yuill, K.H.; Al Kury, L.T.; Howarth, F.C. Characterization of L-type calcium channel activity in atrioventricular nodal myocytes from rats with streptozotocin-induced Diabetes mellitus. Physiol. Rep. 2015, 3, 12632. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Matthes, J.; Schuster, I.; Valdivia, H.H.; Herzig, S.; Richard, S.; Gomez, A.M. Mechanisms of [Ca2+]i transient decrease in cardiomyopathy of db/db type 2 diabetic mice. Diabetes 2006, 55, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.W.; Kiyosue, T.; Shigematsu, S.; Arita, M. Abnormalities of K+ and Ca2+ currents in ventricular myocytes from rats with chronic diabetes. Am. J. Physiol. 1995, 269, H1288–H1296. [Google Scholar] [CrossRef] [PubMed]
- Chattou, S.; Diacono, J.; Feuvray, D. Decrease in sodium-calcium exchange and calcium currents in diabetic rat ventricular myocytes. Acta Physiol. Scand. 1999, 166, 137–144. [Google Scholar] [CrossRef]
- Lu, Z.; Jiang, Y.P.; Xu, X.H.; Ballou, L.M.; Cohen, I.S.; Lin, R.Z. Decreased L-type Ca2+ current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes 2007, 56, 2780–2789. [Google Scholar] [CrossRef]
- Baartscheer, A.; Schumacher, C.A.; Wust, R.C.; Fiolet, J.W.; Stienen, G.J.; Coronel, R.; Zuurbier, C.J. Empagliflozin decreases myocardial cytoplasmic Na+ through inhibition of the cardiac Na+/H+ exchanger in rats and rabbits. Diabetologia 2017, 60, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Laffel, L.M.B.; Tamborlane, W.V.; Yver, A.; Simons, G.; Wu, J.; Nock, V.; Hobson, D.; Hughan, K.S.; Kaspers, S.; Marquard, J. Pharmacokinetic and pharmacodynamic profile of the sodium-glucose co-transporter-2 inhibitor empagliflozin in young people with Type 2 diabetes: A randomized trial. Diabet. Med. 2018, 35, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Duchen, M.R. Calcium microdomains and oxidative stress. Cell Calcium 2006, 40, 561–574. [Google Scholar] [CrossRef]
- Teshima, Y.; Takahashi, N.; Nishio, S.; Saito, S.; Kondo, H.; Fukui, A.; Aoki, K.; Yufu, K.; Nakagawa, M.; Saikawa, T. Production of reactive oxygen species in the diabetic heart. Roles of mitochondria and NADPH oxidase. Circ. J. 2014, 78, 300–306. [Google Scholar] [CrossRef]
- Ono, K.; Iijima, T. Cardiac T-type Ca2+ channels in the heart. J. Mol. Cell. Cardiol. 2010, 48, 65–70. [Google Scholar] [CrossRef]
- Fakhrzadeh, H.; Yamini-Sharif, A.; Sharifi, F.; Tajalizadekhoob, Y.; Mirarefin, M.; Mohammadzadeh, M.; Sadeghian, S.; Badamchizadeh, Z.; Larijani, B. Cardiac autonomic neuropathy measured by heart rate variability and markers of subclinical atherosclerosis in early type 2 diabetes. ISRN Endocrinol. 2012, 2012, 168264. [Google Scholar] [CrossRef]
