Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature

 , , ,
, , , 

Abstract
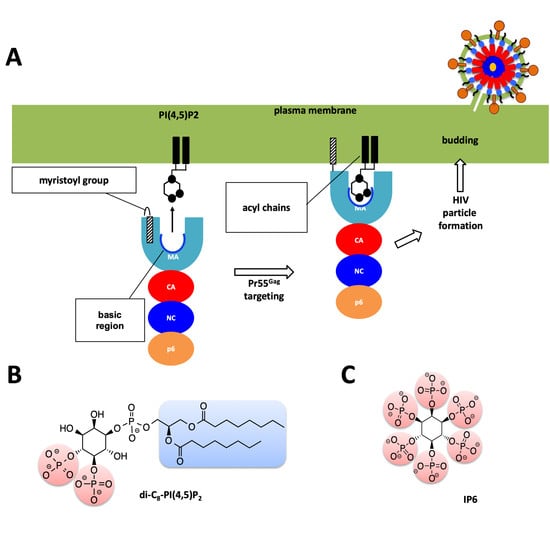
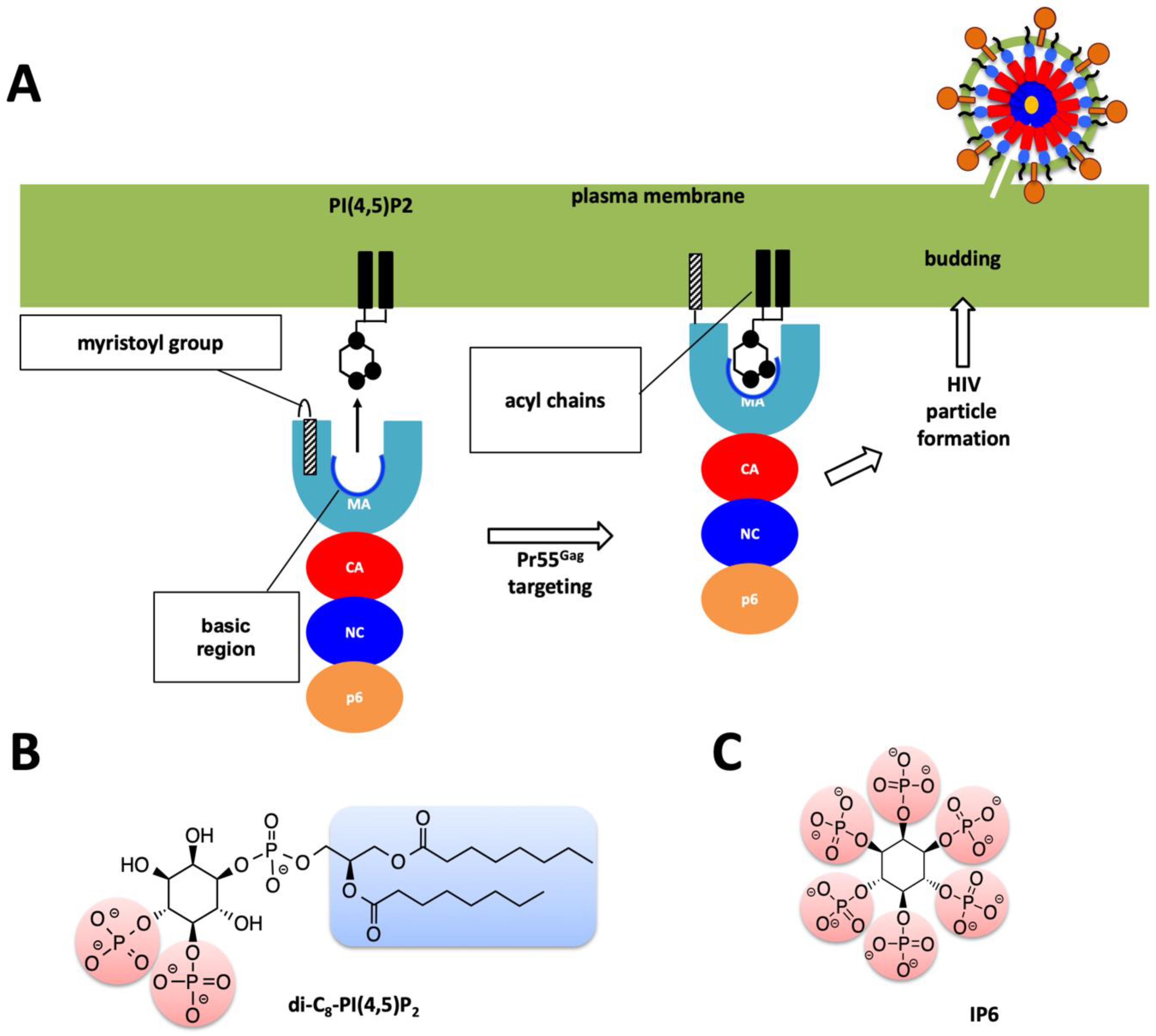
1. Introduction
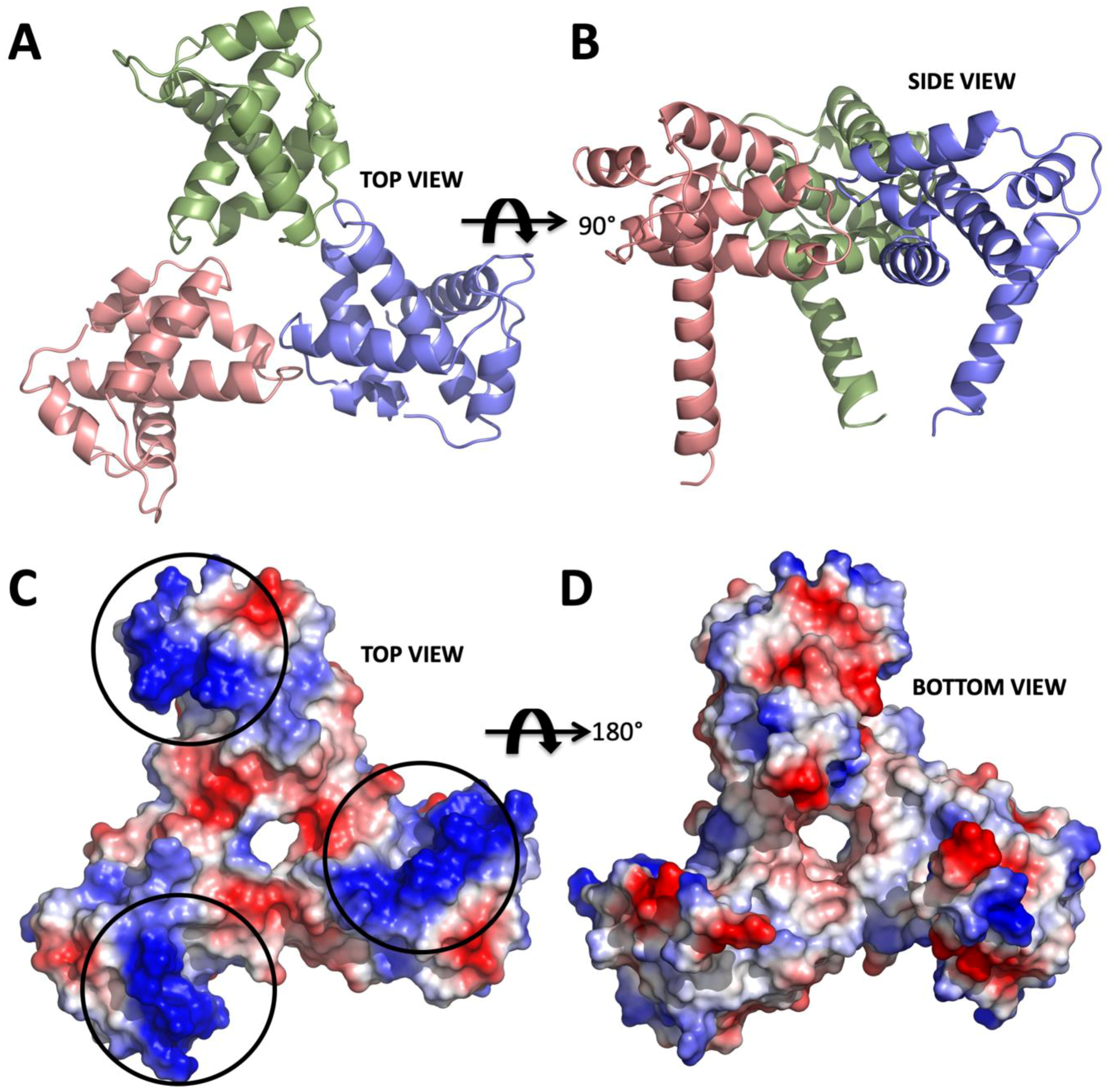
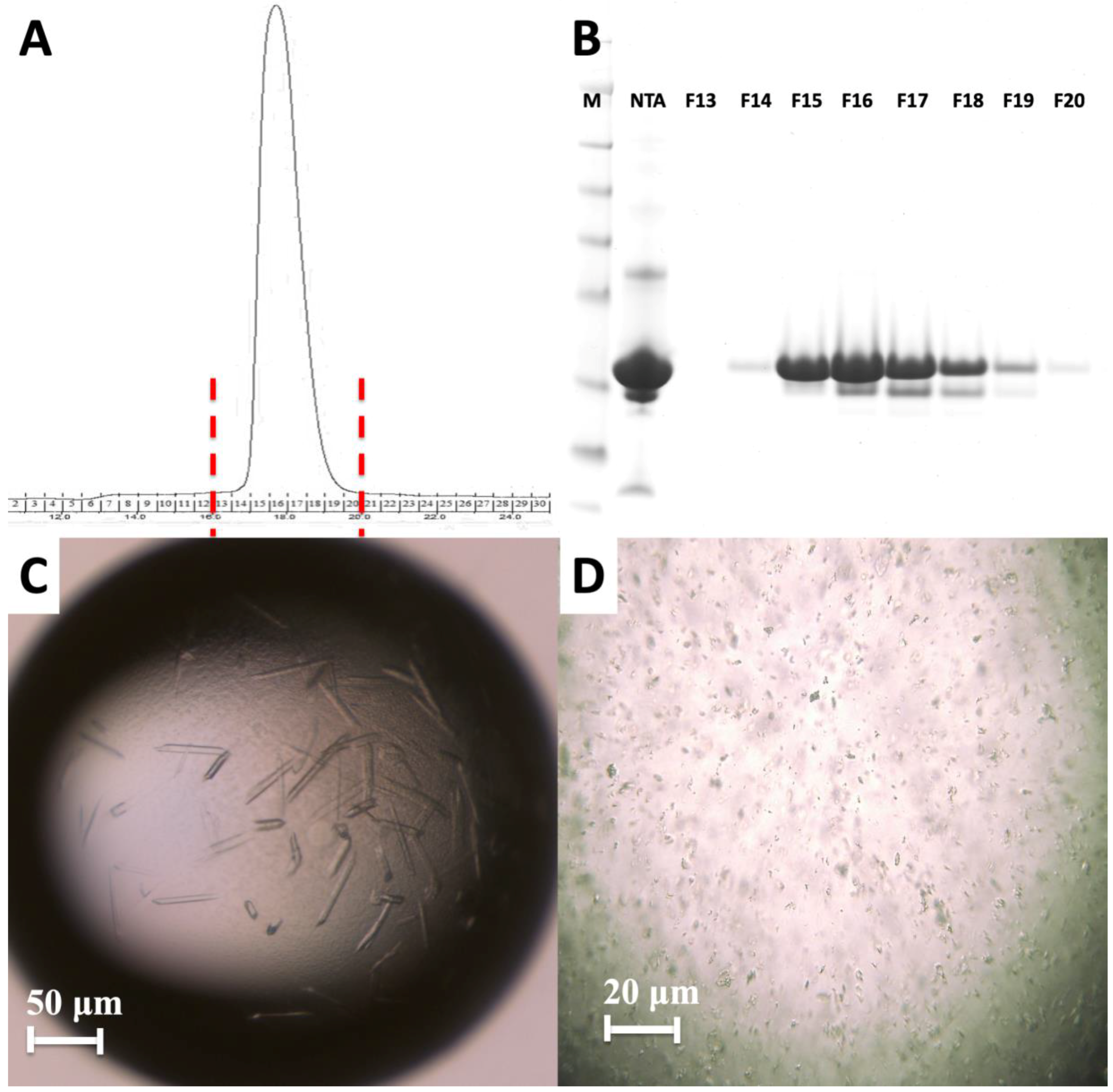
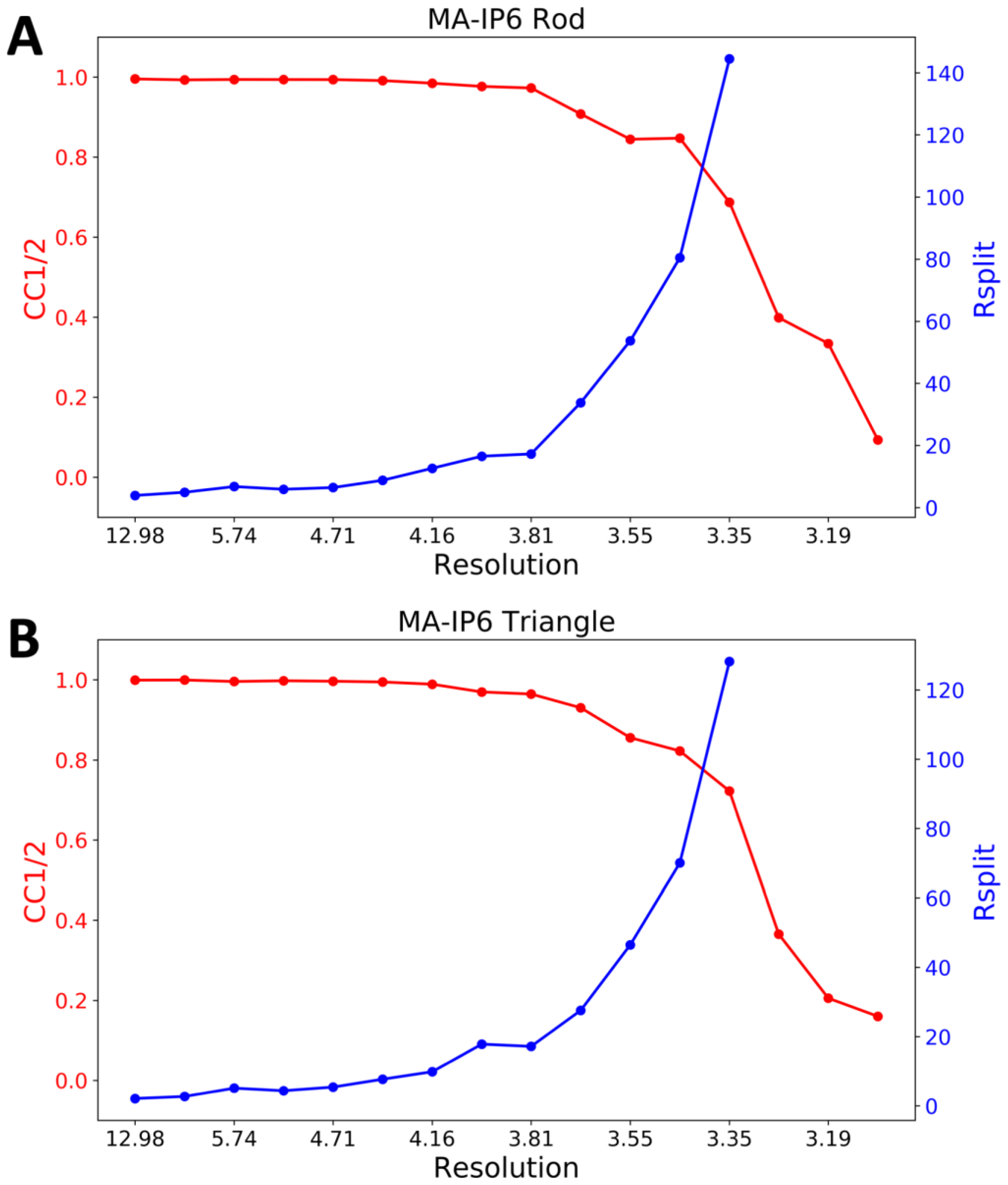
2. Results
3. Discussion
4. Materials and Methods
4.1. Cloning and Overexpression of the HIV-1 Gag MA Domain
4.2. Crystallization of the HIV-1 Gag MA Domain
4.3. XFEL X-Ray Delivery and Detector
4.4. Injection of HIV-1 MA-IP6 Microcrystals into an XFEL and Diffraction Data Collection
4.5. Hit Finding
4.6. Indexing
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gottlieb, M.S.; Schroff, R.; Schanker, H.M.; Weisman, J.D.; Fan, P.T.; Wolf, R.A.; Saxon, A. Pneumocystis carinii pneumonia and mucosal candidiasis in previously healthy homosexual men: Evidence of a new acquired cellular immunodeficiency. N. Engl. J. Med. 1981, 305, 1425–1431. [Google Scholar] [CrossRef]
- Barre-Sinoussi, F.; Chermann, J.; Rey, F.; Nugeyre, M.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vezinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar] [CrossRef]
- Knipe, D.M.; Howley, P.M. Fields’ Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 11072–11081. ISBN 9781451105636. [Google Scholar]
- Hill, C.P.; Worthylake, D.; Bancroft, D.P.; Christensen, A.M.; Sundquist, W.I. Crystal structures of the trimeric human immunodeficiency virus type 1 matrix protein: Implications for membrane association and assembly. Proc. Natl. Acad. Sci. USA 1996, 93, 3099–3104. [Google Scholar] [CrossRef]
