HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem Cell-Derived Neurons


Abstract
1. Introduction
2. Role of HDAC6 in Animal Models Relevant to Nervous System Disorders
3. Studying HDAC Biology in Human Neuronal Cells
4. HDACs and Wnt-GSK3-β-Catenin Biology in Mood Disorders
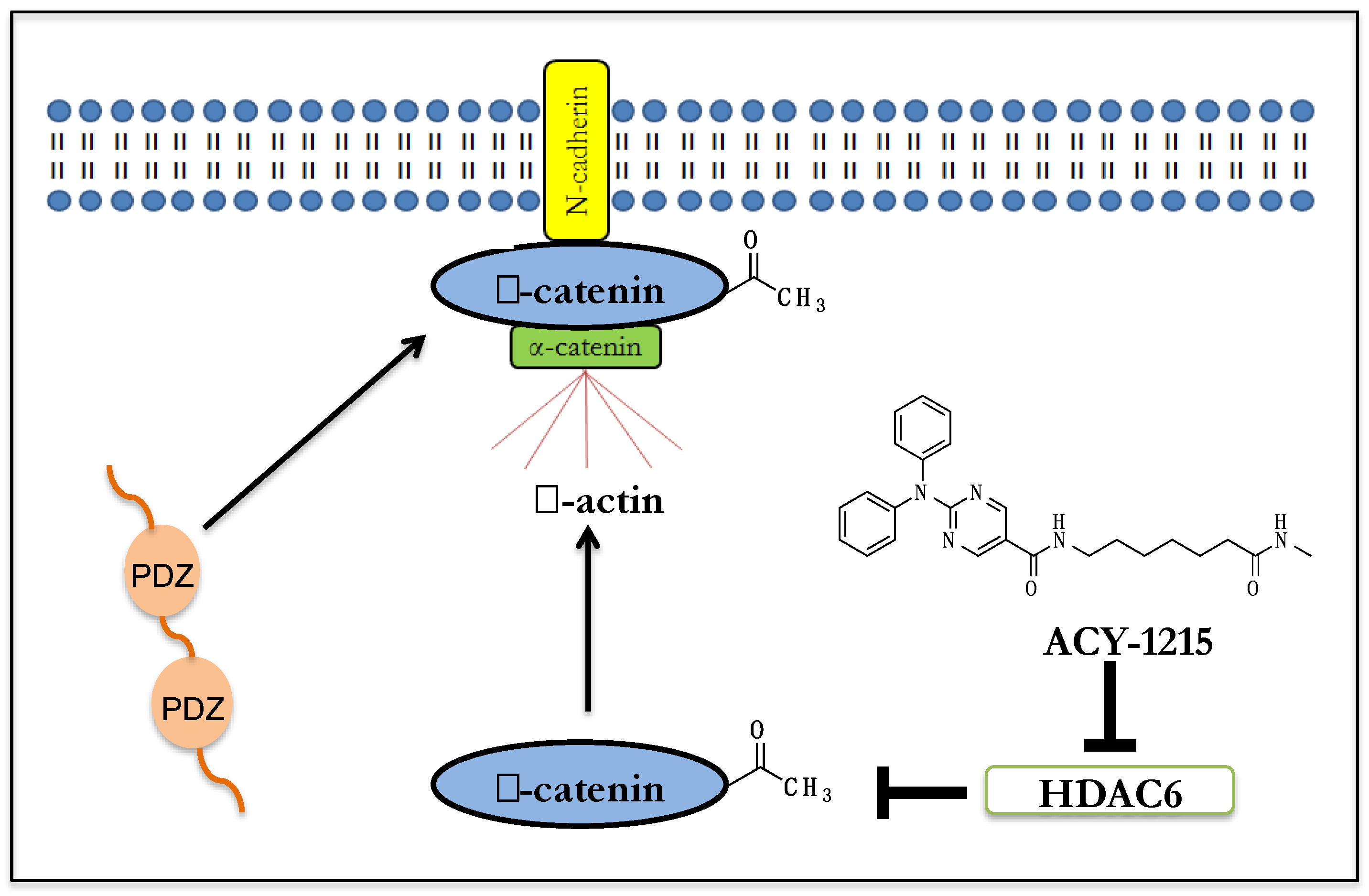
5. Effect of HDAC6 Inhibition on β-Catenin Biology in Human Neuronal Cells
6. β-Catenin’s Function at the Synapse
7. Implications for HDAC6i Mediated β-catenin Membrane Localization
8. HDAC6 Interaction with AKT and Relevance to Neurobiology
9. Summary
Acknowledgments
Conflicts of Interest
References
- Brownell, J.E.; Allis, C.D. Special HATs for special occasions: Linking histone acetylation to chromatin assembly and gene activation. Curr. Opin. Genet. Dev. 1996, 6, 176–184. [Google Scholar] [CrossRef]
- Hassig, C.A.; Schreiber, S.L. Nuclear histone acetylases and deacetylases and transcriptional regulation: HATs off to HDACs. Curr. Opin. Chem. Biol. 1997, 1, 300–308. [Google Scholar] [CrossRef]
- Cho, Y.; Cavalli, V. HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol. 2014, 27, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Mahgoub, M.; Monteggia, L.M. A role for histone deacetylases in the cellular and behavioral mechanisms underlying learning and memory. Learn. Mem. 2014, 21, 564–568. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.M.; Schroeder, F.A.; Perlis, R.H.; Haggarty, S.J. Epigenetic mechanisms in mood disorders: Targeting neuroplasticity. Neuroscience 2014, 264, 112–130. [Google Scholar] [CrossRef]
- Shi, P.; Scott, M.A.; Ghosh, B.; Wan, D.; Wissner-Gross, Z.; Mazitschek, R.; Haggarty, S.J.; Yanik, M.F. Synapse microarray identification of small molecules that enhance synaptogenesis. Nat. Commun. 2011, 2, 510. [Google Scholar] [CrossRef]
- Rumbaugh, G.; Sillivan, S.E.; Ozkan, E.D.; Rojas, C.S.; Hubbs, C.R.; Aceti, M.; Kilgore, M.; Kudugunti, S.; Puthanveettil, S.V.; Sweatt, J.D.; et al. Pharmacological Selectivity Within Class I Histone Deacetylases Predicts Effects on Synaptic Function and Memory Rescue. Neuropsychopharmacology 2015, 40, 2307–2316. [Google Scholar] [CrossRef]
- Covington, H.E.; Maze, I.; LaPlant, Q.C.; Vialou, V.F.; Ohnishi, Y.N.; Berton, O.; Fass, D.M.; Renthal, W.; Rush, A.J.; Wu, E.Y.; et al. Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009, 29, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Richon, V.M.; Webb, Y.; Merger, R.; Sheppard, T.; Jursic, B.; Ngo, L.; Civoli, F.; Breslow, R.; Rifkind, R.A.; Marks, P.A. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc. Natl. Acad. Sci. USA 1996, 93, 5705–5708. [Google Scholar] [CrossRef] [PubMed]
- Fass, D.M.; Reis, S.A.; Ghosh, B.; Hennig, K.M.; Joseph, N.F.; Zhao, W.N.; Nieland, T.J.; Guan, J.S.; Kuhnle, C.E.; Tang, W.; et al. Crebinostat: A novel cognitive enhancer that inhibits histone deacetylase activity and modulates chromatin-mediated neuroplasticity. Neuropharmacology 2013, 64, 81–96. [Google Scholar] [CrossRef] [PubMed]
- el-Beltagi, H.M.; Martens, A.C.; Lelieveld, P.; Haroun, E.A.; Hagenbeek, A. Acetyldinaline: A new oral cytostatic drug with impressive differential activity against leukemic cells and normal stem cells—Preclinical studies in a relevant rat model for human acute myelocytic leukemia. Cancer Res. 1993, 53, 3008–3014. [Google Scholar]
- Moradei, O.M.; Mallais, T.C.; Frechette, S.; Paquin, I.; Tessier, P.E.; Leit, S.M.; Fournel, M.; Bonfils, C.; Trachy-Bourget, M.C.; Liu, J.; et al. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. [Google Scholar] [CrossRef] [PubMed]
- Minami, J.; Suzuki, R.; Mazitschek, R.; Gorgun, G.; Ghosh, B.; Cirstea, D.; Hu, Y.; Mimura, N.; Ohguchi, H.; Cottini, F.; et al. Histone deacetylase 3 as a novel therapeutic target in multiple myeloma. Leukemia 2014, 28, 680–689. [Google Scholar] [CrossRef]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [PubMed]
- Mithraprabhu, S.; Khong, T.; Jones, S.S.; Spencer, A. Histone deacetylase (HDAC) inhibitors as single agents induce multiple myeloma cell death principally through the inhibition of class I HDAC. Br. J. Haematol. 2013, 162, 559–562. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Strebl, M.G.; Campbell, A.J.; Zhao, W.N.; Schroeder, F.A.; Riley, M.M.; Chindavong, P.S.; Morin, T.M.; Haggarty, S.J.; Wagner, F.F.; Ritter, T.; et al. HDAC6 Brain Mapping with [18F]Bavarostat Enabled by a Ru-Mediated Deoxyfluorination. ACS Cent. Sci. 2017, 3, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef]
