Physical Activity Alleviates Cognitive Dysfunction of Alzheimer’s Disease through Regulating the mTOR Signaling Pathway



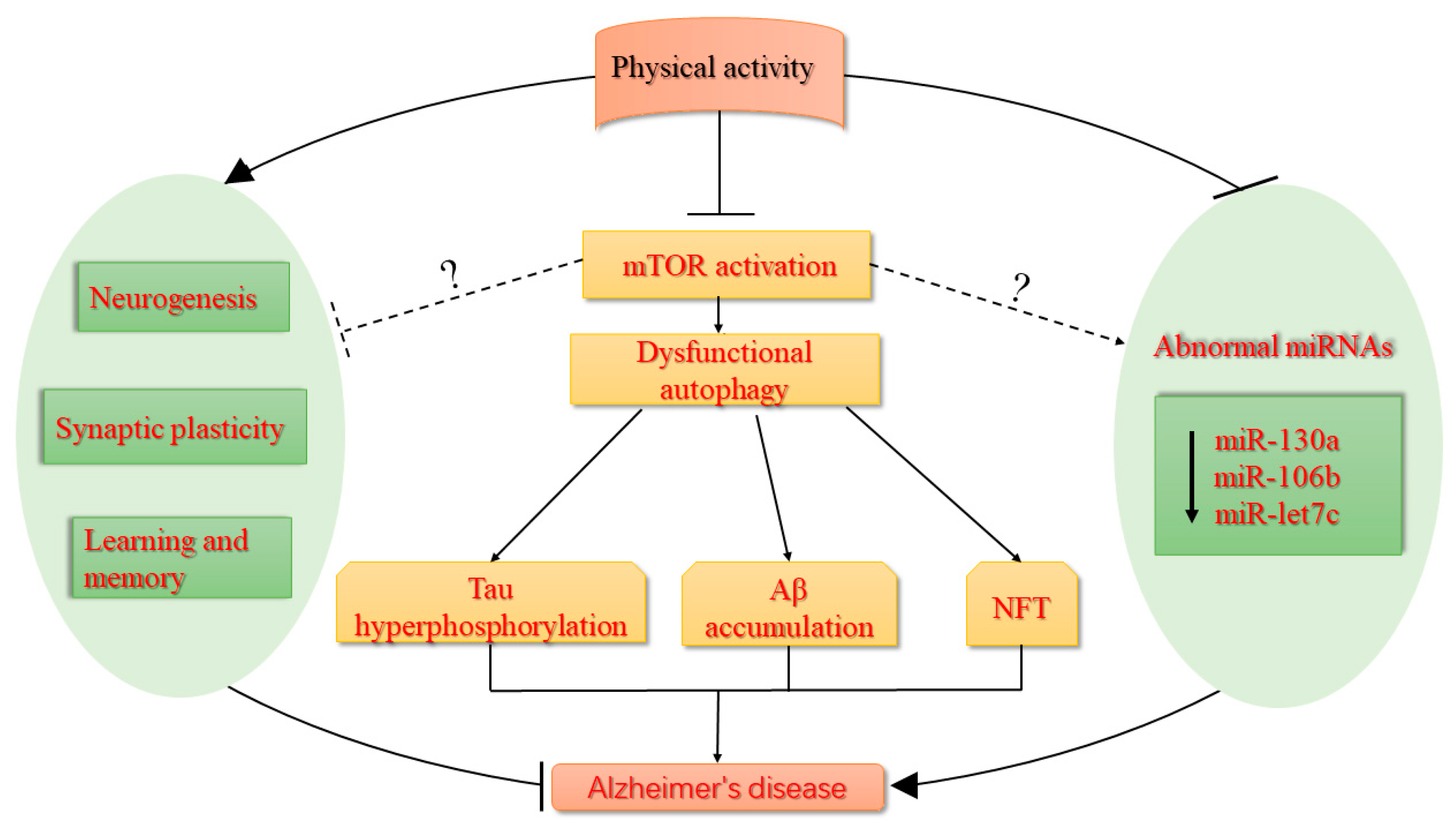{kind=link}
Abstract
:1. Introduction
2. The mTOR Signaling Pathway
2.1. The mTOR Signaling Pathway and Autophagy
2.2. Activated mTOR Signaling Triggers Aβ Generation and Induces the Failure of Aβ Clearance
2.3. mTOR Activation Induces Hyperphosphorylation of Tau Protein
3. The Alteration of miRNAs in AD and Aging-Related Diseases
4. The Role of Physical Activity in AD
4.1. Physical Activity is Beneficial for the Improvement of Learning and Memory Capacity
4.2. Physical Activity Increases Neurogenesis
4.3. Physical Activity Enhances Structural and Synaptic Plasticity in Hippocampus
4.4. Physical Activity Regulates Abnormal miRNAs
5. Clinical Studies of Physical Activity in AD
6. mTOR as a New Target for the Prevention and Treatment of AD During Physical Activity?
