Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources

Abstract
1. Introduction
2. Synchrotron Light Sources
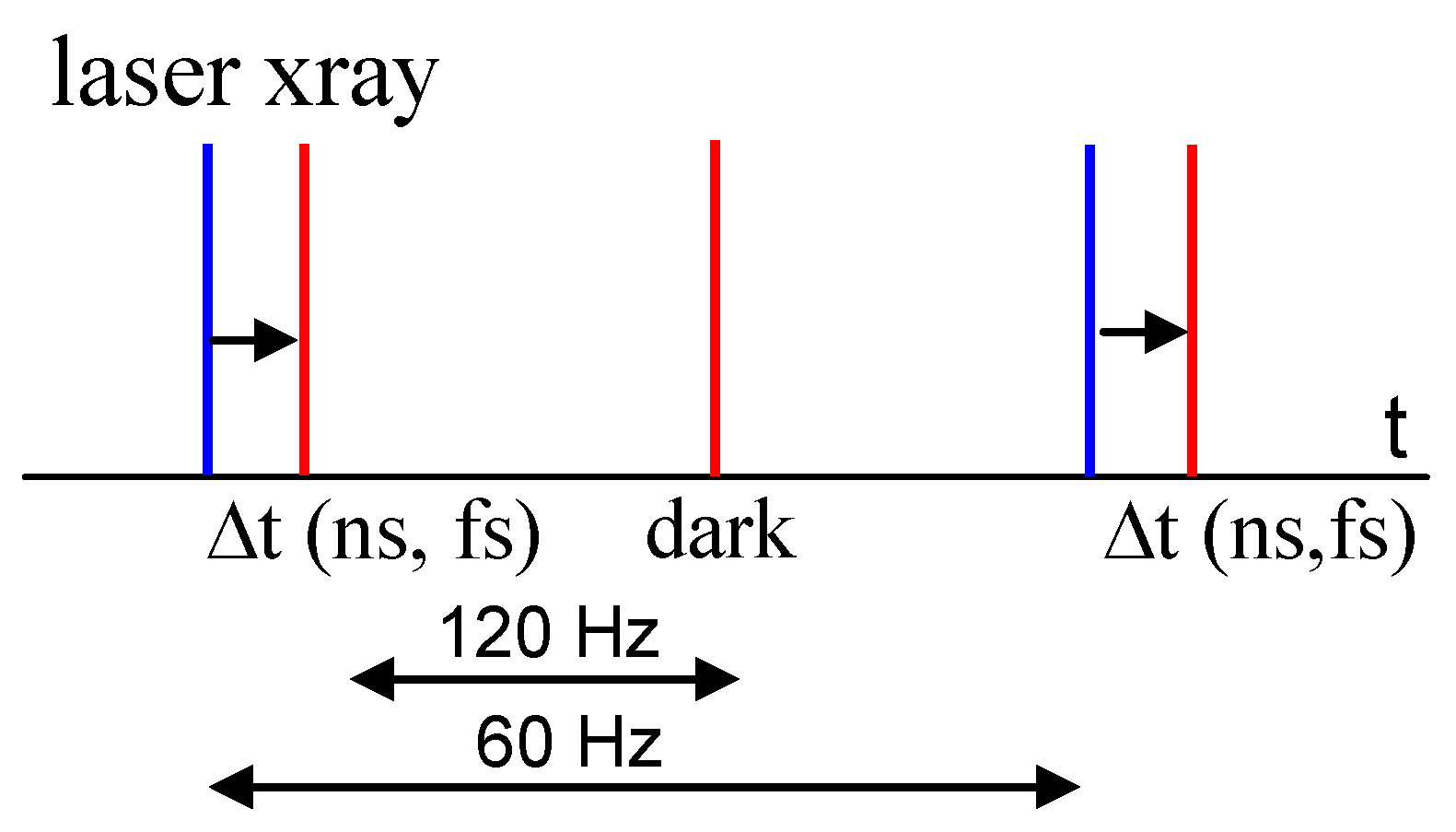
3. Free Electron Lasers
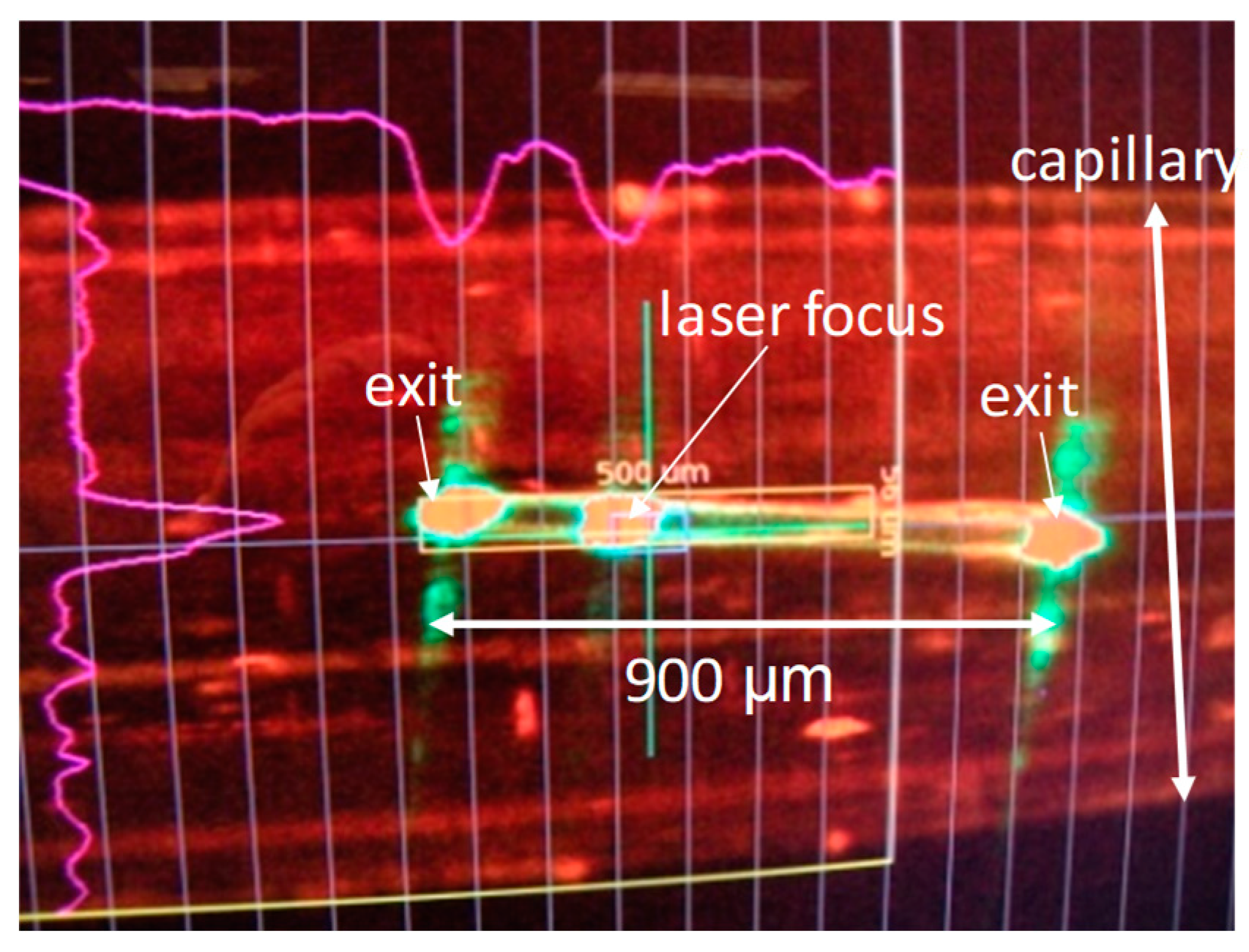
4. Reaction Initiation
5. Data Collection and Structure Determination
5.1. Twinning
5.2. Difference Maps
5.3. Analysis of Time Series
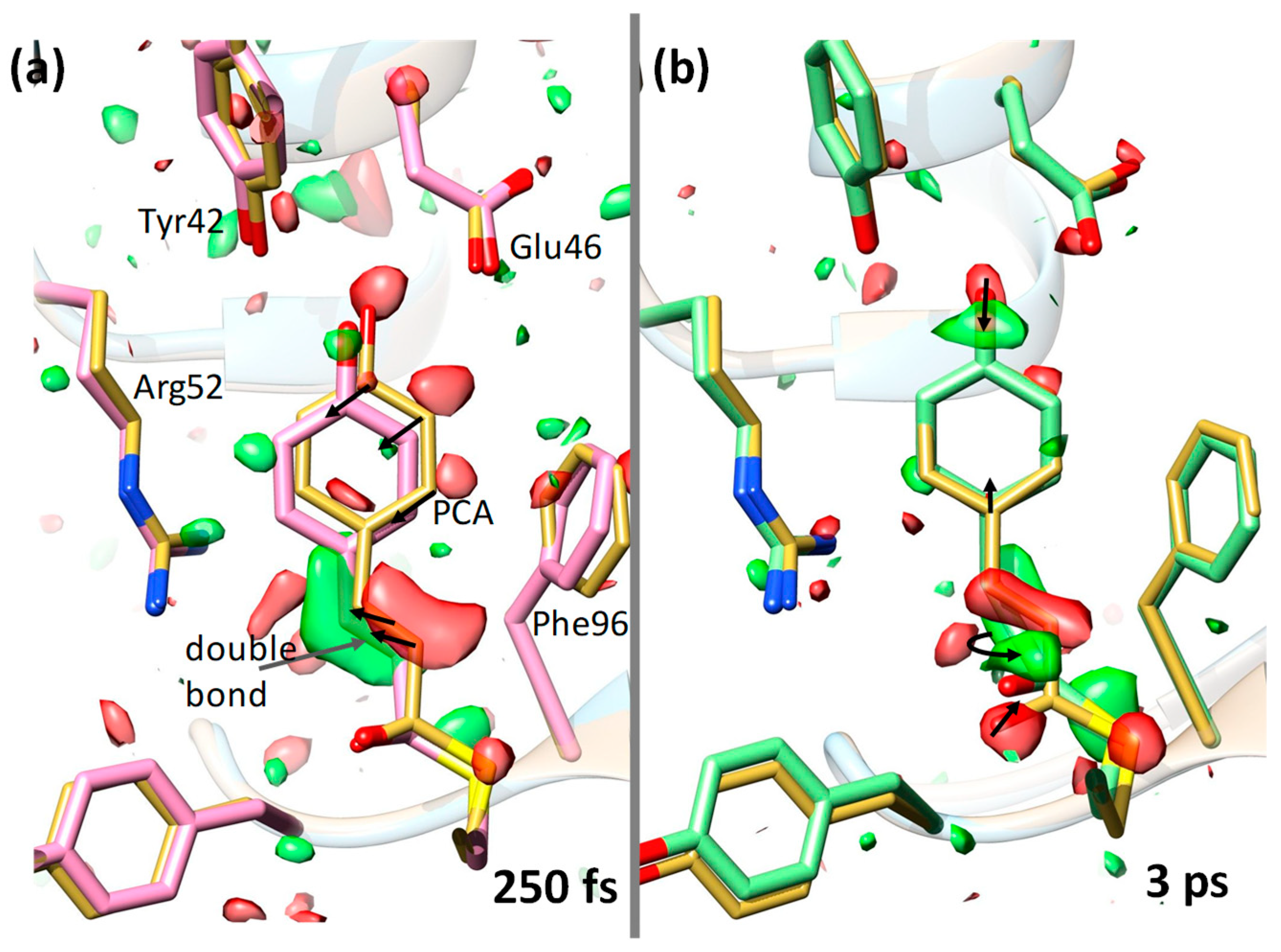
6. The Blue-Light Receptor Photoactive Yellow Protein
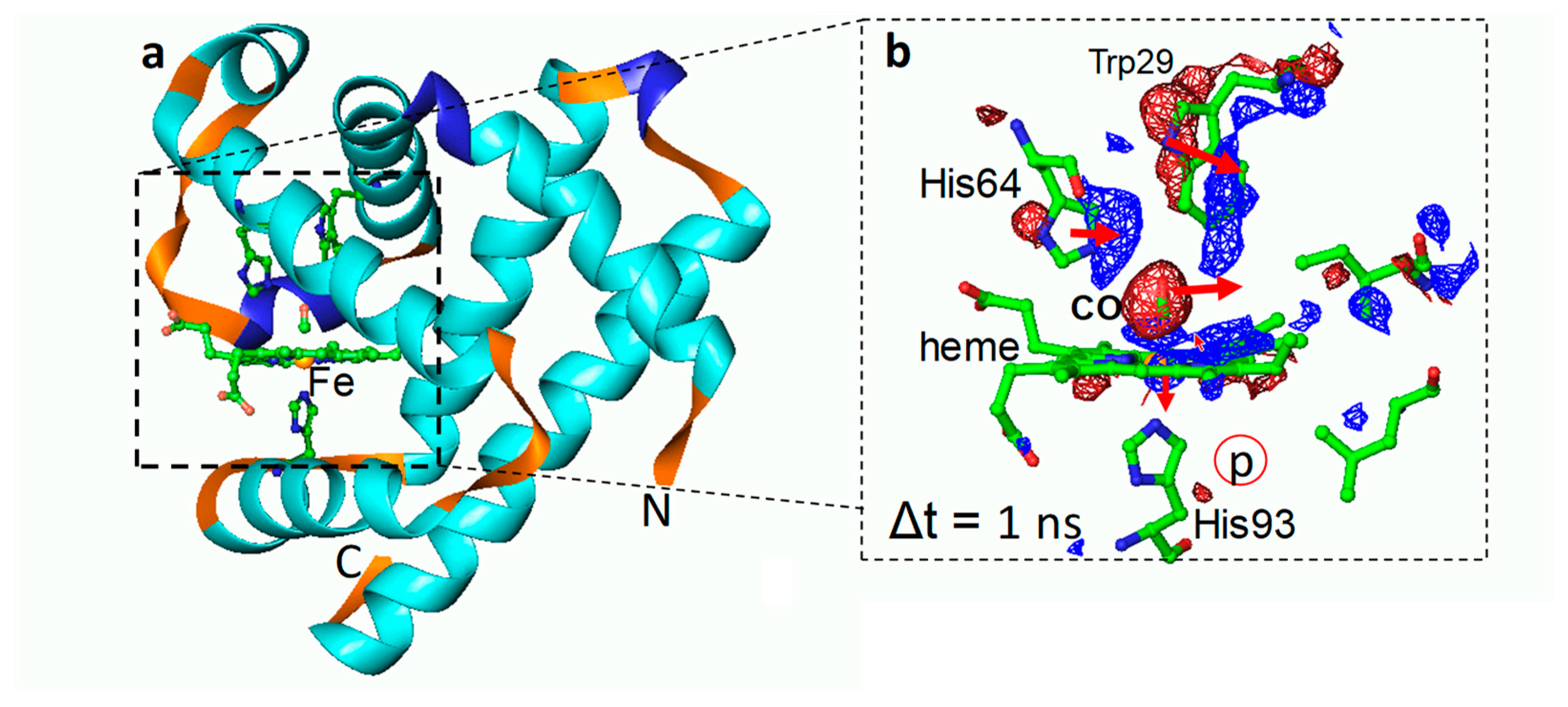
7. Transition Metal-Containing Proteins
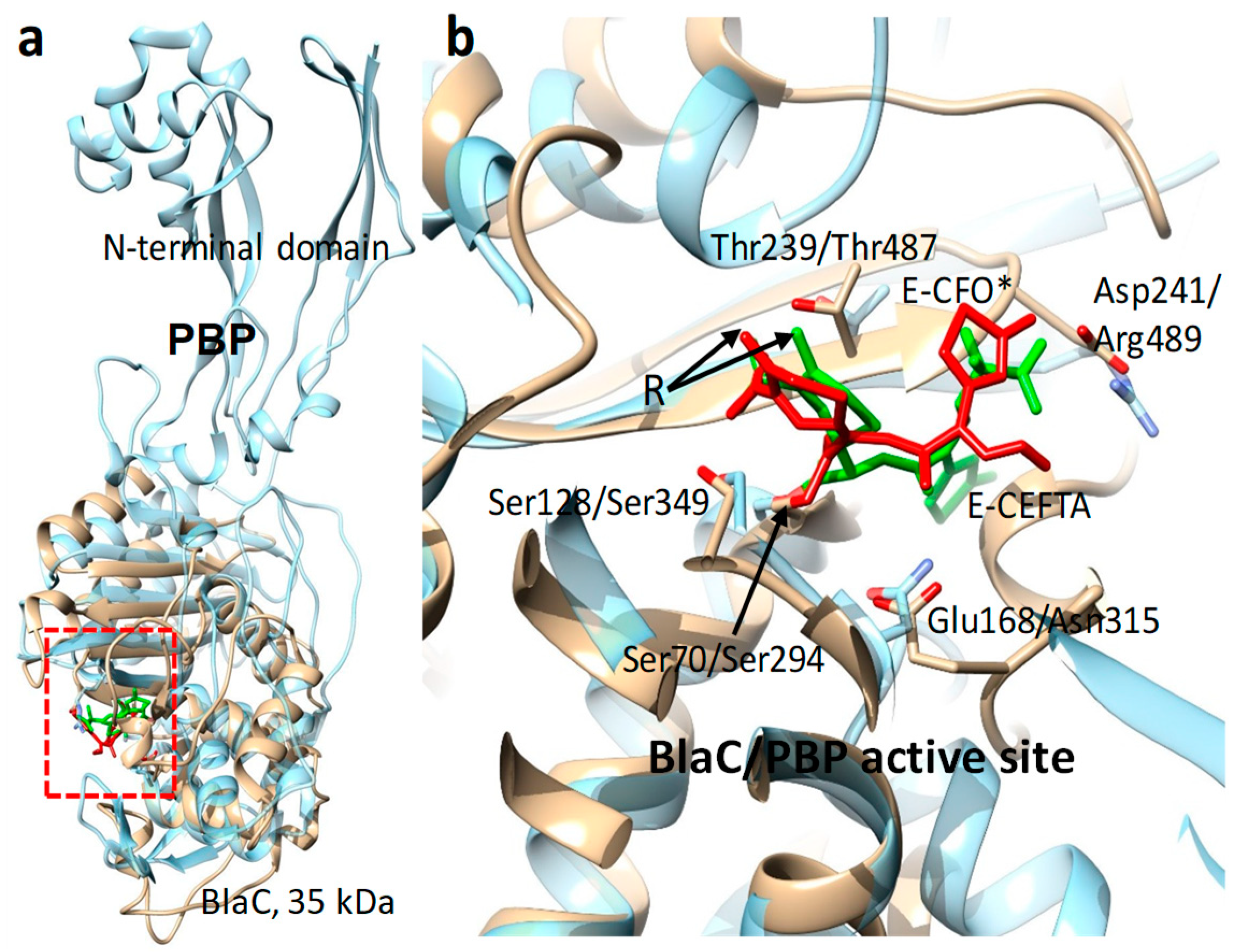
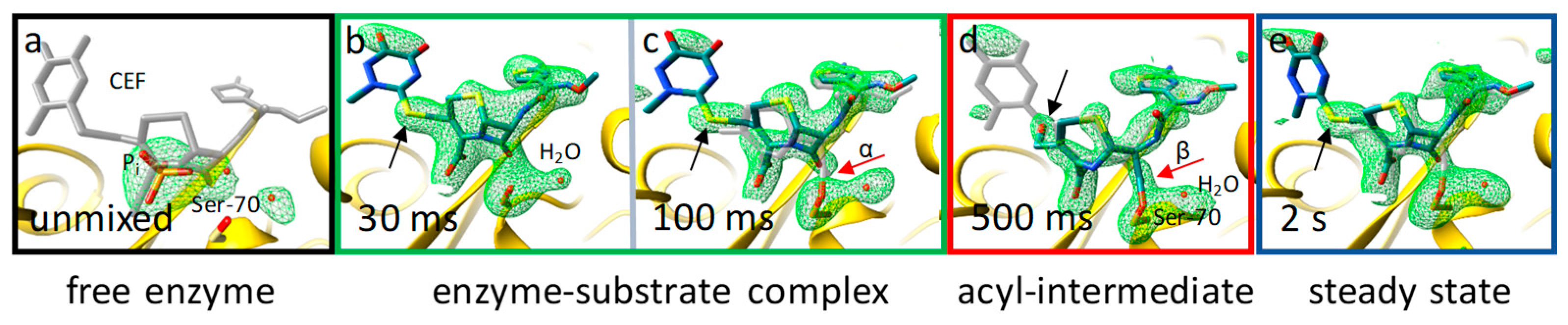
8. Enzymes
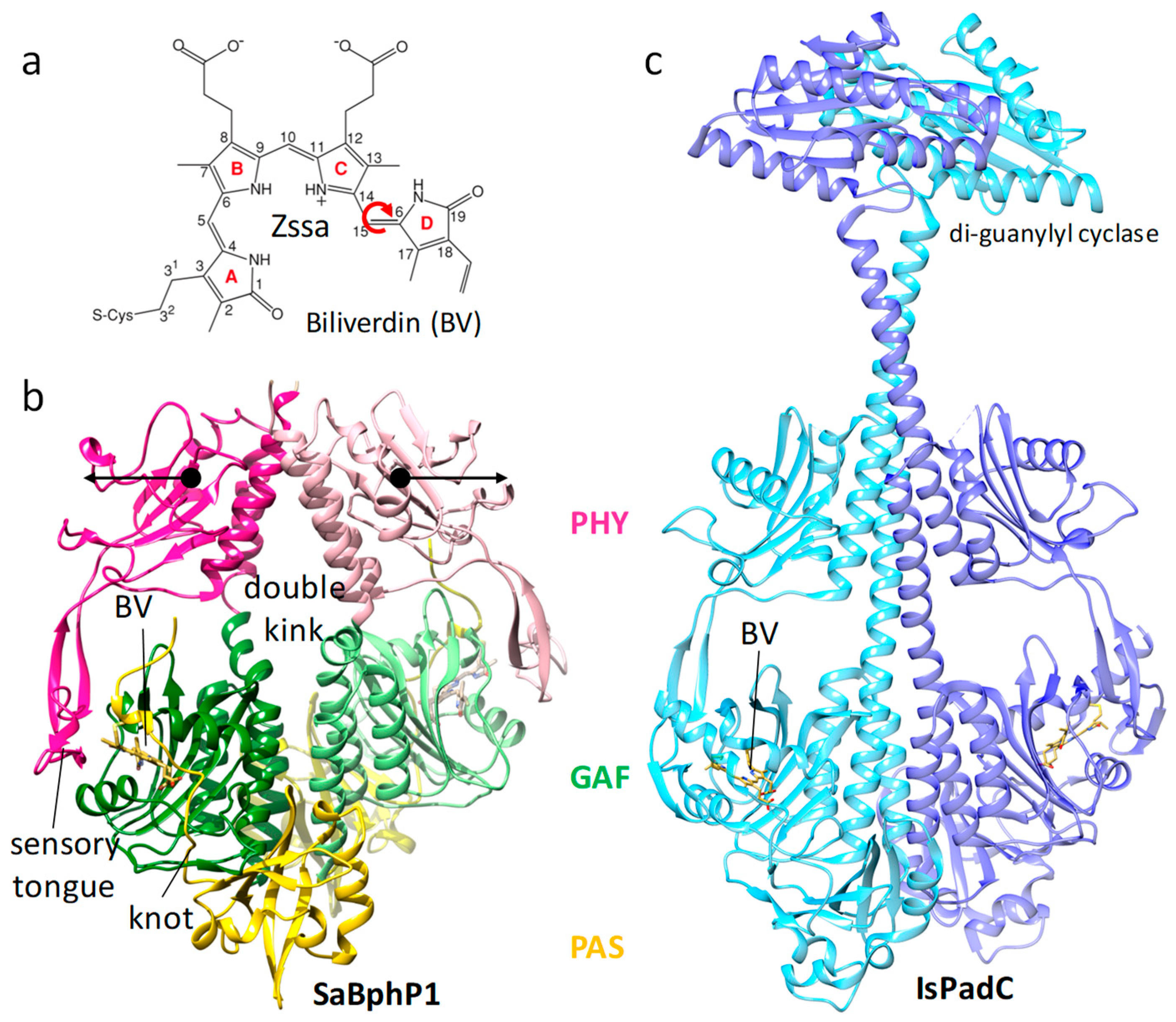
9. Other Light-Controlled Proteins and Enzymes
10. Outlook
Funding
Acknowledgments
Conflicts of Interest
References
- Moffat, K.; Szebenyi, D.; Bilderback, D. X-ray Laue Diffraction from Protein Crystals. Science 1984, 223, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Hajdu, J.; Machin, P.A.; Campbell, J.W.; Greenhough, T.J.; Clifton, I.J.; Zurek, S.; Gover, S.; Johnson, L.N.; Elder, M. Millisecond X-ray diffraction and the first electron density map from Laue photographs of a protein crystal. Nature 1987, 329, 178–181. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I.; Almo, S.C.; Rapp, G.; Wilson, K.; Petratos, K.; Lentfer, A.; Wittinghofer, A.; Kabsch, W.; Pai, E.F.; Petsko, G.A.; et al. Time-Resolved X-ray Crystallographic Study of the Conformational Change in Ha-Ras P21 Protein on Gtp Hydrolysis. Nature 1990, 345, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Tenboer, J.; Basu, S.; Zatsepin, N.; Pande, K.; Milathianaki, D.; Frank, M.; Hunter, M.; Boutet, S.; Williams, G.J.; Koglin, J.E.; et al. Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 2014, 346, 1242–1246. [Google Scholar] [CrossRef] [PubMed]
- Pande, K.; Hutchison, C.D.M.; Groenhof, G.; Aquila, A.; Robinson, J.S.; Tenboer, J.; Basu, S.; Boutet, S.; Deponte, D.; Liang, M.; et al. Femtosecond Structural Dynamics Drives the Trans/Cis Isomerization in Photoactive Yellow Protein. Science 2016, 352, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Barends, T.R.; Foucar, L.; Ardevol, A.; Nass, K.; Aquila, A.; Botha, S.; Doak, R.B.; Falahati, K.; Hartmann, E.; Hilpert, M.; et al. Direct observation of ultrafast collective motions in CO myoglobin upon ligand dissociation. Science 2015. [Google Scholar] [CrossRef]
- Amoros, J.L.; Buerger, M.J.; de Amoros, M.L.C. The Laue Method; Academic Press: Cambridge, MA, USA, 1975. [Google Scholar]
- Buerger, M.J. X-ray Crystallography; John Wiley and Sons: Hoboken, NJ, USA, 1949. [Google Scholar]
- Ren, Z.; Moffat, K. Quantitative-Analysis of Synchrotron Laue Diffraction Patterns in Macromolecular Crystallography. J. Appl. Crystallogr. 1995, 28, 461–481. [Google Scholar] [CrossRef]
- Bourgeois, D. New processing tools for weak and/or spatially overlapped macromolecular diffraction patterns. Acta Crystallogr. Sect. D Biol. Crystallogr. 1999, 55, 1733–1741. [Google Scholar] [CrossRef]
- Kirian, R.A.; Wang, X.; Weierstall, U.; Schmidt, K.E.; Spence, J.C.; Hunter, M.; Fromme, P.; White, T.; Chapman, H.N.; Holton, J. Femtosecond protein nanocrystallography-data analysis methods. Opt. Express 2010, 18, 5713–5723. [Google Scholar] [CrossRef] [PubMed]
- Kirian, R.A.; White, T.A.; Holton, J.M.; Chapman, H.N.; Fromme, P.; Barty, A.; Lomb, L.; Aquila, A.; Maia, F.R.; Martin, A.V.; et al. Structure-factor analysis of femtosecond microdiffraction patterns from protein nanocrystals. Acta Crystallogr. Sect. A Found. Crystallogr. 2011, 67, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I. Experimental phasing of serial femtosecond crystallography data. Iucrj 2017, 4, 516–517. [Google Scholar] [CrossRef] [PubMed]
- Holton, J.M.; Frankel, K.A. The minimum crystal size needed for a complete diffraction data set. Acta Crystallogr. D 2010, 66, 393–408. [Google Scholar] [CrossRef] [PubMed]
- Graber, T.; Anderson, S.; Brewer, H.; Chen, Y.S.; Cho, H.S.; Dashdorj, N.; Henning, R.W.; Kosheleva, I.; Macha, G.; Meron, M.; et al. BioCARS: A synchrotron resource for time-resolved X-ray science. J. Synchrotron. Radiat. 2011, 18, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Bourgeois, D.; Helliwell, J.R.; Moffat, K.; Srajer, V.; Stoddard, B.L. Laue crystallography: Coming of age. J. Synchrotron. Radiat. 1999, 6, 891–917. [Google Scholar] [CrossRef]