- Jochems, J.; Boulden, J.; Lee, B.G.; Blendy, J.A.; Jarpe, M.; Mazitschek, R.; Van Duzer, J.H.; Jones, S.; Berton, O. Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 2014, 39, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Majid, T.; Griffin, D.; Criss, Z.; Jarpe, M.; Pautler, R.G. Pharmocologic treatment with histone deacetylase 6 inhibitor (ACY-738) recovers Alzheimer’s disease phenotype in amyloid precursor protein/presenilin 1 (APP/PS1) mice. Alzheimer’s Dement. 2015, 1, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Rivieccio, M.A.; Brochier, C.; Willis, D.E.; Walker, B.A.; D’Annibale, M.A.; McLaughlin, K.; Siddiq, A.; Kozikowski, A.P.; Jaffrey, S.R.; Twiss, J.L.; et al. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc. Natl. Acad. Sci. USA 2009, 106, 19599–19604. [Google Scholar] [CrossRef]
- Hanson, K.; Tian, N.; Vickers, J.C.; King, A.E. The HDAC6 Inhibitor Trichostatin A Acetylates Microtubules and Protects Axons from Excitotoxin-Induced Degeneration in a Compartmented Culture Model. Front. Neurosci. 2018, 12, 872. [Google Scholar] [CrossRef] [PubMed]
- Ganai, S.A. Small-molecule Modulation of HDAC6 Activity: The Propitious Therapeutic Strategy to Vanquish Neurodegenerative Disorders. Curr. Med. Chem. 2017, 24, 4104–4120. [Google Scholar] [CrossRef]
- Kim, S.H.; Shanware, N.P.; Bowler, M.J.; Tibbetts, R.S. Amyotrophic lateral sclerosis-associated proteins TDP-43 and FUS/TLS function in a common biochemical complex to co-regulate HDAC6 mRNA. J. Biol. Chem. 2010, 285, 34097–34105. [Google Scholar] [CrossRef]
- Fiesel, F.C.; Voigt, A.; Weber, S.S.; Van den Haute, C.; Waldenmaier, A.; Görner, K.; Walter, M.; Anderson, M.L.; Kern, J.V.; Rasse, T.M.; et al. Knockdown of transactive response DNA-binding protein (TDP-43) downregulates histone deacetylase 6. EMBO J. 2010, 29, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Miskiewicz, K.; Jose, L.E.; Yeshaw, W.M.; Valadas, J.S.; Swerts, J.; Munck, S.; Feiguin, F.; Dermaut, B.; Verstreken, P. HDAC6 is a Bruchpilot deacetylase that facilitates neurotransmitter release. Cell Rep. 2014, 8, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Xia, Q.; Wang, H.; Zhang, Y.; Ying, Z.; Wang, G. Loss of TDP-43 Inhibits Amyotrophic Lateral Sclerosis-Linked Mutant SOD1 Aggresome Formation in an HDAC6-Dependent Manner. J. Alzheimer’s Dis. 2015, 45, 373–386. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, X.J.; Li, L.X.; Wang, Y.; Zhong, R.J.; Le, W. Histone deacetylase 6 delays motor neuron degeneration by ameliorating the autophagic flux defect in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci. Bull. 2015, 31, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Gal, J.; Chen, J.; Barnett, K.R.; Yang, L.; Brumley, E.; Zhu, H. HDAC6 regulates mutant SOD1 aggregation through two SMIR motifs and tubulin acetylation. J. Biol. Chem. 2013, 288, 15035–15045. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kawaguchi, Y.; Li, M.; Kapur, M.; Choi, S.J.; Kim, H.J.; Park, S.Y.; Zhu, H.; Yao, T.P. Uncoupling of Protein Aggregation and Neurodegeneration in a Mouse Amyotrophic Lateral Sclerosis Model. Neurodegener. Dis. 2015, 15, 339–349. [Google Scholar] [CrossRef]
- Taes, I.; Timmers, M.; Hersmus, N.; Bento-Abreu, A.; Van Den Bosch, L.; Van Damme, P.; Auwerx, J.; Robberecht, W. Hdac6 deletion delays disease progression in the SOD1G93A mouse model of ALS. Hum. Mol. Genet. 2013, 22, 1783–1790. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K. RNAi versus small molecules: Different mechanisms and specificities can lead to different outcomes. Curr. Opin. Drug Discov. Dev. 2005, 8, 557–566. [Google Scholar]
- Weiss, W.A.; Taylor, S.S.; Shokat, K.M. Recognizing and exploiting differences between RNAi and small-molecule inhibitors. Nat. Chem. Biol. 2007, 3, 739–744. [Google Scholar] [CrossRef]
- Tseng, J.H.; Xie, L.; Song, S.; Xie, Y.; Allen, L.; Ajit, D.; Hong, J.S.; Chen, X.; Meeker, R.B.; Cohen, T.J. The Deacetylase HDAC6 Mediates Endogenous Neuritic Tau Pathology. Cell Rep. 2017, 20, 2169–2183. [Google Scholar] [CrossRef]
- Govindarajan, N.; Rao, P.; Burkhardt, S.; Sananbenesi, F.; Schlüter, O.M.; Bradke, F.; Lu, J.; Fischer, A. Reducing HDAC6 ameliorates cognitive deficits in a mouse model for Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 52–63. [Google Scholar] [CrossRef]