- Freccero, C.; Svensson, H.; Bornmyr, S.; Wollmer, P.; Sundkvist, G. Sympathetic and parasympathetic neuropathy are frequent in both type 1 and type 2 diabetic patients. Diabetes Care 2004, 27, 2936–2941. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Pan, N.H.; Chen, Y.J. Oxidative stress and inflammation modulate peroxisome proliferator-activated receptors with regional discrepancy in diabetic heart. Eur. J. Clin. Investig. 2010, 40, 692–699. [Google Scholar] [CrossRef]
- Hansen, H.H.; Jelsing, J.; Hansen, C.F.; Hansen, G.; Vrang, N.; Mark, M.; Klein, T.; Mayoux, E. The sodium glucose cotransporter type 2 inhibitor empagliflozin preserves beta-cell mass and restores glucose homeostasis in the male zucker diabetic fatty rat. J. Pharmacol. Exp. Ther. 2014, 350, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.C.; Hsu, R.C.; Kao, Y.H.; Liou, J.P.; Lu, Y.Y.; Chen, Y.J. Androgen attenuates cardiac fibroblasts activations through modulations of transforming growth factor-beta and angiotensin II signaling. Int. J. Cardiol. 2014, 176, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Kao, Y.H.; Liou, J.P.; Chung, C.C.; Lien, G.S.; Kuo, C.C.; Chen, S.A.; Chen, Y.J. Histone deacetylase inhibition improved cardiac functions with direct antifibrotic activity in heart failure. Int. J. Cardiol. 2013, 168, 4178–4183. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Kao, Y.H.; Chen, Y.C.; Pan, N.H.; Lin, Y.K.; Chen, Y.J. Cardiac peroxisome-proliferator-activated receptor expression in hypertension co-existing with diabetes. Clin. Sci. 2011, 121, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Suenari, K.; Chen, Y.C.; Kao, Y.H.; Cheng, C.C.; Lin, Y.K.; Chen, Y.J.; Chen, S.A. Discrepant electrophysiological characteristics and calcium homeostasis of left atrial anterior and posterior myocytes. Basic Res. Cardiol. 2011, 106, 65–74. [Google Scholar] [CrossRef]
- Hove-Madsen, L.; Llach, A.; Bayes-Genis, A.; Roura, S.; Rodriguez Font, E.; Aris, A.; Cinca, J. Atrial fibrillation is associated with increased spontaneous calcium release from the sarcoplasmic reticulum in human atrial myocytes. Circulation 2004, 110, 1358–1363. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Chen, Y.C.; Kao, Y.H.; Lin, Y.K.; Yeh, Y.H.; Chen, S.A.; Chen, Y.J. Colchicine modulates calcium homeostasis and electrical property of HL-1 cells. J. Cell. Mol. Med. 2016, 20, 1182–1190. [Google Scholar] [CrossRef]
- Huang, S.Y.; Chen, Y.C.; Kao, Y.H.; Hsieh, M.H.; Lin, Y.K.; Chung, C.C.; Lee, T.I.; Tsai, W.C.; Chen, S.A.; Chen, Y.J. Fibroblast growth factor 23 dysregulates late sodium current and calcium homeostasis with enhanced arrhythmogenesis in pulmonary vein cardiomyocytes. Oncotarget 2016, 7, 69231–69242. [Google Scholar] [CrossRef] [PubMed]