- Chapman, H.N.; Fromme, P.; Barty, A.; White, T.A.; Kirian, R.A.; Aquila, A.; Hunter, M.S.; Schulz, J.; DePonte, D.P.; Weierstall, U.; et al. Femtosecond X-ray protein nanocrystallography. Nature 2011, 470, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.S.; DePonte, D.P.; Shapiro, D.A.; Kirian, R.A.; Wang, X.; Starodub, D.; Marchesini, S.; Weierstall, U.; Doak, R.B.; Spence, J.C.; et al. X-ray diffraction from membrane protein nanocrystals. Biophys. J. 2011, 100, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Lomb, L.; Barends, T.R.; Kassemeyer, S.; Aquila, A.; Epp, S.W.; Erk, B.; Foucar, L.; Hartmann, R.; Rudek, B.; Rolles, D.; et al. Radiation damage in protein serial femtosecond crystallography using an X-ray free-electron laser. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84, 214111. [Google Scholar] [CrossRef]
- Chapman, H.N.; Barty, A.; Bogan, M.J.; Boutet, S.; Frank, M.; Hau-Riege, S.P.; Marchesini, S.; Woods, B.W.; Bajt, S.; Benner, H.; et al. Femtosecond diffractive imaging with a soft-X-ray free-electron laser. Nat. Phys. 2006, 2, 839–843. [Google Scholar] [CrossRef]
- DePonte, D.P.; Weierstall, U.; Schmidt, K.; Warner, J.; Starodub, D.; Spence, J.C.H.; Doak, R.B. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J. Phys. D Appl. Phys. 2008, 41, 195505. [Google Scholar] [CrossRef]
- Barty, A.; Kirian, R.A.; Maia, F.R.N.C.; Hantke, M.; Yoon, C.H.; White, T.A.; Chapman, H. Cheetah: Software for high-throughput reduction and analysis of serial femtosecond X-ray diffraction data. J. Appl. Crystallogr. 2014, 47, 1118–1131. [Google Scholar] [CrossRef]
- White, T.A.; Kirian, R.A.; Martin, A.V.; Aquila, A.; Nass, K.; Barty, A.; Chapman, H.N. CrystFEL: A software suite for snapshot serial crystallography. J. Appl. Crystallogr. 2012, 45, 335–341. [Google Scholar] [CrossRef]
- Weierstall, U.; James, D.; Wang, C.; White, T.A.; Wang, D.; Liu, W.; Spence, J.C.; Bruce Doak, R.; Nelson, G.; Fromme, P.; et al. Lipidic cubic phase injector facilitates membrane protein serial femtosecond crystallography. Nat. Commun. 2014, 5, 3309. [Google Scholar] [CrossRef] [PubMed]
- Nogly, P.; James, D.; Wang, D.; White, T.A.; Zatsepin, N.; Shilova, A.; Nelson, G.; Liu, H.; Johansson, L.; Heymann, M.; et al. Lipidic cubic phase serial millisecond crystallography using synchrotron radiation. IUCrJ 2015, 2, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Nango, E.; Royant, A.; Kubo, M.; Nakane, T.; Wickstrand, C.; Kimura, T.; Tanaka, T.; Tono, K.; Song, C.Y.; Tanaka, R.; et al. A three-dimensional movie of structural changes in bacteriorhodopsin. Science 2016, 354, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Ishchenko, A.; Gati, C.; Cherezov, V. Structural biology of G protein-coupled receptors: New opportunities from XFELs and cryoEM. Curr. Opin. Struct. Biol. 2018, 51, 44–52. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Gati, C.; Han, G.W.; Liu, W.; Zatsepin, N.A.; James, D.; Wang, D.; Nelson, G.; Weierstall, U.; et al. Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 2015, 161, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Mizohata, E.; Nango, E.; Suzuki, M.; Tanaka, T.; Masudala, T.; Tanaka, R.; Shimamura, T.; Tanaka, Y.; Suno, C.; et al. Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 2015, 12, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Nakane, T.; Masuda, T.; Suzuki, M.; Inoue, S.; Song, C.; Tanaka, R.; Nakatsu, T.; Mizohata, E.; Yumoto, F.; et al. Hydroxyethyl cellulose matrix applied to serial crystallography. Sci. Rep. 2017, 7, 703. [Google Scholar] [CrossRef]
- Edlund, P.; Takala, H.; Claesson, E.; Henry, L.; Dods, R.; Lehtivuori, H.; Panman, M.; Pande, K.; White, T.; Nakane, T.; et al. The room temperature crystal structure of a bacterial phytochrome determined by serial femtosecond crystallography. Sci. Rep. 2016, 6, 35279. [Google Scholar] [CrossRef]
- Woitowich, N.C.; Halavaty, A.S.; Waltz, P.; Kupitz, C.; Valera, J.; Tracy, G.; Gallagher, K.D.; Claesson, E.; Nakane, T.; Pandey, S.; et al. Structural basis for light control of cell development revealed by crystal structures of a myxobacterial phytochrome. IUCrJ 2018, 5, 619–634. [Google Scholar] [CrossRef]
- Suga, M.; Akita, F.; Sugahara, M.; Kubo, M.; Nakajima, Y.; Nakane, T.; Yamashita, K.; Umena, Y.; Nakabayashi, M.; Yamane, T.; et al. Light-induced structural changes and the site of O=O bond formation in PSII caught by XFEL. Nature 2017, 543, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.G.; Gati, C.; Laksmono, H.; Dao, E.H.; Gul, S.; Fuller, F.; Kern, J.; Chatterjee, R.; Ibrahim, M.; Brewster, A.S.; et al. Concentric-flow electrokinetic injector enables serial crystallography of ribosome and photosystem II. Nat. Methods 2016, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Greiffenberg, D.; Collaboration, A. The AGIPD detector for the European XFEL. J. Instrum. 2012, 7. [Google Scholar] [CrossRef]