- Selenica, M.L.; Benner, L.; Housley, S.B.; Manchec, B.; Lee, D.C.; Nash, K.R.; Kalin, J.; Bergman, J.A.; Kozikowski, A.; Gordon, M.N.; et al. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimer’s Res. 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Liu, C.; Wu, J.; Tao, J.J.; Sui, X.L.; Yao, Z.G.; Xu, Y.F.; Huang, L.; Zhu, H.; Sheng, S.L.; et al. Tubastatin A/ACY-1215 Improves Cognition in Alzheimer’s Disease Transgenic Mice. J. Alzheimer’s Dis. 2014, 41, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, S.; Feldon, J.; Yee, B.K. Age-dependent phenotypic characteristics of a triple transgenic mouse model of Alzheimer disease. Behav. Neurosci. 2008, 122, 733–747. [Google Scholar] [CrossRef]
- Fan, S.J.; Huang, F.I.; Liou, J.P.; Yang, C.R. The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 655. [Google Scholar] [CrossRef] [PubMed]
- Benoy, V.; Van Helleputte, L.; Prior, R.; d’Ydewalle, C.; Haeck, W.; Geens, N.; Scheveneels, W.; Schevenels, B.; Cader, M.Z.; Talbot, K.; et al. HDAC6 is a therapeutic target in mutant GARS-induced Charcot-Marie-Tooth disease. Brain 2018, 141, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Benoy, V.; Bergman, J.A.; Kalin, J.H.; Frojuello, M.; Vistoli, G.; Haeck, W.; Van Den Bosch, L.; Kozikowski, A.P. Bicyclic-Capped Histone Deacetylase 6 Inhibitors with Improved Activity in a Model of Axonal Charcot-Marie-Tooth Disease. ACS Chem. Neurosci. 2016, 7, 240–258. [Google Scholar] [CrossRef] [PubMed]
- Prior, R.; Van Helleputte, L.; Klingl, Y.E.; Van Den Bosch, L. HDAC6 as a potential therapeutic target for peripheral nerve disorders. Expert Opin. Targets 2018, 22, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Benoy, V.; Vanden Berghe, P.; Jarpe, M.; Van Damme, P.; Robberecht, W.; Van Den Bosch, L. Development of Improved HDAC6 Inhibitors as Pharmacological Therapy for Axonal Charcot-Marie-Tooth Disease. Neurotherapeutics 2017, 14, 417–428. [Google Scholar] [CrossRef]
- Mo, Z.; Zhao, X.; Liu, H.; Hu, Q.; Chen, X.Q.; Pham, J.; Wei, N.; Liu, Z.; Zhou, J.; Burgess, R.W.; et al. Aberrant GlyRS-HDAC6 interaction linked to axonal transport deficits in Charcot-Marie-Tooth neuropathy. Nat. Commun. 2018, 9, 1007. [Google Scholar] [CrossRef] [PubMed]
- Gold, W.A.; Lacina, T.A.; Cantrill, L.C.; Christodoulou, J. MeCP2 deficiency is associated with reduced levels of tubulin acetylation and can be restored using HDAC6 inhibitors. J. Mol. Med. 2015, 93, 63–72. [Google Scholar] [CrossRef]
- Richter-Landsberg, C. Protein aggregate formation in oligodendrocytes: Tau and the cytoskeleton at the intersection of neuroprotection and neurodegeneration. Biol. Chem. 2016, 397, 185–194. [Google Scholar] [CrossRef]
- Richter-Landsberg, C.; Leyk, J. Inclusion body formation, macroautophagy, and the role of HDAC6 in neurodegeneration. Acta Neuropathol. 2013, 126, 793–807. [Google Scholar] [CrossRef]
- Noack, M.; Leyk, J.; Richter-Landsberg, C. HDAC6 inhibition results in tau acetylation and modulates tau phosphorylation and degradation in oligodendrocytes. Glia 2014, 62, 535–547. [Google Scholar] [CrossRef]
- Leyk, J.; Goldbaum, O.; Noack, M.; Richter-Landsberg, C. Inhibition of HDAC6 modifies tau inclusion body formation and impairs autophagic clearance. J. Mol. Neurosci. 2015, 55, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Leyk, J.; Daly, C.; Janssen-Bienhold, U.; Kennedy, B.N.; Richter-Landsberg, C. HDAC6 inhibition by tubastatin A is protective against oxidative stress in a photoreceptor cell line and restores visual function in a zebrafish model of inherited blindness. Cell Death Dis. 2017, 8, e3028. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Evans, S.M. Introduction to special issue on animal models of neuropsychiatric disorders and substance use disorders: Progress and gaps. Exp. Clin. Psychopharmacol. 2017, 25, 61–63. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, T.; Zhou, Y.; Feng, G. Animal models for neuropsychiatric disorders: Prospects for circuit intervention. Curr. Opin. Neurobiol. 2017, 45, 59–65. [Google Scholar] [CrossRef]
- Pound, P.; Ebrahim, S.; Sandercock, P.; Bracken, M.B.; Roberts, I. Where is the evidence that animal research benefits humans? BMJ 2004, 328, 514–517. [Google Scholar] [CrossRef]
- Hackam, D.G.; Redelmeier, D.A. Translation of research evidence from animals to humans. JAMA 2006, 296, 1731–1732. [Google Scholar] [CrossRef] [PubMed]
- Medicine, I.O. Improving the Utility and Translation of Animal Models for Nervous System Disorders: Workshop Summary; The National Academies Press: Washington, DC, USA, 2013. [Google Scholar]
