Mechanistic Insights in NeuroD Potentiation of Mineralocorticoid Receptor Signaling

 , ,
, , 
Abstract
1. Introduction
2. Results
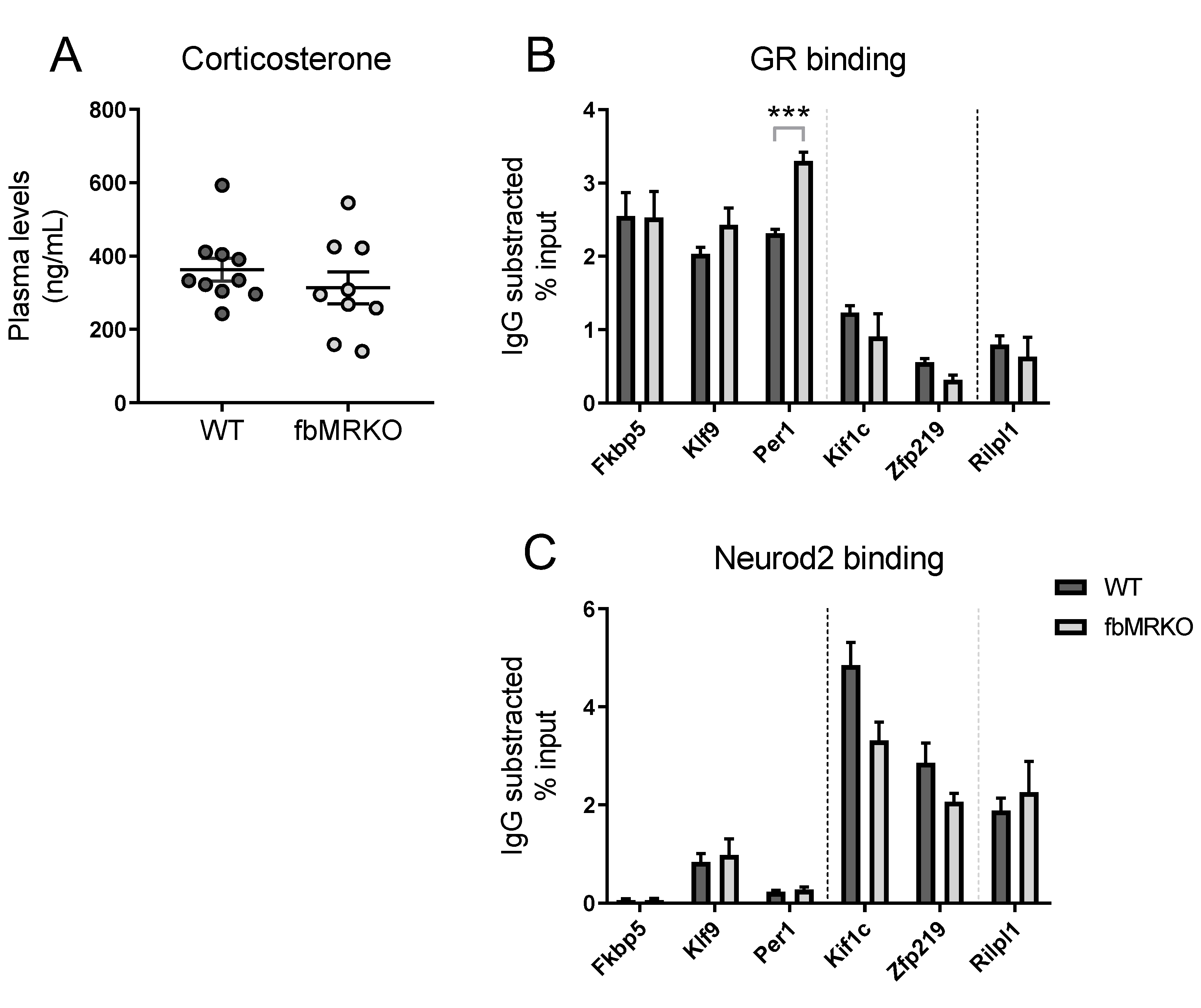
2.1. DNA Binding Assessed by Chromatin Immunoprecipitation
2.1.1. MR Effect on GR Binding