- Navia, M.A.; Fitzgerald, P.M.D.; McKeever, B.M.; Leu, C.-T.; Heimbach, J.C.; Herber, W.K.; Sigal, I.S.; Darke, P.L.; Springer, J.P. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature 1989, 337, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.; Jaskólski, M.; Rao, J.K.M.; Leis, J.; Wlodawer, A. Crystal structure of a retroviral protease proves relationship to aspartic protease family. Nature 1989, 337, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Wlodawer, A.; Miller, M.; Jaskolski, M.; Sathyanarayana, B.; Baldwin, E.; Weber, I.; Selk, L.; Clawson, L.; Schneider, J.; Kent, S. Conserved folding in retroviral proteases: Crystal structure of a synthetic HIV-1 protease. Science 1989, 245, 616–621. [Google Scholar] [CrossRef]
- Miller, M.; Schneider, J.; Sathyanarayana, B.; Toth, M.; Marshall, G.; Clawson, L.; Selk, L.; Kent, S.; Wlodawer, A. Structure of complex of synthetic HIV-1 protease with a substrate-based inhibitor at 2.3 A resolution. Science 1989, 246, 1149–1152. [Google Scholar] [CrossRef] [PubMed]
- Weber, I.; Miller, M.; Jaskolski, M.; Leis, J.; Skalka, A.; Wlodawer, A. Molecular modeling of the HIV-1 protease and its substrate binding site. Science 1989, 243, 928–931. [Google Scholar] [CrossRef] [PubMed]
- Puhl, A.; Garzina Demo, A.; Makarov, V.; Ekins, S. New targets for HIV drug discoverye. Drug Discov. Today 2019. epub. [Google Scholar] [CrossRef]
- Ghanam, R.H.; Samal, A.B.; Fernandez, T.F.; Saad, J.S. Role of the HIV-1 Matrix Protein in Gag Intracellular Trafficking and Targeting to the Plasma Membrane for Virus Assembly. Front. Microbiol. 2012, 3, 55. [Google Scholar] [CrossRef]
- Vlach, J.; Saad, J.S. Structural and molecular determinants of HIV-1 Gag binding to the plasma membrane. Front. Microbiol. 2015, 6, 232. [Google Scholar] [CrossRef] [PubMed]
- Saad, J.S.; Miller, J.; Tai, J.; Kim, A.; Ghanam, R.H.; Summers, M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11364–11369. [Google Scholar] [CrossRef]