- Akhtar, A. The flaws and human harms of animal experimentation. Camb. Q. Healthc. Ethics 2015, 24, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Lal, S.; Li, A.; Dos Remedios, C. Limitations in Translating Animal Studies to Humans in Cardiovascular Disease. J. Cardiovasc. Transl. Res. 2016, 9, 165–166. [Google Scholar] [CrossRef]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Pak, C.; Danko, T.; Zhang, Y.; Aoto, J.; Anderson, G.; Maxeiner, S.; Yi, F.; Wernig, M.; Südhof, T.C. Human Neuropsychiatric Disease Modeling using Conditional Deletion Reveals Synaptic Transmission Defects Caused By Heterozygous Mutations in NRXN1. Cell Stem Cell 2015, 17, 316–328. [Google Scholar] [CrossRef] [PubMed]
- van der Worp, H.B.; Howells, D.W.; Sena, E.S.; Porritt, M.J.; Rewell, S.; O’Collins, V.; Macleod, M.R. Can animal models of disease reliably inform human studies? PLoS Med. 2010, 7, e1000245. [Google Scholar] [CrossRef] [PubMed]
- Rice, J. Animal models: Not close enough. Nature 2012, 484, S9. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Berkovitch, S.S.; Iaconelli, J.; Watmuff, B.; Park, H.; Chattopadhyay, S.; McPhie, D.; Öngür, D.; Cohen, B.M.; Clish, C.B.; et al. Perturbational Profiling of Metabolites in Patient Fibroblasts Implicates α-Aminoadipate as a Potential Biomarker for Bipolar Disorder. Mol. Neuropsychiatry 2016, 2, 97–106. [Google Scholar] [CrossRef]
- Huang, J.H.; Park, H.; Iaconelli, J.; Berkovitch, S.S.; Watmuff, B.; McPhie, D.; Öngür, D.; Cohen, B.M.; Clish, C.B.; Karmacharya, R. Unbiased Metabolite Profiling of Schizophrenia Fibroblasts under Stressful Perturbations Reveals Dysregulation of Plasmalogens and Phosphatidylcholines. J. Proteome Res. 2017, 16, 481–493. [Google Scholar] [CrossRef]
- Wimalasena, N.K.; Le, V.Q.; Wimalasena, K.; Schreiber, S.L.; Karmacharya, R. Gene Expression-Based Screen for Parkinson’s Disease Identifies GW8510 as a Neuroprotective Agent. ACS Chem. Neurosci. 2016, 7, 857–863. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Shi, Y.; Inoue, H.; Wu, J.C.; Yamanaka, S. Induced pluripotent stem cell technology: A decade of progress. Nat. Rev. Drug Discov. 2017, 16, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Karmacharya, R.; Haggarty, S.J. Stem cell models of neuropsychiatric disorders. Mol. Cell. Neurosci. 2016, 73, 1–2. [Google Scholar] [CrossRef]
- Watmuff, B.; Liu, B.; Karmacharya, R. Stem cell-derived neurons in the development of targeted treatment for schizophrenia and bipolar disorder. Pharmacogenomics 2017, 18, 471–479. [Google Scholar] [CrossRef]
- Watmuff, B.; Berkovitch, S.S.; Huang, J.H.; Iaconelli, J.; Toffel, S.; Karmacharya, R. Disease signatures for schizophrenia and bipolar disorder using patient-derived induced pluripotent stem cells. Mol. Cell. Neurosci. 2016, 73, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Berkovitch, S.S.; Iaconelli, J.; Karmacharya, R. Patient-Derived iPSCs as a Model for Schizophrenia. J. Stem Cell Res. Regener. Med. 2015, 2, e001. [Google Scholar]
- Kirwan, P.; Turner-Bridger, B.; Peter, M.; Momoh, A.; Arambepola, D.; Robinson, H.P.; Livesey, F.J. Development and function of human cerebral cortex neural networks from pluripotent stem cells in vitro. Development 2015, 142, 3178–3187. [Google Scholar] [CrossRef]
- Kim, J.Y.; Woo, S.Y.; Hong, Y.B.; Choi, H.; Kim, J.; Mook-Jung, I.; Ha, N.; Kyung, J.; Koo, S.K.; Jung, S.C.; et al. HDAC6 Inhibitors Rescued the Defective Axonal Mitochondrial Movement in Motor Neurons Derived from the Induced Pluripotent Stem Cells of Peripheral Neuropathy Patients with. Stem Cells Int. 2016, 2016, 9475981. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Landucci, E.; Brindisi, M.; Bianciardi, L.; Catania, L.M.; Daga, S.; Croci, S.; Frullanti, E.; Fallerini, C.; Butini, S.; Brogi, S.; et al. iPSC-derived neurons profiling reveals GABAergic circuit disruption and acetylated α-tubulin defect which improves after iHDAC6 treatment in Rett syndrome. Exp. Cell Res. 2018, 368, 225–235. [Google Scholar] [CrossRef]
- Misztak, P.; Pańczyszyn-Trzewik, P.; Sowa-Kućma, M. Histone deacetylases (HDACs) as therapeutic target for depressive disorders. Pharm. Rep. 2018, 70, 398–408. [Google Scholar] [CrossRef]
- Machado-Vieira, R.; Ibrahim, L.; Zarate, C.A. Histone deacetylases and mood disorders: Epigenetic programming in gene-environment interactions. CNS Neurosci. 2011, 17, 699–704. [Google Scholar] [CrossRef]
- Erburu, M.; Muñoz-Cobo, I.; Domínguez-Andrés, J.; Beltran, E.; Suzuki, T.; Mai, A.; Valente, S.; Puerta, E.; Tordera, R.M. Chronic stress and antidepressant induced changes in Hdac5 and Sirt2 affect synaptic plasticity. Eur. Neuropsychopharmacol. 2015, 25, 2036–2048. [Google Scholar] [CrossRef]