- Kondratev, D.; Christ, A.; Gallitelli, M.F. Inhibition of the Na+-H+ exchanger with cariporide abolishes stretch-induced calcium but not sodium accumulation in mouse ventricular myocytes. Cell Calcium 2005, 37, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.S.; Blackwell, D.J.; Gomez-Hurtado, N.; Frisk, M.; Wang, L.; Kim, K.; Dahl, C.P.; Fiane, A.; Tonnessen, T.; Kryshtal, D.O.; et al. Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Circ. Res. 2017, 121, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Viatchenko-Karpinski, S.; Kornyeyev, D.; El-Bizri, N.; Budas, G.; Fan, P.; Jiang, Z.; Yang, J.; Anderson, M.E.; Shryock, J.C.; Chang, C.P.; et al. Intracellular Na+ overload causes oxidation of CaMKII and leads to Ca2+ mishandling in isolated ventricular myocytes. J. Mol. Cell. Cardiol. 2014, 76, 247–256. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Physical Characteristics | Control | DM | Empagliflozin-Treated DM |
|---|---|---|---|
| Baseline FBG (mM) | 4.7 ± 0.2 | 4.7 ± 0.3 | 4.7 ± 0.1 |
| FBG (mM) 2 weeks after streptozotocin | 4.7 | 20.1 ± 0.7 a | 20.1 ± 0.9 a |
| FBG (mM) after empagliflozin treatment | 5.1 ± 0.1 | 20.7 ± 2.0 a | 10.0 ± 1.3 a,b |
| Baseline SBP (mmHg) | 110.1 ± 1.6 | 120.4 ± 4.4 | 114.6 ± 3.9 |
| SBP (mmHg) after treatment | 123.4 ± 7.6 | 122.7 ± 8.4 | 120.2 ± 7.2 |
| Baseline DBP (mmHg) | 64.2 ± 3.2 | 63.9 ± 3.1 | 62.2 ± 3.6 |
| DBP (mmHg) after treatment | 64.2 ± 3.2 | 64.6 ± 3.2 | 63.9 ± 5.5 |
| Baseline HR (bpm) | 414.4 ± 15.9 | 423.8 ± 14.5 | 421.8 ± 14.1 |
| HR (bpm) after treatment | 441.6 ± 10.8 | 345.5 ± 16.0 a | 394.4 ± 16 a,b |
| Baseline BW (gm) | 332.4 ± 4.8 | 323.7 ± 5 | 326.1 ± 7 |
| BW (gm) after treatment | 423.3 ± 10.3 | 278.2 ± 14.9 a | 296.2 ± 11.9 a,b |
| HW (gm) after treatment | 1.3 ± 0.0 | 1.5 ± 0.1 | 1.4 ± 0.1 |
| HW/BW ratio (gm/kg) after treatment | 3.2 ± 0.1 | 5.4 ± 0.3a | 4.4 ± 0.1 a,b |
| Group Studied | LVEDd (mm) | LVESd (mm) | EDV (mL) | ESV (mL) | EF (%) | FS (%) |
|---|---|---|---|---|---|---|
| Control | 7.3 ± 0.2 | 3.5 ± 0.1 | 0.9 ± 0.1 | 0.1 ± 0.01 | 87.7 ± 0.7 | 53.4 ± 1.1 |
| DM | 8.3 ± 0.1 a | 4.7 ± 0.1 a | 1.2 ± 0.1 a | 0.3 ± 0.04 a | 79.0 ± 1.1 a | 42.9 ± 1.1 a |
| Empagliflozin-treated DM | 7.3 ± 0.1 b | 3.7 ± 0.1 b | 0.9 ± 0.0 b | 0.1 ± 0.04 b | 85.4 ± 1.5 b | 49.8 ± 1.7 b |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.-I.; Chen, Y.-C.; Lin, Y.-K.; Chung, C.-C.; Lu, Y.-Y.; Kao, Y.-H.; Chen, Y.-J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. https://doi.org/10.3390/ijms20071680
Lee T-I, Chen Y-C, Lin Y-K, Chung C-C, Lu Y-Y, Kao Y-H, Chen Y-J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. International Journal of Molecular Sciences. 2019; 20(7):1680. https://doi.org/10.3390/ijms20071680
Chicago/Turabian StyleLee, Ting-I, Yao-Chang Chen, Yung-Kuo Lin, Cheng-Chih Chung, Yen-Yu Lu, Yu-Hsun Kao, and Yi-Jen Chen. 2019. "Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats" International Journal of Molecular Sciences 20, no. 7: 1680. https://doi.org/10.3390/ijms20071680
APA StyleLee, T.-I., Chen, Y.-C., Lin, Y.-K., Chung, C.-C., Lu, Y.-Y., Kao, Y.-H., & Chen, Y.-J. (2019). Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. International Journal of Molecular Sciences, 20(7), 1680. https://doi.org/10.3390/ijms20071680