- Dick, R.A.; Vogt, V.M. Membrane interaction of retroviral Gag proteins. Front. Microbiol. 2014, 5, 187. [Google Scholar] [CrossRef]
- Tedbury, P.R.; Freed, E.O. The role of matrix in HIV-1 envelope glycoprotein incorporation. Trends Microbiol. 2014, 22, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.S.; Salzwedel, K.; Freed, E.O. Virus maturation as a new HIV-1 therapeutic target. Expert Opin. Ther. Targets 2009, 13, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Warrell, M.J. Intradermal rabies vaccination: The evolution and future of pre- and post-exposure prophylaxis. Curr. Top. Microbiol. Immunol. 2012, 351, 139–157. [Google Scholar]
- Scarlata, S.; Carter, C. Role of HIV-1 Gag domains in viral assembly. Biochim. Biophys. Acta 2003, 1614, 62–72. [Google Scholar] [CrossRef]
- Tateishi, H.; Anraku, K.; Koga, R.; Okamoto, Y.; Fujita, M.; Otsuka, M. Design and synthesis of lipid-coupled inositol 1,2,3,4,5,6-hexakisphosphate derivatives exhibiting high-affinity binding for the HIV-1 MA domain. Org. Biomol. Chem. 2014, 12, 5006–5022. [Google Scholar] [CrossRef]
- Tateishi, H.; Monde, K.; Anraku, K.; Koga, R.; Hayashi, Y.; Ciftci, H.I.; DeMirci, H.; Higashi, T.; Motoyama, K.; Arima, H.; et al. A clue to unprecedented strategy to HIV eradication: “Lock-in and apoptosis”. Sci. Rep. 2017, 7, 8957. [Google Scholar] [CrossRef]
- Datta, S.A.K.; Zhao, Z.; Clark, P.K.; Tarasov, S.; Alexandratos, J.N.; Campbell, S.J.; Kvaratskhelia, M.; Lebowitz, J.; Rein, A. Interactions between HIV-1 Gag molecules in solution: An inositol phosphate-mediated switch. J. Mol. Biol. 2007, 365, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Gati, C.; Laksmono, H.; Dao, E.H.; Gul, S.; Fuller, F.; Kern, J.; Chatterjee, R.; Ibrahim, M.; Brewster, A.S.; et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods 2016, 13, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Dao, E.H.; Poitevin, F.; Sierra, R.G.; Gati, C.; Rao, Y.; Ciftci, H.I.; Akşit, F.; McGurk, A.; Obrinski, T.; Mgbam, P.; et al. Structure of the 30S ribosomal decoding complex at ambient temperature. RNA 2018, 24, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, M.E.; Poitevin, F.; Sierra, R.G.; Gati, C.; Dao, E.H.; Rao, Y.; Aksit, F.; Ciftci, H.; Corsepius, N.; Greenhouse, R.; et al. Aminoglycoside ribosome interactions reveal novel conformational states at ambient temperature. Nucleic Acids Res. 2018, 46, 9793–9804. [Google Scholar] [CrossRef] [PubMed]
- Fromme, P.; Spence, J.C.H. Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr. Opin. Struct. Biol. 2011, 21, 509–516. [Google Scholar] [CrossRef]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kim, J. Subfemtosecond synchronization of microwave oscillators with mode-locked Er-fiber lasers. Opt. Lett. 2012, 37, 2958–2960. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.S.; Yoon, C.H.; DeMirci, H.; Sierra, R.G.; Dao, E.H.; Ahmadi, R.; Aksit, F.; Aquila, A.L.; Ciftci, H.; Guillet, S.; et al. Selenium single-wavelength anomalous diffraction de novo phasing using an X-ray-free electron laser. Nat. Commun. 2016, 7, 13388. [Google Scholar] [CrossRef]
- Bottieau, E.; Clerinx, J.; de Vega, M.R.; Van den Enden, E.; Colebunders, R.; Van Esbroeck, M.; Vervoort, T.; Van Gompel, A.; Van den Ende, J. Imported Katayama fever: Clinical and biological features at presentation and during treatment. J. Infect. 2006, 52, 339–345. [Google Scholar] [CrossRef]