- Wiedorn, M.O.; Oberthur, D.; Bean, R.; Schubert, R.; Werner, N.; Abbey, B.; Aepfelbacher, M.; Adriano, L.; Allahgholi, A.; Al-Qudami, N.; et al. Megahertz serial crystallography. Nat. Commun. 2018, 9, 4025. [Google Scholar] [CrossRef] [PubMed]
- Grunbein, M.L.; Bielecki, J.; Gorel, A.; Stricker, M.; Bean, R.; Cammarata, M.; Dorner, K.; Frohlich, L.; Hartmann, E.; Hauf, S.; et al. Megahertz data collection from protein microcrystals at an X-ray free-electron laser. Nat. Commun. 2018, 9, 3487. [Google Scholar] [CrossRef] [PubMed]
- Stan, C.A.; Milathianaki, D.; Laksmono, H.; Sierra, R.G.; McQueen, T.A.; Messerschmidt, M.; Williams, G.J.; Koglin, J.E.; Lane, T.J.; Hayes, M.J.; et al. Liquid explosions induced by X-ray laser pulses. Nat. Phys. 2016, 12, 966–971. [Google Scholar] [CrossRef]
- Wiedorn, M.O.; Awel, S.; Morgan, A.J.; Ayyer, K.; Gevorkov, Y.; Fleckenstein, H.; Roth, N.; Adriano, L.; Bean, R.; Beyerlein, K.R.; et al. Rapid sample delivery for megahertz serial crystallography at X-ray FELs. IUCrJ 2018, 5, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Roedig, P.; Vartiainen, I.; Duman, R.; Panneerselvam, S.; Stube, N.; Lorbeer, O.; Warmer, M.; Sutton, G.; Stuart, D.I.; Weckert, E.; et al. A micro-patterned silicon chip as sample holder for macromolecular crystallography experiments with minimal background scattering. Sci. Rep. 2015, 5, 10451. [Google Scholar] [CrossRef] [PubMed]
- Roessler, C.G.; Agarwal, R.; Allaire, M.; Alonso-Mori, R.; Andi, B.; Bachega, J.F.; Bommer, M.; Brewster, A.S.; Browne, M.C.; Chatterjee, R.; et al. Acoustic Injectors for Drop-On-Demand Serial Femtosecond Crystallography. Structure 2016, 24, 631–640. [Google Scholar] [CrossRef]
- Schmidt, M.; Rajagopal, S.; Ren, Z.; Moffat, K. Application of singular value decomposition to the analysis of time-resolved macromolecular X-ray data. Biophys. J. 2003, 84, 2112–2129. [Google Scholar] [CrossRef]
- Schmidt, M. Structure Based Enzyme Kinetics by Time-Resolved X-ray Crystallography. In Ultrashort Laser Pulses in Medicine and Biology; Springer: Berlin, Germany; New York, NY, USA, 2008. [Google Scholar]
- Srajer, V.; Schmidt, M. Watching proteins function with time-resolved X-ray crystallography. J. Phys. D Appl. Phys. 2017, 50, 373001. [Google Scholar] [CrossRef]
- Steinfeld, J.I.; Francisco, J.S.; Hase, W.L. Chemical Kinetics and Dynamics, 2nd ed.; Prentience Hall: Upper Saddle River, NJ, USA, 1985. [Google Scholar]
- Groenhof, G. Introduction to QM/MM Simulations. Methods Mol. Biol. 2013, 924, 43–66. [Google Scholar] [CrossRef] [PubMed]
- Martin-Garcia, J.M.; Conrad, C.E.; Nelson, G.; Stander, N.; Zatsepin, N.A.; Zook, J.; Zhu, L.; Geiger, J.; Chun, E.; Kissick, D.; et al. Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 2017, 4, 439–454. [Google Scholar] [CrossRef] [PubMed]
- Glownia, J.M.; Cryan, J.; Andreasson, J.; Belkacem, A.; Berrah, N.; Blaga, C.I.; Bostedt, C.; Bozek, J.; DiMauro, L.F.; Fang, L.; et al. Time-resolved pump-probe experiments at the LCLS. Opt. Express 2010, 18, 17620–17630. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, N.; Helml, W.; Galler, A.; Bionta, M.R.; Grunert, J.; Molodtsov, S.L.; Ferguson, K.R.; Schorb, S.; Swiggers, M.L.; Carron, S.; et al. Sub-femtosecond precision measurement of relative X-ray arrival time for free-electron lasers. Nat. Photonics 2014, 8, 706–709. [Google Scholar] [CrossRef]
- Harmand, M.; Coffee, R.; Bionta, M.R.; Chollet, M.; French, D.; Zhu, D.; Fritz, D.M.; Lemke, H.T.; Medvedev, N.; Ziaja, B.; et al. Achieving few-femtosecond time-sorting at hard X-ray free-electron lasers. Nat. Photonics 2013, 7, 215–218. [Google Scholar] [CrossRef]
- Powell, H.R. X-ray data processing. Biosci. Rep 2017, 37. [Google Scholar] [CrossRef]
- Hart, P.; Boutet, S.; Gabriella, C.; Dubrovin, M.; Duda, B.; Fritz, D.; Haller, G.; Herbst, R.; Herrmann, S.; Kenney, C.; et al. The CSPAD megapixel X-ray camera at LCLS. Proc. SPIE 2012, 8504, 85040C. [Google Scholar]
- Bionta, M.R.; Lemke, H.T.; Cryan, J.P.; Glownia, J.M.; Bostedt, C.; Cammarata, M.; Castagna, J.C.; Ding, Y.; Fritz, D.M.; Fry, A.R.; et al. Spectral encoding of X-ray/optical relative delay. Opt. Express 2011, 19, 21855–21865. [Google Scholar] [CrossRef] [PubMed]