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hyman, B.T.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Carrillo, M.C.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012, 8, 1–13. [Google Scholar] [CrossRef]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef]
- Karran, E.; De Strooper, B. The amyloid cascade hypothesis: Are we poised for success or failure? J. Neurochem. 2016, 139, 237–252. [Google Scholar] [CrossRef]
- Krishnamurthy, P.K.; Johnson, G.V. Mutant (R406W) human tau is hyperphosphorylated and does not efficiently bind microtubules in a neuronal cortical cell model. J. Biol. Chem. 2004, 279, 7893–7900. [Google Scholar] [CrossRef]
- Leoni, V. The effect of apolipoprotein E (ApoE) genotype on biomarkers of amyloidogenesis, tau pathology and neurodegeneration in Alzheimer’s disease. Clin. Chem. Lab. Med. 2011, 49, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, M.T.; Bruno, M.A.; Ducatenzeiler, A.; Klein, W.L.; Cuello, A.C. Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer’s disease. Neurobiol. Aging 2012, 33, 1329–1342. [Google Scholar] [CrossRef]
- Chen, N.; Karantza-Wadsworth, V. Role and regulation of autophagy in cancer. Biochim. Biophys. Acta 2009, 1793, 1516–1523. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Bergamini, E.; Brunk, U.T.; Droge, W.; Ffrench, M.; Terman, A. Autophagy and aging: The importance of maintaining “clean” cells. Autophagy 2005, 1, 131–140. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K. Regulation of the aging process by autophagy. Trends Mol. Med. 2009, 15, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Marino, G.; Kroemer, G. Autophagy and aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, X.; Le, W. Autophagy dysfunction in Alzheimer’s disease. Neurodegener. Dis. 2010, 7, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Cuervo, A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010, 13, 805–811. [Google Scholar] [CrossRef]
- Kumar, A.; Dhawan, A.; Kadam, A.; Shinde, A. Autophagy and Mitochondria: Targets in Neurodegenerative Disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Ryskalin, L.; Fornai, F. Interdependency Between Autophagy and Synaptic Vesicle Trafficking: Implications for Dopamine Release. Front. Mol. Neurosci. 2018, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Gambardella, S.; Frati, A.; Fornai, F. Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia. Int. J. Mol. Sci. 2018, 19, 2765. [Google Scholar] [CrossRef]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A.; Ojala, J.; Haapasalo, A.; Soininen, H.; Hiltunen, M. Impaired autophagy and APP processing in Alzheimer’s disease: The potential role of Beclin 1 interactome. Prog. Neurobiol. 2013, 106–107, 33–54. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The role of autophagy-lysosome pathway in neurodegeneration associated with Parkinson’s disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef]
- Kou, X.; Chen, N. Resveratrol as a natural autophagy regulator for prevention and treatment of Alzheimer’s disease. Nutrients 2017, 9, 927. [Google Scholar]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy--a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Hung, C.M.; Garcia-Haro, L.; Sparks, C.A.; Guertin, D.A. mTOR-dependent cell survival mechanisms. Cold Spring Harb. Perspect. Biol. 2012, 4, a008771. [Google Scholar] [CrossRef]
- Parsons, R.G.; Gafford, G.M.; Helmstetter, F.J. Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J. Neurosci. 2006, 26, 12977–12983. [Google Scholar] [CrossRef]
- Gouras, G.K. mTOR: At the crossroads of aging, chaperones, and Alzheimer’s disease. J. Neurochem. 2013, 124, 747–748. [Google Scholar] [CrossRef] [PubMed]
- O’neill, C. PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp. Gerontol. 2013, 48, 647–653. [Google Scholar] [CrossRef]
- Gharibi, B.; Farzadi, S.; Ghuman, M.; Hughes, F.J. Inhibition of Akt/mTOR attenuates age-related changes in mesenchymal stem cells. Stem Cells 2014, 32, 2256–2266. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chu, X.; Yin, M.; Liu, X.; Yuan, H.; Niu, Y.; Fu, L. mTOR and autophagy in normal brain aging and caloric restriction ameliorating age-related cognition deficits. Behav. Brain Res. 2014, 264, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Paccalin, M.; Pain-Barc, S.; Pluchon, C.; Paul, C.; Besson, M.N.; Carret-Rebillat, A.S.; Rioux-Bilan, A.; Gil, R.; Hugon, J. Activated mTOR and PKR kinases in lymphocytes correlate with memory and cognitive decline in Alzheimer’s disease. Dement. Geriatr. Cogn. Disord. 2006, 22, 320–326. [Google Scholar] [CrossRef]
- Cai, Z.; Zhao, B.; Li, K.; Zhang, L.; Li, C.; Quazi, S.H.; Tan, Y. Mammalian target of rapamycin: A valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J. Neurosci. Res. 2012, 90, 1105–1118. [Google Scholar] [CrossRef]