- Schlichting, I.; Miao, J. Emerging opportunities in structural biology with X-ray free-electron lasers. Curr. Opin. Struct. Biol. 2012, 22, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Bogan, M.J. X-ray free electron lasers motivate bioanalytical characterization of protein nanocrystals: Serial femtosecond crystallography. Anal. Chem. 2013, 85, 3464–3471. [Google Scholar] [CrossRef]
- Helliwell, J.R. Biochemistry. How to solve protein structures with an X-ray laser. Science 2013, 339, 146–147. [Google Scholar] [CrossRef] [PubMed]
- DePonte, D.P.; Doak, R.B.; Hunter, M.; Liu, Z.; Weierstall, U.; Spence, J.C.H. SEM imaging of liquid jets. Micron 2009, 40, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Barty, A.; Caleman, C.; Aquila, A.; Timneanu, N.; Lomb, L.; White, T.A.; Andreasson, J.; Arnlund, D.; Bajt, S.; Barends, T.R.M.; et al. Self-terminating diffraction gates femtosecond X-ray nanocrystallography measurements. Nat. Photonics 2012, 6, 35–40. [Google Scholar] [CrossRef]
- Sierra, R.G.; Laksmono, H.; Kern, J.; Tran, R.; Hattne, J.; Alonso-Mori, R.; Lassalle-Kaiser, B.; Glöckner, C.; Hellmich, J.; Schafer, D.W.; et al. Nanoflow electrospinning serial femtosecond crystallography. Acta Crystallogr. D. Biol. Crystallogr. 2012, 68, 1584–1587. [Google Scholar] [CrossRef]
- Weierstall, U.; Spence, J.C.H.; Doak, R.B. Injector for scattering measurements on fully solvated biospecies. Rev. Sci. Instrum. 2012, 83, 035108. [Google Scholar] [CrossRef] [PubMed]
- Neutze, R.; Wouts, R.; van der Spoel, D.; Weckert, E.; Hajdu, J. Potential for biomolecular imaging with femtosecond X-ray pulses. Nature 2000, 406, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Boutet, S.; Lomb, L.; Williams, G.J.; Barends, T.R.M.; Aquila, A.; Doak, R.B.; Weierstall, U.; DePonte, D.P.; Steinbrener, J.; Shoeman, R.L.; et al. High-resolution protein structure determination by serial femtosecond crystallography. Science 2012, 337, 362–364. [Google Scholar] [CrossRef] [PubMed]