- White, T.A.; Mariani, V.; Brehm, W.; Yefanov, O.; Barty, A.; Beyerlein, K.R.; Chervinskii, F.; Galli, L.; Gati, C.; Nakane, T.; et al. Recent developments in CrystFEL. J. Appl. Crystallogr. 2016, 49, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Brehm, W.; Diederichs, K. Breaking the indexing ambiguity in serial crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2014, 70, 101–109. [Google Scholar] [CrossRef]
- Efremov, R.; Moukhametzianov, R.; Buldt, G.; Gordeliy, V. Physical detwinning of hemihedrally twinned hexagonal crystals of bacteriorhodopsin. Biophys. J. 2004, 87, 3608–3613. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Srajer, V.; Henning, R.; Ihee, H.; Purwar, N.; Tenboer, J.; Tripathi, S. Protein energy landscapes determined by five-dimensional crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 2534–2542. [Google Scholar] [CrossRef] [PubMed]
- Genick, U.K.; Borgstahl, G.E.; Ng, K.; Ren, Z.; Pradervand, C.; Burke, P.M.; Srajer, V.; Teng, T.Y.; Schildkamp, W.; McRee, D.E.; et al. Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science 1997, 275, 1471–1475. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Srajer, V.; Purwar, N.; Henning, R.; Schmidt, M. pH dependence of the photoactive yellow protein photocycle investigated by time-resolved crystallography. Biophys. J. 2012, 102, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; Moffat, K. The difference Fourier technique in protein crystallography: Errors and their treatment. Acta Crystallogr. B 1971, 27, 1414–1420. [Google Scholar] [CrossRef]
- Schmidt, M.; Srajer, V.; Purwar, N.; Tripathi, S. The kinetic dose limit in room-temperature time-resolved macromolecular crystallography. J. Synchrotron. Radiat. 2012, 19, 264–273. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Olmos, J.L., Jr.; Pandey, S.; Martin-Garcia, J.M.; Calvey, G.; Katz, A.; Knoska, J.; Kupitz, C.; Hunter, M.S.; Liang, M.; Oberthuer, D.; et al. Enzyme intermediates captured "on the fly" by mix-and-inject serial crystallography. BMC Biol. 2018, 16, 59. [Google Scholar] [CrossRef] [PubMed]
- Ihee, H.; Rajagopal, S.; Srajer, V.; Pahl, R.; Anderson, S.; Schmidt, M.; Schotte, F.; Anfinrud, P.A.; Wulff, M.; Moffat, K. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc. Natl. Acad. Sci. USA 2005, 102, 7145–7150. [Google Scholar] [CrossRef]
- Schmidt, M.; Nienhaus, K.; Pahl, R.; Krasselt, A.; Anderson, S.; Parak, F.; Nienhaus, G.U.; Srajer, V. Ligand migration pathway and protein dynamics in myoglobin: A time-resolved crystallographic study on L29W MbCO. Proc. Natl. Acad. Sci. USA 2005, 102, 11704–11709. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.O.; Lee, J.H.; Kim, J.; Schmidt, M.; Moffat, K.; Srajer, V.; Ihee, H. Volume-conserving trans-cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography. Nat. Chem. 2013, 5, 212–220. [Google Scholar] [CrossRef]
- Rajagopal, S.; Schmidt, M.; Anderson, S.; Ihee, H.; Moffat, K. Analysis of experimental time-resolved crystallographic data by singular value decomposition. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Pahl, R.; Srajer, V.; Anderson, S.; Ren, Z.; Ihee, H.; Rajagopal, S.; Moffat, K. Protein kinetics: Structures of intermediates and reaction mechanism from time-resolved X-ray data. Proc. Natl. Acad. Sci. USA 2004, 101, 4799–4804. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M. Time-Resolved Macromolecular Crystallography at Modern X-ray Sources. Methods Mol. Biol. 2017, 1607, 273–294. [Google Scholar] [CrossRef] [PubMed]
- Levantino, M.; Schiro, G.; Lemke, H.T.; Cottone, G.; Glownia, J.M.; Zhu, D.; Chollet, M.; Ihee, H.; Cupane, A.; Cammarata, M. Ultrafast myoglobin structural dynamics observed with an X-ray free-electron laser. Nat. Commun. 2015, 6, 6772. [Google Scholar] [CrossRef] [PubMed]
- Arnlund, D.; Johansson, L.C.; Wickstrand, C.; Barty, A.; Williams, G.J.; Malmerberg, E.; Davidsson, J.; Milathianaki, D.; DePonte, D.P.; Shoeman, R.L.; et al. Visualizing a protein quake with time-resolved X-ray scattering at a free-electron laser. Nat. Methods 2014, 11, 923–926. [Google Scholar] [CrossRef] [PubMed]
- Fung, R.; Hanna, A.M.; Vendrell, O.; Ramakrishna, S.; Seideman, T.; Santra, R.; Ourmazd, A.; Ourmazd, A. Dynamics from noisy data with extreme timing uncertainty. Nature 2016, 532, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.E. Isolation and characterization of soluble cytochromes, ferredoxins and other chromophoric proteins from the halophilic phototrophic bacterium Ectothiorhodospira halophila. Biochim. Biophys. Acta 1985, 806, 175–183. [Google Scholar] [CrossRef]
- Schmidt, M. A short history of structure based research on the photocycle of photoactive yellow protein. Struct. Dyn. 2017, 4, 032201. [Google Scholar] [CrossRef] [PubMed]