- Pozueta, J.; Lefort, R.; Shelanski, M.L. Synaptic changes in Alzheimer’s disease and its models. Neuroscience 2013, 251, 51–65. [Google Scholar] [CrossRef]
- Lafay-Chebassier, C.; Paccalin, M.; Page, G.; Barc-Pain, S.; Perault-Pochat, M.C.; Gil, R.; Pradier, L.; Hugon, J. mTOR/p70S6k signalling alteration by Abeta exposure as well as in APP-PS1 transgenic models and in patients with Alzheimer’s disease. J. Neurochem. 2005, 94, 215–225. [Google Scholar] [CrossRef]
- Kaliman, P.; Parrizas, M.; Lalanza, J.F.; Camins, A.; Escorihuela, R.M.; Pallas, M. Neurophysiological and epigenetic effects of physical exercise on the aging process. Ageing Res. Rev. 2011, 10, 475–486. [Google Scholar] [CrossRef]
- Marques-Aleixo, I.; Santos-Alves, E.; Balca, M.M.; Moreira, P.I.; Oliveira, P.J.; Magalhaes, J.; Ascensao, A. Physical exercise mitigates doxorubicin-induced brain cortex and cerebellum mitochondrial alterations and cellular quality control signaling. Mitochondrion 2016, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Rees, P.S.; Davidson, S.M.; Harding, S.E.; McGregor, C.; Elliot, P.M.; Yellon, D.M.; Hausenloy, D.J. The mitochondrial permeability transition pore as a target for cardioprotection in hypertrophic cardiomyopathy. Cardiovasc. Drugs Ther. 2013, 27, 235–237. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Ihasz, F.; Koltai, E.; Goto, S.; Taylor, A.W.; Boldogh, I. The redox-associated adaptive response of brain to physical exercise. Free Radic. Res. 2014, 48, 84–92. [Google Scholar] [CrossRef] [PubMed]
- van Praag, H.; Fleshner, M.; Schwartz, M.W.; Mattson, M.P. Exercise, energy intake, glucose homeostasis, and the brain. J. Neurosci. 2014, 34, 15139–15149. [Google Scholar] [CrossRef] [PubMed]
- Kou, X.; Li, J.; Liu, X.; Chang, J.; Zhao, Q.; Jia, S.; Fan, J.; Chen, N. Swimming attenuates d-galactose-induced brain aging via suppressing miR-34a-mediated autophagy impairment and abnormal mitochondrial dynamics. J. Appl. Physiol. 2017, 122, 1462–1469. [Google Scholar] [CrossRef]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef]
- Connolly, E.; Braunstein, S.; Formenti, S.; Schneider, R.J. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells. Mol. Cell. Biol. 2006, 26, 3955–3965. [Google Scholar] [CrossRef] [PubMed]
- Meske, V.; Albert, F.; Ohm, T.G. Coupling of mammalian target of rapamycin with phosphoinositide 3-kinase signaling pathway regulates protein phosphatase 2A- and glycogen synthase kinase-3 -dependent phosphorylation of tau. J. Biol. Chem. 2008, 283, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, A.; Maldonado, M.A.; Majumder, S.; Medina, D.X.; Holbein, W.; Magri, A.; Oddo, S. Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J. Biol. Chem. 2011, 286, 8924–8932. [Google Scholar] [CrossRef]
- Mizushima, N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010, 22, 132–139. [Google Scholar] [CrossRef]
- Shirooie, S.; Nabavi, S.F.; Dehpour, A.R.; Belwal, T.; Habtemariam, S.; Arguelles, S.; Sureda, A.; Daglia, M.; Tomczyk, M.; Sobarzo-Sanchez, E.; et al. Targeting mTORs by omega-3 fatty acids: A possible novel therapeutic strategy for neurodegeneration? Pharmacol. Res. 2018, 135, 37–48. [Google Scholar] [CrossRef]
- Caccamo, A.; Magri, A.; Medina, D.X.; Wisely, E.V.; Lopez-Aranda, M.F.; Silva, A.J.; Oddo, S. mTOR regulates tau phosphorylation and degradation: Implications for Alzheimer’s disease and other tauopathies. Aging Cell 2013, 12, 370–380. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Cammalleri, M.; Lutjens, R.; Berton, F.; King, A.R.; Simpson, C.; Francesconi, W.; Sanna, P.P. Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc. Natl. Acad. Sci. USA 2003, 100, 14368–14373. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. mTOR: On target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013, 19, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Zemke, D.; Azhar, S.; Majid, A. The mTOR pathway as a potential target for the development of therapies against neurological disease. Drug News Perspect. 2007, 20, 495–499. [Google Scholar] [CrossRef]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.; Mohan, P.; Coffey, E.E.; Kompella, U.; Mitchell, C.H.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.S.; Stavrides, P.; Mohan, P.S.; Kaushik, S.; Kumar, A.; Ohno, M.; Schmidt, S.D.; Wesson, D.W.; Bandyopadhyay, U.; Jiang, Y.; et al. Therapeutic effects of remediating autophagy failure in a mouse model of Alzheimer disease by enhancing lysosomal proteolysis. Autophagy 2011, 7, 788–789. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhang, S.; Zhang, X.; Li, T.; Tang, Y.; Liu, H.; Yang, W.; Le, W. Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Bordi, M.; Berg, M.J.; Mohan, P.S.; Peterhoff, C.M.; Alldred, M.J.; Che, S.; Ginsberg, S.D.; Nixon, R.A. Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 2016, 12, 2467–2483. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and tau: Effects on cognitive impairments. J. Biol. Chem. 2010, 285, 13107–13120. [Google Scholar] [CrossRef] [PubMed]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Pierce, A.; Podlutskaya, N.; Halloran, J.J.; Hussong, S.A.; Lin, P.Y.; Burbank, R.; Hart, M.J.; Galvan, V. Over-expression of heat shock factor 1 phenocopies the effect of chronic inhibition of TOR by rapamycin and is sufficient to ameliorate Alzheimer’s-like deficits in mice modeling the disease. J. Neurochem. 2013, 124, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.J.; Wang, D.C.; Chen, S.S. Amyloid-beta interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J. Neurosci. Res. 2009, 87, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Song, H.; Byun, J.; Park, K.S.; Jang, H.C.; Park, Y.J.; Mook-Jung, I. Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes 2012, 61, 3126–3138. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Limanaqi, F.; Frati, A.; Busceti, C.L.; Fornai, F. mTOR-Related Brain Dysfunctions in Neuropsychiatric Disorders. Int. J. Mol. Sci. 2018, 19, 2226. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Monteggia, L.M. mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 2013, 16, 1537–1543. [Google Scholar] [CrossRef]
- Fan, Q.W.; Weiss, W.A. Inhibition of PI3K-Akt-mTOR signaling in glioblastoma by mTORC1/2 inhibitors. Methods Mol. Biol. 2012, 821, 349–359. [Google Scholar] [PubMed]
- Ryskalin, L.; Lazzeri, G.; Flaibani, M.; Biagioni, F.; Gambardella, S.; Frati, A.; Fornai, F. mTOR-Dependent Cell Proliferation in the Brain. BioMed Res. Int. 2017, 2017, 7082696. [Google Scholar] [CrossRef] [PubMed]
- Wong, M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed. J. 2013, 36, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Majumder, S.; Richardson, A.; Strong, R.; Oddo, S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS ONE 2011, 6, e25416. [Google Scholar] [CrossRef] [PubMed]
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937. [Google Scholar] [CrossRef]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. tau is essential to beta -amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Gotz, J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Rodrigues, S.; Sampaio-Marques, B.; Gomes, P.; Neves-Carvalho, A.; Dioli, C.; Soares-Cunha, C.; Mazuik, B.F.; Takashima, A.; Ludovico, P.; et al. Dysregulation of autophagy and stress granule-related proteins in stress-driven tau pathology. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Griffin, R.J.; Moloney, A.; Kelliher, M.; Johnston, J.A.; Ravid, R.; Dockery, P.; O’Connor, R.; O’Neill, C. Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J. Neurochem. 2005, 93, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.J.; Bjorkdahl, C.; Zhang, H.; Zhou, X.; Winblad, B. p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimers Dis. 2008, 14, 385–392. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Selman, C.; Tullet, J.M.; Wieser, D.; Irvine, E.; Lingard, S.J.; Choudhury, A.I.; Claret, M.; Al-Qassab, H.; Carmignac, D.; Ramadani, F.; et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 2009, 326, 140–144. [Google Scholar] [CrossRef]
- Khurana, V.; Lu, Y.; Steinhilb, M.L.; Oldham, S.; Shulman, J.M.; Feany, M.B. TOR-mediated cell-cycle activation causes neurodegeneration in a Drosophila tauopathy model. Curr. Biol. 2006, 16, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Chen, G.; He, W.; Xiao, M.; Yan, L.J. Activation of mTOR: A culprit of Alzheimer’s disease? Neuropsychiatr. Dis. Treat. 2015, 11, 1015–1030. [Google Scholar] [CrossRef]
- Spilman, P.; Podlutskaya, N.; Hart, M.J.; Debnath, J.; Gorostiza, O.; Bredesen, D.; Richardson, A.; Strong, R.; Galvan, V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS ONE 2010, 5, e9979. [Google Scholar] [CrossRef]
- Delay, C.; Mandemakers, W.; Hebert, S.S. MicroRNAs in Alzheimer’s disease. Neurobiol. Dis. 2012, 46, 285–290. [Google Scholar] [CrossRef]
- Fan, J.; Kou, X.; Yang, Y.; Chen, N. MicroRNA-regulated proinflammatory cytokines in sarcopenia. Med. Inflamm. 2016, 2016, 1438686. [Google Scholar] [CrossRef]
- Kumar, S.; Reddy, P.H. MicroRNA-455-3p as a Potential Biomarker for Alzheimer’s Disease: An Update. Front. Aging Neurosci. 2018, 11, 41. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, N. Regulatory role of microRNAs in muscle atrophy during exercise intervention. Int. J. Mol. Sci. 2018, 19, 405. [Google Scholar] [CrossRef]