- Redecke, L.; Nass, K.; DePonte, D.P.; White, T.A.; Rehders, D.; Barty, A.; Stellato, F.; Liang, M.; Barends, T.R.M.; Boutet, S.; et al. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science 2013, 339, 227–230. [Google Scholar] [CrossRef]
- Kern, J.; Alonso-Mori, R.; Hellmich, J.; Tran, R.; Hattne, J.; Laksmono, H.; Glöckner, C.; Echols, N.; Sierra, R.G.; Sellberg, J.; et al. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc. Natl. Acad. Sci. USA 2012, 109, 9721–9726. [Google Scholar] [CrossRef]
- Demirci, H.; Sierra, R.G.; Laksmono, H.; Shoeman, R.L.; Botha, S.; Barends, T.R.M.; Nass, K.; Schlichting, I.; Doak, R.B.; Gati, C.; et al. Serial femtosecond X-ray diffraction of 30S ribosomal subunit microcrystals in liquid suspension at ambient temperature using an X-ray free-electron laser. Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 2013, 69, 1066–1069. [Google Scholar] [CrossRef]
- Nannenga, B.L.; Gonen, T. MicroED opens a new era for biological structure determination. Curr. Opin. Struct. Biol. 2016, 40, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.O.; Gristick, H.B.; Freund, N.T.; Escolano, A.; Lyubimov, A.Y.; Hartweger, H.; West, A.P.; Cohen, A.E.; Nussenzweig, M.C.; Bjorkman, P.J. Structural characterization of a highly-potent V3-glycan broadly neutralizing antibody bound to natively-glycosylated HIV-1 envelope. Nat. Commun. 2018, 9, 1251. [Google Scholar] [CrossRef]