- Perman, B.; Srajer, V.; Ren, Z.; Teng, T.Y.; Pradervand, C.; Ursby, T.; Bourgeois, D.; Schotte, F.; Wulff, M.; Kort, R.; et al. Energy transduction on the nanosecond time scale: Early structural events in a xanthopsin photocycle. Science 1998, 279, 1946–1950. [Google Scholar] [CrossRef] [PubMed]
- Schotte, F.; Cho, H.S.; Kaila, V.R.; Kamikubo, H.; Dashdorj, N.; Henry, E.R.; Graber, T.J.; Henning, R.; Wulff, M.; Hummer, G.; et al. Watching a signaling protein function in real time via 100-ps time-resolved Laue crystallography. Proc. Natl. Acad. Sci. USA 2012, 109, 19256–19261. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, W.W.; Hoff, W.D.; Armitage, J.P.; Hellingwerf, K.J. The Eubacterium Ectothiorhodospira-Halophila Is Negatively Phototactic, with a Wavelength Dependence That Fits the Absorption-Spectrum of the Photoactive Yellow Protein. J. Bacteriol. 1993, 175, 3096–3104. [Google Scholar] [CrossRef] [PubMed]
- Kort, R.; Vonk, H.; Xu, X.; Hoff, W.D.; Crielaard, W.; Hellingwerf, K.J. Evidence for trans-cis isomerization of the p-coumaric acid chromophore as the photochemical basis of the photocycle of photoactive yellow protein. FEBS Lett. 1996, 382, 73–78. [Google Scholar] [CrossRef]
- van Thor, J.J.; Warren, M.M.; Lincoln, C.N.; Chollet, M.; Lemke, H.T.; Fritz, D.M.; Schmidt, M.; Tenboer, J.; Ren, Z.; Srajer, V.; et al. Signal to noise considerations for single crystal femtosecond time resolved crystallography of the Photoactive Yellow Protein. Faraday Discuss. 2014, 171, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Hutchison, C.D.M.; van Thor, J.J. Populations and coherence in femtosecond time resolved X-ray crystallography of the photoactive yellow protein. Int. Rev. Phys. Chem. 2017, 36, 117–143. [Google Scholar] [CrossRef]
- Prokhorenko, V.I.; Nagy, A.M.; Waschuk, S.A.; Brown, L.S.; Birge, R.R.; Miller, R.J. Coherent control of retinal isomerization in bacteriorhodopsin. Science 2006, 313, 1257–1261. [Google Scholar] [CrossRef] [PubMed]
- Austin, R.H.; Beeson, K.W.; Eisenstein, L.; Frauenfelder, H.; Gunsalus, I.C. Dynamics of ligand binding to myoglobin. Biochemistry 1975, 14, 5355–5373. [Google Scholar] [CrossRef] [PubMed]
- Ostermann, A.; Waschipky, R.; Parak, F.G.; Nienhaus, G.U. Ligand binding and conformational motions in myoglobin. Nature 2000, 404, 205–208. [Google Scholar] [CrossRef]
- Srajer, V.; Teng, T.Y.; Ursby, T.; Pradervand, C.; Ren, Z.; Adachi, S.; Schildkamp, W.; Bourgeois, D.; Wulff, M.; Moffat, K. Photolysis of the carbon monoxide complex of myoglobin: Nanosecond time-resolved crystallography. Science 1996, 274, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, I.; Berendzen, J.; Phillips, G.N.; Sweet, R.M. Crystal-Structure of Photolyzed Carbonmonoxy-Myoglobin. Nature 1994, 371, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Schotte, F.; Lim, M.; Jackson, T.A.; Smirnov, A.V.; Soman, J.; Olson, J.S.; Phillips, G.N., Jr.; Wulff, M.; Anfinrud, P.A. Watching a protein as it functions with 150-ps time-resolved X-ray crystallography. Science 2003, 300, 1944–1947. [Google Scholar] [CrossRef]
- Bourgeois, D.; Vallone, B.; Schotte, F.; Arcovito, A.; Miele, A.E.; Sciara, G.; Wulff, M.; Anfinrud, P.; Brunori, M. Complex landscape of protein structural dynamics unveiled by nanosecond Laue crystallography. Proc. Natl. Acad. Sci. USA 2003, 100, 8704–8709. [Google Scholar] [CrossRef]
- Lim, M.; Jackson, T.A.; Anfinrud, P.A. Binding of CO to myoglobin from a heme pocket docking site to form nearly linear Fe-C-O. Science 1995, 269, 962–966. [Google Scholar] [CrossRef]
- Genberg, L.; Richard, L.; McLendon, G.; Miller, R.J. Direct observation of global protein motion in hemoglobin and myoglobin on picosecond time scales. Science 1991, 251, 1051–1054. [Google Scholar] [CrossRef] [PubMed]
- Einsle, O.; Messerschmidt, A.; Stach, P.; Bourenkov, G.P.; Bartunik, H.D.; Huber, R.; Kroneck, P.M.H. Structure of cytochrome c nitrite reductase. Nature 1999, 400, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Youngblut, M.; Judd, E.T.; Srajer, V.; Sayyed, B.; Goelzer, T.; Elliott, S.J.; Schmidt, M.; Pacheco, A.A. Laue crystal structure of Shewanella oneidensis cytochrome c nitrite reductase from a high-yield expression system. J. Biol. Inorg. Chem. 2012, 17, 647–662. [Google Scholar] [CrossRef]