- Müller, M.; Perrone, G.; Kuiperij, H.B.; Verbeek, M.M. Expression of five miRNA targets in hippocampus and cerebrospinal fluid in Alzheimer’s disease. Alzheimers Dement. J. Alzheimers Assoc. 2012, 8, P273. [Google Scholar] [CrossRef]
- Kapsimali, M.; Kloosterman, W.P.; de Bruijn, E.; Rosa, F.; Plasterk, R.H.; Wilson, S.W. MicroRNAs show a wide diversity of expression profiles in the developing and mature central nervous system. Genome Biol. 2007, 8, R173. [Google Scholar] [CrossRef] [PubMed]
- Hackl, M.; Brunner, S.; Fortschegger, K.; Schreiner, C.; Micutkova, L.; Muck, C.; Laschober, G.T.; Lepperdinger, G.; Sampson, N.; Berger, P.; et al. miR-17, miR-19b, miR-20a, and miR-106a are downregulated in human aging. Aging Cell 2010, 9, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Luo, H.; Chen, Y.; Wu, Q.; Xiong, Y.; Zhu, J.; Diao, Y.; Wu, Z.; Miao, J.; Wan, J. MicroRNAs 99b-5p/100-5p Regulated by Endoplasmic Reticulum Stress are Involved in Abeta-Induced Pathologies. Front. Aging Neurosci. 2015, 7, 210. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Lee, Y.S.; Malhotra, A.; Kim, H.K.; Matecic, M.; Evans, C.; Jensen, R.V.; Moskaluk, C.A.; Dutta, A. miR-99 family of MicroRNAs suppresses the expression of prostate-specific antigen and prostate cancer cell proliferation. Cancer Res. 2011, 71, 1313–1324. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Luo, X.Q.; Han, B.W.; Duan, F.T.; Wei, P.P.; Chen, Y.Q. MicroRNA-100/99a, deregulated in acute lymphoblastic leukaemia, suppress proliferation and promote apoptosis by regulating the FKBP51 and IGF1R/mTOR signalling pathways. Br. J. Cancer 2013, 109, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zeng, Q.; Xu, W.; Jiao, L.; Chen, Y.; Zhang, Z.; Wu, C.; Jin, T.; Pan, A.; Wei, R.; et al. miRNA-100 inhibits human bladder urothelial carcinogenesis by directly targeting mTOR. Mol. Cancer Ther. 2013, 12, 207–219. [Google Scholar] [CrossRef]
- Higaki, S.; Muramatsu, M.; Matsuda, A.; Matsumoto, K.; Satoh, J.I.; Michikawa, M.; Niida, S. Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS ONE 2018, 13, e0196929. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Liu, P.; Wang, Z.; Fang, S.; Liu, Y.; Wang, J.; Liu, W.; Wang, N.; Chen, L.; Wang, J.; Zhang, H.; Wang, L. Inhibition of MicroRNA-96 Ameliorates Cognitive Impairment and Inactivation Autophagy Following Chronic Cerebral Hypoperfusion in the Rat. Cell Physiol. Biochem. 2018, 49, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Denk, J.; Boelmans, K.; Siegismund, C.; Lassner, D.; Arlt, S.; Jahn, H. MicroRNA Profiling of CSF Reveals Potential Biomarkers to Detect Alzheimer’s Disease. PLoS ONE 2015, 10, e0126423. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed]
- Kocerha, J.; Kauppinen, S.; Wahlestedt, C. microRNAs in CNS disorders. Neuromol. Med. 2009, 11, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Schratt, G. microRNAs at the synapse. Nat. Rev. Neurosci. 2009, 10, 842–849. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Abdelmohsen, K.; Gorospe, M.; Ejiogu, N.; Zonderman, A.B.; Evans, M.K. microRNA expression patterns reveal differential expression of target genes with age. PLoS ONE 2010, 5, e10724. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, H.; Zhang, C.; Jing, Y.; Wang, C.; Liu, C.; Zhang, R.; Wang, J.; Zhang, J.; Zen, K.; et al. Investigation of microRNA expression in human serum during the aging process. J. Gerontol. A Biol. Sci. Med. Sci. 2015, 70, 102–109. [Google Scholar] [CrossRef]
- Wang, W.X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Al Hashimi, A.; Girard, J.; Delay, C.; Hebert, S.S. In vivo regulation of amyloid precursor protein neuronal splicing by microRNAs. J. Neurochem. 2011, 116, 240–247. [Google Scholar] [CrossRef]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef]
- Hebert, S.S.; Horre, K.; Nicolai, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef]
- Kumar, S.; Vijayan, M.; Reddy, P.H. MicroRNA-455-3p as a potential peripheral biomarker for Alzheimer’s disease. Hum. Mol. Genet. 2017, 26, 3808–3822. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. miR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Schafer, D. No old man ever forgot where he buried his treasure: Concepts of cognitive impairment in old age circa 1700. J. Am. Geriatr. Soc. 2005, 53, 2023–2027. [Google Scholar] [CrossRef]
- Williams, A.F.; Gagnon, J. Neuronal cell Thy-1 glycoprotein: Homology with immunoglobulin. Science 1982, 216, 696–703. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Buttini, M.; Yu, G.Q.; Shockley, K.; Huang, Y.; Jones, B.; Masliah, E.; Mallory, M.; Yeo, T.; Longo, F.M.; Mucke, L. Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid beta peptides but not on plaque formation. J. Neurosci. 2002, 22, 10539–10548. [Google Scholar] [CrossRef] [PubMed]