- Phiel, C.J.; Zhang, F.; Huang, E.Y.; Guenther, M.G.; Lazar, M.A.; Klein, P.S. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J. Biol. Chem. 2001, 276, 36734–36741. [Google Scholar] [CrossRef]
- Covington, H.E.; Maze, I.; Vialou, V.; Nestler, E.J. Antidepressant action of HDAC inhibition in the prefrontal cortex. Neuroscience 2015, 298, 329–335. [Google Scholar] [CrossRef]
- Schroeder, F.A.; Lewis, M.C.; Fass, D.M.; Wagner, F.F.; Zhang, Y.L.; Hennig, K.M.; Gale, J.; Zhao, W.N.; Reis, S.; Barker, D.D.; et al. A selective HDAC 1/2 inhibitor modulates chromatin and gene expression in brain and alters mouse behavior in two mood-related tests. PLoS ONE 2013, 8, e71323. [Google Scholar] [CrossRef]
- Meylan, E.M.; Halfon, O.; Magistretti, P.J.; Cardinaux, J.R. The HDAC inhibitor SAHA improves depressive-like behavior of CRTC1-deficient mice: Possible relevance for treatment-resistant depression. Neuropharmacology 2016, 107, 111–121. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Hong, G.; Pang, M.; Xu, H.; Li, H.; Tian, F.; Fang, R.; Yao, Y.; Liu, J. Antidepressant-like effects of sodium butyrate and its possible mechanisms of action in mice exposed to chronic unpredictable mild stress. Neurosci. Lett. 2016, 618, 159–166. [Google Scholar] [CrossRef]
- Golden, S.A.; Christoffel, D.J.; Heshmati, M.; Hodes, G.E.; Magida, J.; Davis, K.; Cahill, M.E.; Dias, C.; Ribeiro, E.; Ables, J.L.; et al. Epigenetic regulation of RAC1 induces synaptic remodeling in stress disorders and depression. Nat. Med. 2013, 19, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Fukada, M.; Hanai, A.; Nakayama, A.; Suzuki, T.; Miyata, N.; Rodriguiz, R.M.; Wetsel, W.C.; Yao, T.P.; Kawaguchi, Y. Loss of deacetylation activity of Hdac6 affects emotional behavior in mice. PLoS ONE 2012, 7, e30924. [Google Scholar] [CrossRef] [PubMed]
- Maguschak, K.A.; Ressler, K.J. The dynamic role of beta-catenin in synaptic plasticity. Neuropharmacology 2012, 62, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Gould, T.D.; Einat, H.; O’Donnell, K.C.; Picchini, A.M.; Schloesser, R.J.; Manji, H.K. Beta-catenin overexpression in the mouse brain phenocopies lithium-sensitive behaviors. Neuropsychopharmacology 2007, 32, 2173–2183. [Google Scholar] [CrossRef]
- Gould, T.D.; O’Donnell, K.C.; Picchini, A.M.; Dow, E.R.; Chen, G.; Manji, H.K. Generation and behavioral characterization of beta-catenin forebrain-specific conditional knock-out mice. Behav. Brain Res. 2008, 189, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Valvezan, A.J.; Klein, P.S. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front. Mol. Neurosci. 2012, 5, 1. [Google Scholar] [CrossRef]
- Dias, C.; Feng, J.; Sun, H.; Shao, N.Y.; Mazei-Robison, M.S.; Damez-Werno, D.; Scobie, K.; Bagot, R.; LaBonte, B.; Ribeiro, E.; et al. beta-catenin mediates stress resilience through Dicer1/microRNA regulation. Nature 2014, 516, 51–55. [Google Scholar] [CrossRef]
- Mulligan, K.A.; Cheyette, B.N. Neurodevelopmental Perspectives on Wnt Signaling in Psychiatry. Mol. Neuropsychiatry 2017, 2, 219–246. [Google Scholar] [CrossRef]
- Li, X.; Jope, R.S. Is glycogen synthase kinase-3 a central modulator in mood regulation? Neuropsychopharmacology 2010, 35, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Karege, F.; Perroud, N.; Burkhardt, S.; Fernandez, R.; Ballmann, E.; La Harpe, R.; Malafosse, A. Protein levels of β-catenin and activation state of glycogen synthase kinase-3β in major depression. A study with postmortem prefrontal cortex. J. Affect. Disord. 2012, 136, 185–188. [Google Scholar] [CrossRef]
- Gould, T.D.; Dow, E.R.; O’Donnell, K.C.; Chen, G.; Manji, H.K. Targeting signal transduction pathways in the treatment of mood disorders: Recent insights into the relevance of the Wnt pathway. CNS Neurol. Disord. Drug Targets 2007, 6, 193–204. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.C.; Gould, T.D. The behavioral actions of lithium in rodent models: Leads to develop novel therapeutics. Neurosci. Biobehav. Rev. 2007, 31, 932–962. [Google Scholar] [CrossRef]
- Bersudsky, Y.; Shaldubina, A.; Belmaker, R.H. Lithium’s effect in forced-swim test is blood level dependent but not dependent on weight loss. Behav Pharm. 2007, 18, 77–80. [Google Scholar] [CrossRef]
- O’Brien, W.T.; Harper, A.D.; Jove, F.; Woodgett, J.R.; Maretto, S.; Piccolo, S.; Klein, P.S. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J. Neurosci. 2004, 24, 6791–6798. [Google Scholar] [CrossRef]