- Kupitz, C.; Basu, S.; Grotjohann, I.; Fromme, R.; Zatsepin, N.A.; Rendek, K.N.; Hunter, M.S.; Shoeman, R.L.; White, T.A.; Wang, D.; et al. Serial time-resolved crystallography of photosystem II using a femtosecond X-ray laser. Nature 2014, 513, 5. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Ibrahim, M.; Chatterjee, R.; Gul, S.; Fuller, F.; Koroidov, S.; Brewster, A.S.; Tran, R.; Alonso-Mori, R.; Kroll, T.; et al. Structure of photosystem II and substrate binding at room temperature. Nature 2016, 540, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Kubo, M.; Baba, S.; Yamashita, K.; Hirata, K.; Ueno, G.; Nomura, T.; Kimura, T.; Shinzawa-Itoh, K.; Baba, J.; et al. A nanosecond time-resolved XFEL analysis of structural changes associated with CO release from cytochrome c oxidase. Sci. Adv. 2017, 3, e1603042. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Meier, B.; Parak, F. X-ray structure of the cambialistic superoxide dismutase from Propionibacterium shermanii active with Fe or Mn. J. Biol. Inorg. Chem. 1996, 1, 532–541. [Google Scholar] [CrossRef]
- Sheng, Y.; Abreu, I.A.; Cabelli, D.E.; Maroney, M.J.; Miller, A.F.; Teixeira, M.; Valentine, J.S. Superoxide dismutases and superoxide reductases. Chem. Rev. 2014, 114, 3854–3918. [Google Scholar] [CrossRef]
- Mangani, S. Textbook of Metalloproteins, Metal Ion Routes between Organisms and the Inorganic World; World Scientific Publishing: Singapore, 2020. [Google Scholar] [CrossRef]
- Taberman, H. Radiation Damage in Macromolecular Crystallography—An Experimentalist’s View. Crystals 2018, 8, 157. [Google Scholar] [CrossRef]
- Nass, K.; Foucar, L.; Barends, T.R.; Hartmann, E.; Botha, S.; Shoeman, R.L.; Doak, R.B.; Alonso-Mori, R.; Aquila, A.; Bajt, S.; et al. Indications of radiation damage in ferredoxin microcrystals using high-intensity X-FEL beams. J. Synchrotron. Radiat. 2015, 22, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 A. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Suga, M.; Akita, F.; Hirata, K.; Ueno, G.; Murakami, H.; Nakajima, Y.; Shimizu, T.; Yamashita, K.; Yamamoto, M.; Ago, H.; et al. Native structure of photosystem II at 1.95 A resolution viewed by femtosecond X-ray pulses. Nature 2015, 517, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Chatterjee, R.; Young, I.D.; Fuller, F.D.; Lassalle, L.; Ibrahim, M.; Gul, S.; Fransson, T.; Brewster, A.S.; Alonso-Mori, R.; et al. Structures of the intermediates of Kok’s photosynthetic water oxidation clock. Nature 2018, 563, 421–425. [Google Scholar] [CrossRef]
- Beyerlein, K.R.; Dierksmeyer, D.; Mariani, V.; Kuhn, M.; Sarrou, I.; Ottaviano, A.; Awel, S.; Knoska, J.; Fuglerud, S.; Jonsson, O.; et al. Mix-and-diffuse serial synchrotron crystallography. IUCrJ 2017, 4, 769–777. [Google Scholar] [CrossRef]
- Stellato, F.; Oberthuer, D.; Mengning, L.; Bean, R.; Gati, C.; Yefanov, O.; Barty, A.; Burkhardt, A.; Fischer, P.; Galli, L.; et al. Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ 2014, 1, 204–212. [Google Scholar] [CrossRef]
- Meents, A.; Wiedorn, M.O.; Srajer, V.; Henning, R.; Sarrou, I.; Bergtholdt, J.; Barthelmess, M.; Reinke, P.Y.A.; Dierksmeyer, D.; Tolstikova, A.; et al. Pink-beam serial crystallography. Nat. Commun. 2017, 8, 1281. [Google Scholar] [CrossRef] [PubMed]
- Goelder, M.; Givens, R. Dynamic Studies in Biology: Phototriggers, Photoswitches and Caged Biomolecules; Wiley VCH: Hoboken, NJ, USA, 2005. [Google Scholar]
- Stoddard, B.L.; Cohen, B.E.; Brubaker, M.; Mesecar, A.D.; Koshland, D.E., Jr. Millisecond Laue structures of an enzyme-product complex using photocaged substrate analogs. Nat. Struct. Biol. 1998, 5, 891–897. [Google Scholar] [CrossRef]
- Schlichting, I.; Rapp, G.; John, J.; Wittinghofer, A.; Pai, E.F.; Goody, R.S. Biochemical and crystallographic characterization of a complex of c-Ha-ras p21 and caged GTP with flash photolysis. Proc. Natl. Acad. Sci. USA 1989, 86, 7687–7690. [Google Scholar] [CrossRef] [PubMed]
- Moglich, A.; Moffat, K. Engineered photoreceptors as novel optogenetic tools. Photochem. Photobiol. Sci. 2010, 9, 1286–1300. [Google Scholar] [CrossRef]
- Moffat, K. Time-resolved crystallography and protein design: Signalling photoreceptors and optogenetics. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.A. The Biology of Cancer; Garland Science: New York, NY, USA, 2007. [Google Scholar]
- Wyckoff, H.W.; Doscher, M.; Tsernoglou, D.; Inagami, T.; Johnson, L.N.; Hardman, K.D.; Allewell, N.M.; Kelly, D.M.; Richards, F.M. Design of a diffractometer and flow cell system for X-ray analysis of crystalline proteins with applications to the crystal chemistry of ribonuclease-S. J. Mol. Biol. 1967, 27, 563–578. [Google Scholar] [CrossRef]
- Perry, S.L.; Guha, S.; Pawate, A.S.; Henning, R.; Kosheleva, I.; Srajer, V.; Kenis, P.J.; Ren, Z. In situ serial Laue diffraction on a microfluidic crystallization device. J. Appl. Crystallogr. 2014, 47, 1975–1982. [Google Scholar] [CrossRef] [PubMed]