- Raber, J.; Wong, D.; Yu, G.Q.; Buttini, M.; Mahley, R.W.; Pitas, R.E.; Mucke, L. Apolipoprotein E and cognitive performance. Nature 2000, 404, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Churchill, J.D.; Galvez, R.; Colcombe, S.; Swain, R.A.; Kramer, A.F.; Greenough, W.T. Exercise, experience and the aging brain. Neurobiol. Aging 2002, 23, 941–955. [Google Scholar] [CrossRef]
- Heyn, P.; Abreu, B.C.; Ottenbacher, K.J. The effects of exercise training on elderly persons with cognitive impairment and dementia: A meta-analysis. Arch. Phys. Med. Rehabil. 2004, 85, 1694–1704. [Google Scholar] [CrossRef]
- Groot, C.; Hooghiemstra, A.M.; Raijmakers, P.G.; van Berckel, B.N.; Scheltens, P.; Scherder, E.J.; van der Flier, W.M.; Ossenkoppele, R. The effect of physical activity on cognitive function in patients with dementia: A meta-analysis of randomized control trials. Ageing Res. Rev. 2016, 25, 13–23. [Google Scholar] [CrossRef]
- Yuede, C.M.; Zimmerman, S.D.; Dong, H.; Kling, M.J.; Bero, A.W.; Holtzman, D.M.; Timson, B.F.; Csernansky, J.G. Effects of voluntary and forced exercise on plaque deposition, hippocampal volume, and behavior in the Tg2576 mouse model of Alzheimer’s disease. Neurobiol. Dis. 2009, 35, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Nichol, K.; Deeny, S.P.; Seif, J.; Camaclang, K.; Cotman, C.W. Exercise improves cognition and hippocampal plasticity in APOE epsilon4 mice. Alzheimers Dement. 2009, 5, 287–294. [Google Scholar] [CrossRef]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar] [CrossRef]
- Nichol, K.E.; Parachikova, A.I.; Cotman, C.W. Three weeks of running wheel exposure improves cognitive performance in the aged Tg2576 mouse. Behav. Brain Res. 2007, 184, 124–132. [Google Scholar] [CrossRef]
- Tapia-Rojas, C.; Aranguiz, F.; Varela-Nallar, L.; Inestrosa, N.C. Voluntary Running Attenuates Memory Loss, Decreases Neuropathological Changes and Induces Neurogenesis in a Mouse Model of Alzheimer’s Disease. Brain Pathol. 2016, 26, 62–74. [Google Scholar] [CrossRef]
- Erickson, K.I.; Gildengers, A.G.; Butters, M.A. Physical activity and brain plasticity in late adulthood. Dialogues Clin. Neurosci. 2013, 15, 99–108. [Google Scholar]
- Colcombe, S.; Kramer, A.F. Fitness effects on the cognitive function of older adults: A meta-analytic study. Psychol. Sci. 2003, 14, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Mirochnic, S.; Wolf, S.; Staufenbiel, M.; Kempermann, G. Age effects on the regulation of adult hippocampal neurogenesis by physical activity and environmental enrichment in the APP23 mouse model of Alzheimer disease. Hippocampus 2009, 19, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Patten, A.R.; Yau, S.Y.; Fontaine, C.J.; Meconi, A.; Wortman, R.C.; Christie, B.R. The Benefits of Exercise on Structural and Functional Plasticity in the Rodent Hippocampus of Different Disease Models. Brain Plast. 2015, 1, 97–127. [Google Scholar] [CrossRef] [PubMed]
- Barry, A.E.; Klyubin, I.; Mc Donald, J.M.; Mably, A.J.; Farrell, M.A.; Scott, M.; Walsh, D.M.; Rowan, M.J. Alzheimer’s disease brain-derived amyloid-beta-mediated inhibition of LTP in vivo is prevented by immunotargeting cellular prion protein. J. Neurosci. 2011, 31, 7259–7263. [Google Scholar] [CrossRef] [PubMed]
- Jo, J.; Whitcomb, D.J.; Olsen, K.M.; Kerrigan, T.L.; Lo, S.C.; Bru-Mercier, G.; Dickinson, B.; Scullion, S.; Sheng, M.; Collingridge, G.; et al. Abeta(1-42) inhibition of LTP is mediated by a signaling pathway involving caspase-3, Akt1 and GSK-3beta. Nat. Neurosci. 2011, 14, 545–547. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, R.M.; Ohle, R.; Kelly, A.M. The effects of forced exercise on hippocampal plasticity in the rat: A comparison of LTP, spatial- and non-spatial learning. Behav. Brain Res. 2007, 176, 362–366. [Google Scholar] [CrossRef]
- Farmer, J.; Zhao, X.; van Praag, H.; Wodtke, K.; Gage, F.H.; Christie, B.R. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neuroscience 2004, 124, 71–79. [Google Scholar] [CrossRef] [PubMed]
- van Praag, H.; Christie, B.R.; Sejnowski, T.J.; Gage, F.H. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 13427–13431. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Lopez-Ramos, J.C.; Gimenez-Llort, L.; Revilla, S.; Guerra, R.; Gruart, A.; Laferla, F.M.; Cristofol, R.; Delgado-Garcia, J.M.; Sanfeliu, C. Physical exercise protects against Alzheimer’s disease in 3xTg-AD mice. J. Alzheimers Dis. 2011, 24, 421–454. [Google Scholar] [CrossRef]
- Kumar, A.; Rani, A.; Tchigranova, O.; Lee, W.H.; Foster, T.C. Influence of late-life exposure to environmental enrichment or exercise on hippocampal function and CA1 senescent physiology. Neurobiol. Aging 2012, 33, 828.e1–828.e17. [Google Scholar] [CrossRef]