- Valenta, T.; Hausmann, G.; Basler, K. The many faces and functions of beta-catenin. EMBO J. 2012, 31, 2714–2736. [Google Scholar] [CrossRef] [PubMed]
- Bielen, H.; Houart, C. The Wnt cries many: Wnt regulation of neurogenesis through tissue patterning, proliferation, and asymmetric cell division. Dev. Neurobiol. 2014, 74, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nusse, R.; Clevers, H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Iaconelli, J.; Huang, J.H.; Berkovitch, S.S.; Chattopadhyay, S.; Mazitschek, R.; Schreiber, S.L.; Haggarty, S.J.; Karmacharya, R. HDAC6 inhibitors modulate Lys49 acetylation and membrane localization of beta-catenin in human iPSC-derived neuronal cells. ACS Chem. Boil. 2015, 10, 883–890. [Google Scholar] [CrossRef]
- Wolf, D.; Rodova, M.; Miska, E.A.; Calvet, J.P.; Kouzarides, T. Acetylation of beta-catenin by CREB-binding protein (CBP). J. Boil. Chem. 2002, 277, 25562–25567. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999, 59, 1811–1815. [Google Scholar] [PubMed]
- Li, Y.; Zhang, X.; Polakiewicz, R.D.; Yao, T.P.; Comb, M.J. HDAC6 is required for epidermal growth factor-induced beta-catenin nuclear localization. J. Boil. Chem. 2008, 283, 12686–12690. [Google Scholar] [CrossRef]
- Jiang, S.; Zhang, M.; Sun, J.; Yang, X. Casein kinase 1α: Biological mechanisms and theranostic potential. Cell Commun. Signal. 2018, 16, 23. [Google Scholar] [CrossRef] [PubMed]
- Amit, S.; Hatzubai, A.; Birman, Y.; Andersen, J.S.; Ben-Shushan, E.; Mann, M.; Ben-Neriah, Y.; Alkalay, I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: A molecular switch for the Wnt pathway. Genes Dev. 2002, 16, 1066–1076. [Google Scholar] [CrossRef]
- Maher, M.T.; Mo, R.; Flozak, A.S.; Peled, O.N.; Gottardi, C.J. Beta-catenin phosphorylated at serine 45 is spatially uncoupled from beta-catenin phosphorylated in the GSK3 domain: Implications for signaling. PLoS ONE 2010, 5, e10184. [Google Scholar] [CrossRef]
- Winer, I.S.; Bommer, G.T.; Gonik, N.; Fearon, E.R. Lysine residues Lys-19 and Lys-49 of beta-catenin regulate its levels and function in T cell factor transcriptional activation and neoplastic transformation. J. Boil. Chem. 2006, 281, 26181–26187. [Google Scholar] [CrossRef] [PubMed]
- Caron, C.; Boyault, C.; Khochbin, S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. BioEssays 2005, 27, 408–415. [Google Scholar] [CrossRef]
- Nihira, N.T.; Ogura, K.; Shimizu, K.; North, B.J.; Zhang, J.; Gao, D.; Inuzuka, H.; Wei, W. Acetylation-dependent regulation of MDM2 E3 ligase activity dictates its oncogenic function. Sci. Signal. 2017, 10, eaai8026. [Google Scholar] [CrossRef]
- Lalonde, J.; Reis, S.A.; Sivakumaran, S.; Holland, C.S.; Wesseling, H.; Sauld, J.F.; Alural, B.; Zhao, W.N.; Steen, J.A.; Haggarty, S.J. Chemogenomic analysis reveals key role for lysine acetylation in regulating Arc stability. Nat. Commun. 2017, 8, 1659. [Google Scholar] [CrossRef]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef]
- Ge, X.; Jin, Q.; Zhang, F.; Yan, T.; Zhai, Q. PCAF acetylates {beta}-catenin and improves its stability. Mol. Biol. Cell. 2009, 20, 419–427. [Google Scholar] [CrossRef]
- Seong, E.; Yuan, L.; Arikkath, J. Cadherins and catenins in dendrite and synapse morphogenesis. Cell Adhes. Migr. 2015, 9, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Ochs, S.M.; Dorostkar, M.M.; Aramuni, G.; Schön, C.; Filser, S.; Pöschl, J.; Kremer, A.; Van Leuven, F.; Ovsepian, S.V.; Herms, J. Loss of neuronal GSK3β reduces dendritic spine stability and attenuates excitatory synaptic transmission via β-catenin. Mol. Psychiatry 2015, 20, 482–489. [Google Scholar] [CrossRef]
- Bian, W.J.; Miao, W.Y.; He, S.J.; Qiu, Z.; Yu, X. Coordinated Spine Pruning and Maturation Mediated by Inter-Spine Competition for Cadherin/Catenin Complexes. Cell 2015, 162, 808–822. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.R.; He, S.; Marie, H.; Zeng, S.Y.; Ma, J.; Tan, Z.J.; Lee, S.Y.; Malenka, R.C.; Yu, X. Coordinated changes in dendritic arborization and synaptic strength during neural circuit development. Neuron 2009, 61, 71–84. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Amadei, C.; Downey, K.; Jope, R.S. Ketamine-induced inhibition of glycogen synthase kinase-3 contributes to the augmentation of α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) receptor signaling. Bipolar Disord. 2016, 18, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.J.; Fuchikami, M.; Dwyer, J.M.; Lepack, A.E.; Duman, R.S.; Aghajanian, G.K. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology 2013, 38, 2268–2277. [Google Scholar] [CrossRef]