- Helliwell, J.R.; Nieh, Y.P.; Raftery, J.; Cassetta, A.; Habash, J.; Carr, P.D.; Ursby, T.; Wulff, M.; Thompson, A.W.; Niemann, A.C.; et al. Time-resolved structures of hydroxymethylbilane synthase (Lys59Gln mutant) as it is loaded with substrate in the crystal determined by Laue diffraction. J. Chem. Soc. Faraday Trans. 1998, 94, 2615–2622. [Google Scholar] [CrossRef]
- Kim, T.H.; Mehrabi, P.; Ren, Z.; Sljoka, A.; Ing, C.; Bezginov, A.; Ye, L.; Pomes, R.; Prosser, R.S.; Pai, E.F. The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Blake, C.C.; Koenig, D.F.; Mair, G.A.; North, A.C.; Phillips, D.C.; Sarma, V.R. Structure of hen egg-white lysozyme. A three-dimensional Fourier synthesis at 2 Angstrom resolution. Nature 1965, 206, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Sainsbury, S.; Bird, L.; Rao, V.; Shepherd, S.M.; Stuart, D.I.; Hunter, W.N.; Owens, R.J.; Ren, J. Crystal structures of penicillin-binding protein 3 from Pseudomonas aeruginosa: Comparison of native and antibiotic-bound forms. J. Mol. Biol. 2011, 405, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Fair, R.J.; Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 2014, 6, 25–64. [Google Scholar] [CrossRef]
- Bassetti, M.; Merelli, M.; Temperoni, C.; Astilean, A. New antibiotics for bad bugs: Where are we? Ann. Clin. Microbiol. Antimicrob. 2013, 12, 22. [Google Scholar] [CrossRef]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. [Google Scholar] [CrossRef]
- Kong, K.F.; Schneper, L.; Mathee, K. Beta-lactam antibiotics: From antibiosis to resistance and bacteriology. APMIS 2010, 118, 1–36. [Google Scholar] [CrossRef] [PubMed]
- Ambler, R.P. The structure of beta-lactamases. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1980, 289, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Kupitz, C.; Olmos, J.L., Jr.; Holl, M.; Tremblay, L.; Pande, K.; Pandey, S.; Oberthur, D.; Hunter, M.; Liang, M.; Aquila, A.; et al. Structural enzymology using X-ray free electron lasers. Struct. Dyn. 2017, 4, 044003. [Google Scholar] [CrossRef] [PubMed]
- Carslaw, H.S.; Jaeger, J.C. Conduction Heat in Solids, 2nd ed.; Clarendon Press: Oxford, UK, 1959. [Google Scholar]
- Calvey, G.D.; Katz, A.M.; Schaffer, C.B.; Pollack, L. Mixing injector enables time-resolved crystallography with high hit rate at X-ray free electron lasers. Struct. Dyn. 2016, 3, 054301. [Google Scholar] [CrossRef] [PubMed]
- Kiener, P.A.; Waley, S.G. Reversible inhibitors of penicillinases. Biochem. J. 1978, 169, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhou, X.E.; Gao, X.; He, Y.; Liu, W.; Ishchenko, A.; Barty, A.; White, T.A.; Yefanov, O.; Han, G.W.; et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015, 523, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Wald, G. Molecular basis of visual excitation. Science 1968, 162, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Ernst, O.P.; Lodowski, D.T.; Elstner, M.; Hegemann, P.; Brown, L.S.; Kandori, H. Microbial and animal rhodopsins: Structures, functions, and molecular mechanisms. Chem. Rev. 2014, 114, 126–163. [Google Scholar] [CrossRef] [PubMed]
- Stoeckenius, W.; Lozier, R.H.; Bogomolni, R.A. Bacteriorhodopsin and the purple membrane of halobacteria. Biochim. Biophys. Acta 1979, 505, 215–278. [Google Scholar] [CrossRef]
- Kuhlbrandt, W. Bacteriorhodopsin—The movie. Nature 2000, 406, 569–570. [Google Scholar] [CrossRef] [PubMed]
- Landau, E.M.; Rosenbusch, J.P. Lipidic cubic phases: A novel concept for the crystallization of membrane proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 14532–14535. [Google Scholar] [CrossRef]
- Pebay-Peyroula, E.; Rummel, G.; Rosenbusch, J.P.; Landau, E.M. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science 1997, 277, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Luecke, H.; Schobert, B.; Richter, H.T.; Cartailler, J.P.; Lanyi, J.K. Structure of bacteriorhodopsin at 1.55 A resolution. J. Mol. Biol. 1999, 291, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Nogly, P.; Weinert, T.; James, D.; Carbajo, S.; Ozerov, D.; Furrer, A.; Gashi, D.; Borin, V.; Skopintsev, P.; Jaeger, K.; et al. Retinal isomerization in bacteriorhodopsin captured by a femtosecond X-ray laser. Science 2018, 361. [Google Scholar] [CrossRef]
- Butler, W.L.; Norris, K.H.; Siegelman, H.W.; Hendricks, S.B. Detection, assay, and preliminary purification of the pigment controlling photoresponsive development of plants. Proc. Natl. Acad. Sci. USA 1959, 89, 6. [Google Scholar] [CrossRef]