- Liang, M.; Williams, G.J.; Messerschmidt, M.; Seibert, M.M.; Montanez, P.A.; Hayes, M.; Milathianaki, D.; Aquila, A.; Hunter, M.S.; Koglin, J.E.; et al. The Coherent X-ray Imaging instrument at the Linac Coherent Light Source. J. Synchrotron Radiat. 2015, 22, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Mariani, V.; Morgan, A.; Yoon, C.H.; Lane, T.J.; White, T.A.; O’grady, C.; Kuhn, M.; Aplin, S.; Koglin, J.; Barty, A.; et al. OnDA: Online data analysis and feedback for serial X-ray imaging. J. Appl. Crystallogr. 2016. [Google Scholar] [CrossRef]
- Hart, P.; Boutet, S.; Carini, G.; Dragone, A.; Duda, B.; Freytag, D.; Haller, G.; Herbst, R.; Herrmann, S.; Kenney, C.; et al. The Cornell-SLAC pixel array detector at LCLS. In Proceedings of the 2012 IEEE Nuclear Science Symposium and Medical Imaging Conference Record (NSS/MIC), Anaheim, CA, USA, 29 October–3 November 2012; IEEE: Piscataway, NJ, USA, 2012; pp. 538–541. [Google Scholar]
- Damiani, D.; Dubrovin, M.; Gaponenko, I.; Kroeger, W.; Lane, T.J.; Mitra, A.; O’Grady, C.P.; Salnikov, A.; Sanchez-Gonzalez, A.; Schneider, D.; et al. Linac Coherent Light Source data analysis using psana. J. Appl. Crystallogr. 2016, 49, 672–679. [Google Scholar] [CrossRef]
- Thayer, J.; Damiani, D.; Ford, C.; Dubrovin, M.; Gaponenko, I.; O’Grady, C.P.; Kroeger, W.; Pines, J.; Lane, T.J.; Salnikov, A.; et al. Data systems for the Linac coherent light source. Adv. Struct. Chem. Imaging 2017, 3, 3. [Google Scholar] [CrossRef]
- White, T.A.; Mariani, V.; Brehm, W.; Yefanov, O.; Barty, A.; Beyerlein, K.R.; Chervinskii, F.; Galli, L.; Gati, C.; Nakane, T.; et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 2016, 49, 680–689. [Google Scholar] [CrossRef]
- White, T.A.; Kirian, R.A.; Martin, A.V.; Aquila, A.; Nass, K.; Barty, A.; Chapman, H.N. CrystFEL: A software suite for snapshot serial crystallography. J. Appl. Crystallogr. 2012, 45, 335–341. [Google Scholar] [CrossRef]