- Pilar-Cuellar, F.; Vidal, R.; Pazos, A. Subchronic treatment with fluoxetine and ketanserin increases hippocampal brain-derived neurotrophic factor, beta-catenin and antidepressant-like effects. Br. J. Pharmacol. 2012, 165, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Madsen, T.M.; Newton, S.S.; Eaton, M.E.; Russell, D.S.; Duman, R.S. Chronic electroconvulsive seizure up-regulates β-catenin expression in rat hippocampus: Role in adult neurogenesis. Biol. Psychiatry 2003, 54, 1006–1014. [Google Scholar] [CrossRef]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef]
- Toto, A.; Pedersen, S.W.; Karlsson, O.A.; Moran, G.E.; Andersson, E.; Chi, C.N.; Strømgaard, K.; Gianni, S.; Jemth, P. Ligand binding to the PDZ domains of postsynaptic density protein 95. Protein Eng. Des. Sel. 2016, 29, 169–175. [Google Scholar] [CrossRef]
- Manjunath, G.P.; Ramanujam, P.L.; Galande, S. Structure function relations in PDZ-domain-containing proteins: Implications for protein networks in cellular signalling. J. Biosci. 2018, 43, 155–171. [Google Scholar] [CrossRef] [PubMed]
- Gujral, T.S.; Karp, E.S.; Chan, M.; Chang, B.H.; MacBeath, G. Family-wide investigation of PDZ domain-mediated protein-protein interactions implicates beta-catenin in maintaining the integrity of tight junctions. Chem. Biol. 2013, 20, 816–827. [Google Scholar] [CrossRef]
- Iaconelli, J.; Lalonde, J.; Watmuff, B.; Liu, B.; Mazitschek, R.; Haggarty, S.J.; Karmacharya, R. Lysine Deacetylation by HDAC6 Regulates the Kinase Activity of AKT in Human Neural Progenitor Cells. ACS Chem. Boil. 2017, 12, 2139–2148. [Google Scholar] [CrossRef] [PubMed]
- Jochems, J.; Teegarden, S.L.; Chen, Y.; Boulden, J.; Challis, C.; Ben-Dor, G.A.; Kim, S.F.; Berton, O. Enhancement of stress resilience through histone deacetylase 6-mediated regulation of glucocorticoid receptor chaperone dynamics. Biol. Psychiatry 2015, 77, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Karmacharya, R.; Sliwoski, G.R.; Lundy, M.Y.; Suckow, R.F.; Cohen, B.M.; Buttner, E.A. Clozapine interaction with phosphatidyl inositol 3-kinase (PI3K)/insulin-signaling pathway in Caenorhabditis elegans. Neuropsychopharmacology 2009, 34, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Del’Guidice, T.; Beaulieu, J.M. Selective disruption of dopamine D2-receptors/beta-arrestin2 signaling by mood stabilizers. J. Recept. Signal Transduct. Res. 2015, 35, 224–232. [Google Scholar] [CrossRef]
- Gao, Y.; Peterson, S.; Masri, B.; Hougland, M.T.; Adham, N.; Gyertyán, I.; Kiss, B.; Caron, M.G.; El-Mallakh, R.S. Cariprazine exerts antimanic properties and interferes with dopamine D2 receptor β-arrestin interactions. Pharmacol. Res. Perspect. 2015, 3, e00073. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, D.; Nebhan, C.A.; Hu, L.; Anderson, B.; Webb, D.J. An APPL1/Akt signaling complex regulates dendritic spine and synapse formation in hippocampal neurons. Mol. Cell. Neurosci. 2011, 46, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Pillai, V.B.; Sundaresan, N.R.; Gupta, M.P. Regulation of Akt signaling by sirtuins: Its implication in cardiac hypertrophy and aging. Circ. Res. 2014, 114, 368–378. [Google Scholar] [CrossRef]
- Ramakrishnan, G.; Davaakhuu, G.; Kaplun, L.; Chung, W.C.; Rana, A.; Atfi, A.; Miele, L.; Tzivion, G. Sirt2 deacetylase is a novel AKT binding partner critical for AKT activation by insulin. J. Boil. Chem. 2014, 289, 6054–6066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shemezis, J.R.; McQuinn, E.R.; Wang, J.; Sverdlov, M.; Chenn, A. AKT activation by N-cadherin regulates beta-catenin signaling and neuronal differentiation during cortical development. Neural Dev. 2013, 8, 7. [Google Scholar] [CrossRef]
- Jansen, L.A.; Mirzaa, G.M.; Ishak, G.E.; O’Roak, B.J.; Hiatt, J.B.; Roden, W.H.; Gunter, S.A.; Christian, S.L.; Collins, S.; Adams, C.; et al. PI3K/AKT pathway mutations cause a spectrum of brain malformations from megalencephaly to focal cortical dysplasia. Brain 2015, 138, 1613–1628. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.; Yuan, J.; Huang, L.; Xiang, X.; Zhu, H.; Chen, F.; Chen, Y.; Lin, J.; Feng, H. Valproic Acid Arrests Proliferation but Promotes Neuronal Differentiation of Adult Spinal NSPCs from SCI Rats. Neurochem. Res. 2015, 40, 1472–1486. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC7 | HDAC8 | HDAC9 | Selectivity | Reference | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| SAHA | 0.0013 | 0.0016 | 0.005 | _ | 3.6 | 0.0016 | _ | 0.48 | _ | 1,2,3,6,8 | [11] |