- Auldridge, M.E.; Forest, K.T. Bacterial phytochromes: More than meets the light. Crit. Rev. Biochem. Mol. 2011, 46, 67–88. [Google Scholar] [CrossRef] [PubMed]
- Batschauer, A. Photoreceptors of higher plants. Planta 1998, 206, 479–492. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.J.; Vener, A.V.; Vierstra, R.D. Bacteriophytochromes: Phytochrome-like photoreceptors from nonphotosynthetic eubacteria. Science 1999, 286, 2517–2520. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Brunzelle, J.S.; Forest, K.T.; Vierstra, R.D. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature 2005, 438, 325–331. [Google Scholar] [CrossRef]
- Gourinchas, G.; Etzl, S.; Gobl, C.; Vide, U.; Madl, T.; Winkler, A. Long-range allosteric signaling in red light-regulated diguanylyl cyclases. Sci. Adv. 2017, 3, e1602498. [Google Scholar] [CrossRef] [PubMed]
- Otero, L.H.; Klinke, S.; Rinaldi, J.; Velazquez-Escobar, F.; Mroginski, M.A.; Fernandez Lopez, M.; Malamud, F.; Vojnov, A.A.; Hildebrandt, P.; Goldbaum, F.A.; et al. Structure of the Full-Length Bacteriophytochrome from the Plant Pathogen Xanthomonas campestris Provides Clues to its Long-Range Signaling Mechanism. J. Mol. Biol. 2016, 428, 3702–3720. [Google Scholar] [CrossRef] [PubMed]
- Burgie, E.S.; Bussell, A.N.; Walker, J.M.; Dubiel, K.; Vierstra, R.D. Crystal structure of the photosensing module from a red/far-red light-absorbing plant phytochrome. Proc. Natl. Acad. Sci. USA 2014, 111, 10179–10184. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.R.; Zhang, J.; von Stetten, D.; Gunther, M.; Murgida, D.H.; Mroginski, M.A.; Walker, J.M.; Forest, K.T.; Hildebrandt, P.; Vierstra, R.D. Mutational analysis of Deinococcus radiodurans bacteriophytochrome reveals key amino acids necessary for the photochromicity and proton exchange cycle of phytochromes. J. Biol. Chem. 2008, 283, 12212–12226. [Google Scholar] [CrossRef] [PubMed]
- Takala, H.; Bjorling, A.; Berntsson, O.; Lehtivuori, H.; Niebling, S.; Hoernke, M.; Kosheleva, I.; Henning, R.; Menzel, A.; Ihalainen, J.A.; et al. Signal amplification and transduction in phytochrome photosensors. Nature 2014, 509, 245–248. [Google Scholar] [CrossRef]
- Burgie, E.S.; Zhang, J.; Vierstra, R.D. Crystal Structure of Deinococcus Phytochrome in the Photoactivated State Reveals a Cascade of Structural Rearrangements during Photoconversion. Structure 2016, 24, 448–457. [Google Scholar] [CrossRef]
- Stagno, J.R.; Liu, Y.; Bhandari, Y.R.; Conrad, C.E.; Panja, S.; Swain, M.; Fan, L.; Nelson, G.; Li, C.; Wendel, D.R.; et al. Structures of riboswitch RNA reaction states by mix-and-inject XFEL serial crystallography. Nature 2017, 541, 242–246. [Google Scholar] [CrossRef]
- Toh, K.C.; Stojkovic, E.A.; van Stokkum, I.H.M.; Moffat, K.; Kennis, J.T.M. Fluorescence quantum yield and photochemistry of bacteriophytochrome constructs. Phys. Chem. Chem. Phys. 2011, 13, 11985–11997. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science 2009, 324, 804–807. [Google Scholar] [CrossRef] [PubMed]
- Sahl, S.J.; Hell, S.W.; Jakobs, S. Fluorescence nanoscopy in cell biology. Nat. Rev. Mol. Cell Biol. 2017, 18, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Stauch, B.; Johansson, L.; Ishchenko, A.; Han, G.W.; Batyuk, A.; Cherezov, V. Advances in Structure Determination of G Protein-Coupled Receptors by SFX. In X-ray Free Electron Lasers; Boutet, S., Fromme, P., Hunter, M., Eds.; Springer Nature: Cham, Switzerland, 2018; pp. 301–329. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delay | Mixing Time | Crystal Size (μm3) | Diffusion Coefficient (cm2·s−1) | Diffusion Time | Substrate after Mixing | Time to Reach Stoichiometric Concentration |
|---|---|---|---|---|---|---|
| 30 ms | 5 ms | 10 × 10 × 3 | 2.3 × 10−6 | 3.5 ms | 240 mmol/L | 0.24 ms |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, M. Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources. Int. J. Mol. Sci. 2019, 20, 1401. https://doi.org/10.3390/ijms20061401
Schmidt M. Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources. International Journal of Molecular Sciences. 2019; 20(6):1401. https://doi.org/10.3390/ijms20061401
Chicago/Turabian StyleSchmidt, Marius. 2019. "Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources" International Journal of Molecular Sciences 20, no. 6: 1401. https://doi.org/10.3390/ijms20061401
APA StyleSchmidt, M. (2019). Time-Resolved Macromolecular Crystallography at Pulsed X-ray Sources. International Journal of Molecular Sciences, 20(6), 1401. https://doi.org/10.3390/ijms20061401