- White, T.A.; Barty, A.; Stellato, F.; Holton, J.M.; Kirian, R.A.; Zatsepin, N.A.; Chapman, H.N. Crystallographic data processing for free-electron laser sources. Acta Crystallogr. D. Biol. Crystallogr. 2013, 69, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peak Finding Algorithm | Peak Criteria | Min. Peaks |
|---|---|---|
| Adaptive | Amax_thr 300, Atot_thr 600, Peak size 2–30, Son_min 10, Rank 3, Radius 3, Dr 2 | 15 |
| Indexing Algorithm | Integration Radii | Unit Cell Tolerance |
|---|---|---|
| Mosflm, dirax | 3,4,5 | Axes lengths = 5% Axes angles = 1.5° |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
I Ciftci, H.; G Sierra, R.; Yoon, C.H.; Su, Z.; Tateishi, H.; Koga, R.; Kotaro, K.; Yumoto, F.; Senda, T.; Liang, M.; et al. Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature. Int. J. Mol. Sci. 2019, 20, 1675. https://doi.org/10.3390/ijms20071675
I Ciftci H, G Sierra R, Yoon CH, Su Z, Tateishi H, Koga R, Kotaro K, Yumoto F, Senda T, Liang M, et al. Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature. International Journal of Molecular Sciences. 2019; 20(7):1675. https://doi.org/10.3390/ijms20071675
Chicago/Turabian StyleI Ciftci, Halil, Raymond G Sierra, Chun Hong Yoon, Zhen Su, Hiroshi Tateishi, Ryoko Koga, Koiwai Kotaro, Fumiaki Yumoto, Toshiya Senda, Mengling Liang, and et al. 2019. "Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature" International Journal of Molecular Sciences 20, no. 7: 1675. https://doi.org/10.3390/ijms20071675
APA StyleI Ciftci, H., G Sierra, R., Yoon, C. H., Su, Z., Tateishi, H., Koga, R., Kotaro, K., Yumoto, F., Senda, T., Liang, M., Wakatsuki, S., Otsuka, M., Fujita, M., & DeMirci, H. (2019). Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature. International Journal of Molecular Sciences, 20(7), 1675. https://doi.org/10.3390/ijms20071675