| Crebinostat | 0.0007 | 0.001 | 0.002 | _ | _ | 0.009 | _ | _ | _ | 1,2,3,6 | [12] |
| CI-994 | 0.05 | 0.19 | 0.55 | _ | _ | _ | _ | _ | _ | 1,2,3 | [13] |
| Cpd-60 | 0.001 | 0.008 | 0.458 | _ | _ | _ | _ | _ | _ | 1,2 | [14] |
| BG-45 | 2 | 2.2 | 0.289 | _ | _ | >20 | _ | _ | _ | 3 | [15] |
| Tubacin | 0.028 | 0.042 | 0.275 | 17 | 1.5 | 0.016 | 8.5 | 0.17 | _ | 6 | [16] |
| Tubastatin A | 3.2 | 3.5 | 4.9 | _ | _ | 0.018 | _ | _ | _ | 6 | [17] |
| ACY-738 | 0.094 | 0.128 | 0.218 | _ | _ | 0.0017 | _ | _ | _ | 6 | [18] |
| ACY-775 | 2.123 | 2.57 | 1.12 | _ | _ | 0.0075 | _ | _ | _ | 6 | [18] |
| ACY-1215 | 0.058 | 0.048 | 0.051 | 7 | 5 | 0.004 | 1.4 | 0.1 | 10 | 6 | [19] |
| Bavarostat | >1000 | >1000 | >1000 | 11.3 | 19 | 0.06 | 4.7 | 8.5 | 5.2 | 6 | [20] |
| PCI-34051 | >50 | >50 | 6.8 | _ | _ | 2.9 | _ | 0.01 | >50 | 8 | [21] |
| Study Model | HDAC6 Modulation | Effect of HDAC6 Inhibition | Citation |
|---|---|---|---|
| Mouse cortical neurons | Genetic and pharmacological (Trichostatin A) | Increased survival and regeneration in setting of oxidative stress. | Rivieccio et al. (2009) [24] |
| Mouse cortical neurons | Trichostatin A Scriptaid | Protection of kainic acid-induced axonal degeneration. | Hanson et al. (2018) [25] |
| SOD1G93A ALS mouse model | HDAC6 deletion | Buildup of SOD1G93A aggregates but only mild effects on motor function. | Lee et al. (2015) [33] |
| SOD1G93A ALS mouse model | HDAC6 deletion | Decrease in disease progression and prolonged survival. | Taes et al. (2013) [34] |
| APP/PS1 mouse model | HDAC6 deletion | Improvement in memory function | Govindarajan et al. (2013) [38] |
| rTg4510 mouse model | Tubastatin A | Improvement in memory function and lower tau levels | Selenica et al. (2014) [39] |
| AD mouse model | Tubastatin A ACY-1215 | Improvement in behavior and decrease in amyloid β and hyperphosphorylated tau. | Zhang et al. (2014) [40] |
| AD mouse model | MPT0G211 | Improvement in learning and memory and decrease in tau phosphorylation. | Fan et al. (2018) [42] |
| Charcot-Marie-Tooth HSPB1 mouse model | ACY-738 ACY-775 | Rescue of axonal transport deficits | Benoy et al. (2017) [43] |
| Charcot-Marie-Tooth GARS mouse model | Tubastatin A | Improved deficits in axonal transport & motor functioning | Shen et al. (2016) [44] |
| Cortical neurons from MECP2T158A mouse model | Tubastatin A | Increased α-tubulin acetylation | Gold et al. (2015) [48] |
| Rett syndrome patient fibroblast | Tubastatin A | Ameliorated microtubule defects | Gold et al. (2015) [48] |
| Cultured rat oligodendrocytes | Tubastatin A shRNA | Reduced microtubule binding activity of tau. Reduced protein aggregation. | Noack et al. (2014) [51] Leyk et al. (2015) [52] |
| dyeucd6 zebrafish model | Tubastatin A | Rescued visual function and retinal morphology | Leyk et al. (2017) [53] |
| Motor neurons from iPSCs of CMT2F and dHMN2B patients | CHEMICAL X4 CHEMICAL X9 | Reversed axonal movement defects of mitochondria | Kim et al. (2015) [75] |
| Motor neurons from iPSCs of FUS-ALS patients | ACY-738 Tubastatin A Antisense oligos | Restore axonal transport defects and increase mitochondria-ER overlay | Guo et al. (2017) [76] |
| Neurons from iPSCs of Rett syndrome patients with MECP2 mutations | ACY-1215 | Reversal of decrease in α-tubulin acetylation | Landucci et al. (2018) [77] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iaconelli, J.; Xuan, L.; Karmacharya, R. HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem Cell-Derived Neurons. Int. J. Mol. Sci. 2019, 20, 1605. https://doi.org/10.3390/ijms20071605
Iaconelli J, Xuan L, Karmacharya R. HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem Cell-Derived Neurons. International Journal of Molecular Sciences. 2019; 20(7):1605. https://doi.org/10.3390/ijms20071605
Chicago/Turabian StyleIaconelli, Jonathan, Lucius Xuan, and Rakesh Karmacharya. 2019. "HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem Cell-Derived Neurons" International Journal of Molecular Sciences 20, no. 7: 1605. https://doi.org/10.3390/ijms20071605
APA StyleIaconelli, J., Xuan, L., & Karmacharya, R. (2019). HDAC6 Modulates Signaling Pathways Relevant to Synaptic Biology and Neuronal Differentiation in Human Stem Cell-Derived Neurons. International Journal of Molecular Sciences, 20(7), 1605. https://doi.org/10.3390/ijms20071605