- Dao, A.T.; Zagaar, M.A.; Levine, A.T.; Salim, S.; Eriksen, J.L.; Alkadhi, K.A. Treadmill exercise prevents learning and memory impairment in Alzheimer’s disease-like pathology. Curr. Alzheimer Res. 2013, 10, 507–515. [Google Scholar] [CrossRef]
- Nielsen, S.; Akerstrom, T.; Rinnov, A.; Yfanti, C.; Scheele, C.; Pedersen, B.K.; Laye, M.J. The miRNA plasma signature in response to acute aerobic exercise and endurance training. PLoS ONE 2014, 9, e87308. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.D.; Zeng, K.; Liu, W.L.; Gao, Y.G.; Gong, C.S.; Zhang, C.X.; Chen, Y.Q. Effect of aerobic exercise on miRNA-TLR4 signaling in atherosclerosis. Int. J. Sports Med. 2014, 35, 344–350. [Google Scholar] [CrossRef]
- Fernandes, T.; Hashimoto, N.Y.; Magalhaes, F.C.; Fernandes, F.B.; Casarini, D.E.; Carmona, A.K.; Krieger, J.E.; Phillips, M.I.; Oliveira, E.M. Aerobic exercise training-induced left ventricular hypertrophy involves regulatory MicroRNAs, decreased angiotensin-converting enzyme-angiotensin ii, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1-7). Hypertension 2011, 58, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Bates, D.J.; An, J.; Terry, D.A.; Wang, E. Upregulation of key microRNAs, and inverse downregulation of their predicted oxidative phosphorylation target genes, during aging in mouse brain. Neurobiol. Aging 2011, 32, 944–955. [Google Scholar] [CrossRef]
- Larson, E.B.; Wang, L.; Bowen, J.D.; McCormick, W.C.; Teri, L.; Crane, P.; Kukull, W. Exercise is associated with reduced risk for incident dementia among persons 65 years of age and older. Ann. Intern. Med. 2006, 144, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Deeny, S.P.; Poeppel, D.; Zimmerman, J.B.; Roth, S.M.; Brandauer, J.; Witkowski, S.; Hearn, J.W.; Ludlow, A.T.; Contreras-Vidal, J.L.; Brandt, J.; et al. Exercise, APOE, and working memory: MEG and behavioral evidence for benefit of exercise in epsilon4 carriers. Biol. Psychol. 2008, 78, 179–187. [Google Scholar] [CrossRef]
- Etnier, J.L.; Caselli, R.J.; Reiman, E.M.; Alexander, G.E.; Sibley, B.A.; Tessier, D.; McLemore, E.C. Cognitive performance in older women relative to ApoE-epsilon4 genotype and aerobic fitness. Med. Sci. Sports Exerc. 2007, 39, 199–207. [Google Scholar] [CrossRef]
- Rovio, S.; Kareholt, I.; Helkala, E.L.; Viitanen, M.; Winblad, B.; Tuomilehto, J.; Soininen, H.; Nissinen, A.; Kivipelto, M. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet Neurol. 2005, 4, 705–711. [Google Scholar] [CrossRef]
- Schuit, A.J.; Feskens, E.J.; Launer, L.J.; Kromhout, D. Physical activity and cognitive decline, the role of the apolipoprotein e4 allele. Med. Sci. Sports Exerc. 2001, 33, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Laurin, D.; Verreault, R.; Lindsay, J.; MacPherson, K.; Rockwood, K. Physical activity and risk of cognitive impairment and dementia in elderly persons. Arch. Neurol. 2001, 58, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Verghese, J.; Lipton, R.B.; Katz, M.J.; Hall, C.B.; Derby, C.A.; Kuslansky, G.; Ambrose, A.F.; Sliwinski, M.; Buschke, H. Leisure activities and the risk of dementia in the elderly. N. Engl. J. Med. 2003, 348, 2508–2516. [Google Scholar] [CrossRef]
- Akbaraly, T.N.; Portet, F.; Fustinoni, S.; Dartigues, J.F.; Artero, S.; Rouaud, O.; Touchon, J.; Ritchie, K.; Berr, C. Leisure activities and the risk of dementia in the elderly: Results from the Three-City Study. Neurology 2009, 73, 854–861. [Google Scholar] [CrossRef]
- Cai, Z.; Yan, L.J.; Li, K.; Quazi, S.H.; Zhao, B. Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromol. Med. 2012, 14, 1–14. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.; Kiely, A.P.; Coakley, M.F.; Manning, S.; Long-Smith, C.M. Insulin and IGF-1 signalling: Longevity, protein homoeostasis and Alzheimer’s disease. Biochem. Soc. Trans. 2012, 40, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Kang, E.B.; Cho, J.Y. Effect of treadmill exercise on PI3K/AKT/mTOR, autophagy, and tau hyperphosphorylation in the cerebral cortex of NSE/htau23 transgenic mice. J. Exerc. Nutr. Biochem. 2015, 19, 199–209. [Google Scholar] [CrossRef]
- Tramutola, A.; Triplett, J.C.; Di Domenico, F.; Niedowicz, D.M.; Murphy, M.P.; Coccia, R.; Perluigi, M.; Butterfield, D.A. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): Analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J. Neurochem. 2015, 133, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Fang, C.X.; Aberle, N.S.; Ren, B.H.; Ceylan-Isik, A.F.; Ren, J. Inhibition of PI-3 kinase/Akt/mTOR, but not calcineurin signaling, reverses insulin-like growth factor I-induced protection against glucose toxicity in cardiomyocyte contractile function. J. Endocrinol. 2005, 186, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Yates, S.C.; Zafar, A.; Hubbard, P.; Nagy, S.; Durant, S.; Bicknell, R.; Wilcock, G.; Christie, S.; Esiri, M.M.; Smith, A.D.; Nagy, Z. Dysfunction of the mTOR pathway is a risk factor for Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Um, H.S.; Kang, E.B.; Koo, J.H.; Kim, H.T.; Jin, L.; Kim, E.J.; Yang, C.H.; An, G.Y.; Cho, I.H.; Cho, J.Y. Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci. Res. 2011, 69, 161–173. [Google Scholar] [CrossRef]
- Jeong, J.H.; Kang, E.B. Effects of treadmill exercise on PI3K/AKT/GSK-3beta pathway and tau protein in high-fat diet-fed rats. J. Exerc. Nutr. Biochem. 2018, 22, 9–14. [Google Scholar] [CrossRef]
- Jin, X.; Townley, R.; Shapiro, L. Structural insight into AMPK regulation: ADP comes into play. Structure 2007, 15, 1285–1295. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef]
- Ju, T.C.; Chen, H.M.; Lin, J.T.; Chang, C.P.; Chang, W.C.; Kang, J.J.; Sun, C.P.; Tao, M.H.; Tu, P.H.; Chang, C.; et al. Nuclear translocation of AMPK-alpha1 potentiates striatal neurodegeneration in Huntington’s disease. J. Cell Biol. 2011, 194, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Chandakkar, P.; Zhao, H.; d’Abramo, C.; Davies, P.; Marambaud, P. Novel synthetic small-molecule activators of AMPK as enhancers of autophagy and amyloid-beta peptide degradation. FASEB J. 2011, 25, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Greco, S.J.; Sarkar, S.; Johnston, J.M.; Tezapsidis, N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 2009, 380, 98–104. [Google Scholar] [CrossRef]
- Won, J.S.; Im, Y.B.; Kim, J.; Singh, A.K.; Singh, I. Involvement of AMP-activated-protein-kinase (AMPK) in neuronal amyloidogenesis. Biochem. Biophys. Res. Commun. 2010, 399, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Thornton, C.; Bright, N.J.; Sastre, M.; Muckett, P.J.; Carling, D. AMP-activated protein kinase (AMPK) is a tau kinase, activated in response to amyloid beta-peptide exposure. Biochem. J. 2011, 434, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Goedert, M. Phosphorylation of microtubule-associated protein tau by AMPK-related kinases. J. Neurochem. 2012, 120, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Rönnemaa, E.; Zethelius, B.; Sundelöf, J.; Sundström, J.; Degerman-Gunnarsson, M.; Berne, C.; Lannfelt, L.; Kilander, L. Impaired insulin secretion increases the risk of Alzheimer disease. Neurology 2008, 71, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Gibas, M.K.; Gibas, K.J. Induced and controlled dietary ketosis as a regulator of obesity and metabolic syndrome pathologies. Diabetes Metab. Syndr. 2017, 11, S385–S390. [Google Scholar] [CrossRef]
- Halikas, A.; Gibas, K.J. AMPK induced memory improvements in the diabetic population: A Case Study. Diabetes Metab. Syndr. 2018, 12, 1141–1146. [Google Scholar] [CrossRef]
- Thompson, H.J.; Jiang, W.; Zhu, Z. Candidate mechanisms accounting for effects of physical activity on breast carcinogenesis. IUBMB Life 2009, 61, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef]
- Dethlefsen, C.; Lillelund, C.; Midtgaard, J.; Andersen, C.; Pedersen, B.K.; Christensen, J.F.; Hojman, P. Exercise regulates breast cancer cell viability: Systemic training adaptations versus acute exercise responses. Breast Cancer Res. Treat. 2016, 159, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pinilla, F.; Vaynman, S.; Ying, Z. Brain-derived neurotrophic factor functions as a metabotrophin to mediate the effects of exercise on cognition. Eur. J. Neurosci. 2008, 28, 2278–2287. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kou, X.; Chen, D.; Chen, N. Physical Activity Alleviates Cognitive Dysfunction of Alzheimer’s Disease through Regulating the mTOR Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 1591. https://doi.org/10.3390/ijms20071591
Kou X, Chen D, Chen N. Physical Activity Alleviates Cognitive Dysfunction of Alzheimer’s Disease through Regulating the mTOR Signaling Pathway. International Journal of Molecular Sciences. 2019; 20(7):1591. https://doi.org/10.3390/ijms20071591
Chicago/Turabian StyleKou, Xianjuan, Dandan Chen, and Ning Chen. 2019. "Physical Activity Alleviates Cognitive Dysfunction of Alzheimer’s Disease through Regulating the mTOR Signaling Pathway" International Journal of Molecular Sciences 20, no. 7: 1591. https://doi.org/10.3390/ijms20071591
APA StyleKou, X., Chen, D., & Chen, N. (2019). Physical Activity Alleviates Cognitive Dysfunction of Alzheimer’s Disease through Regulating the mTOR Signaling Pathway. International Journal of Molecular Sciences, 20(7), 1591. https://doi.org/10.3390/ijms20071591