2.1.2. MR Effect on Neurod2 Binding
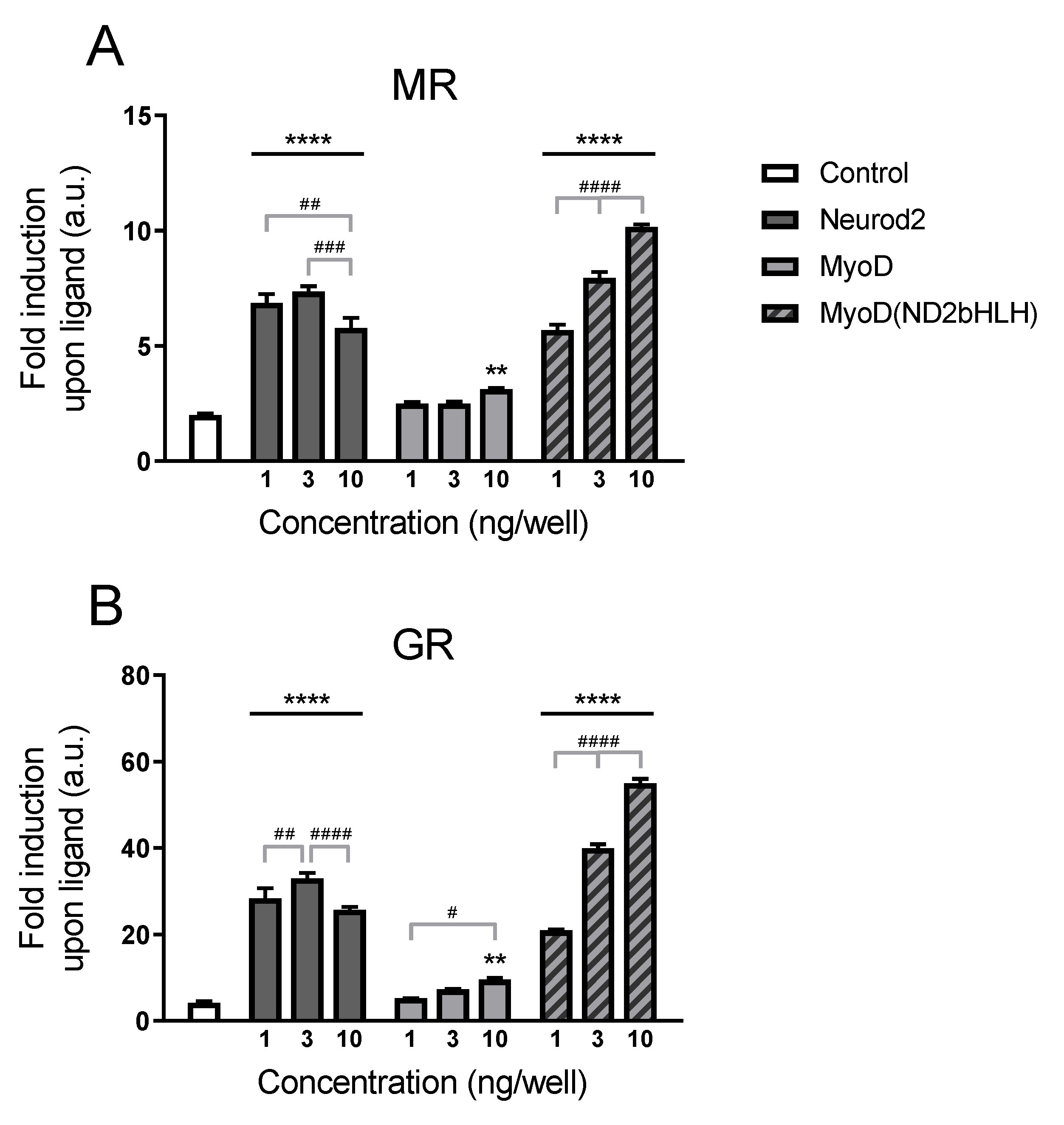
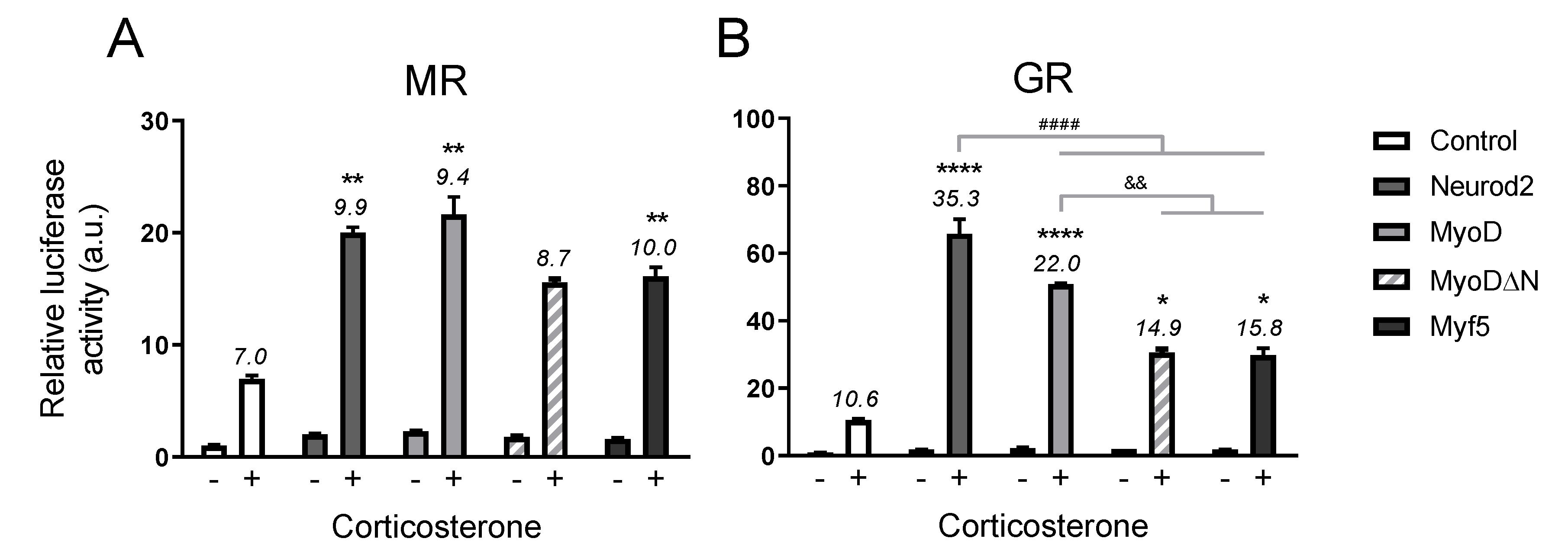
2.2. Structure–Function Relationship
2.2.1. Transcriptional Potentiation by MyoD
2.2.2. Activation Domain Not Crucial for Potentiation
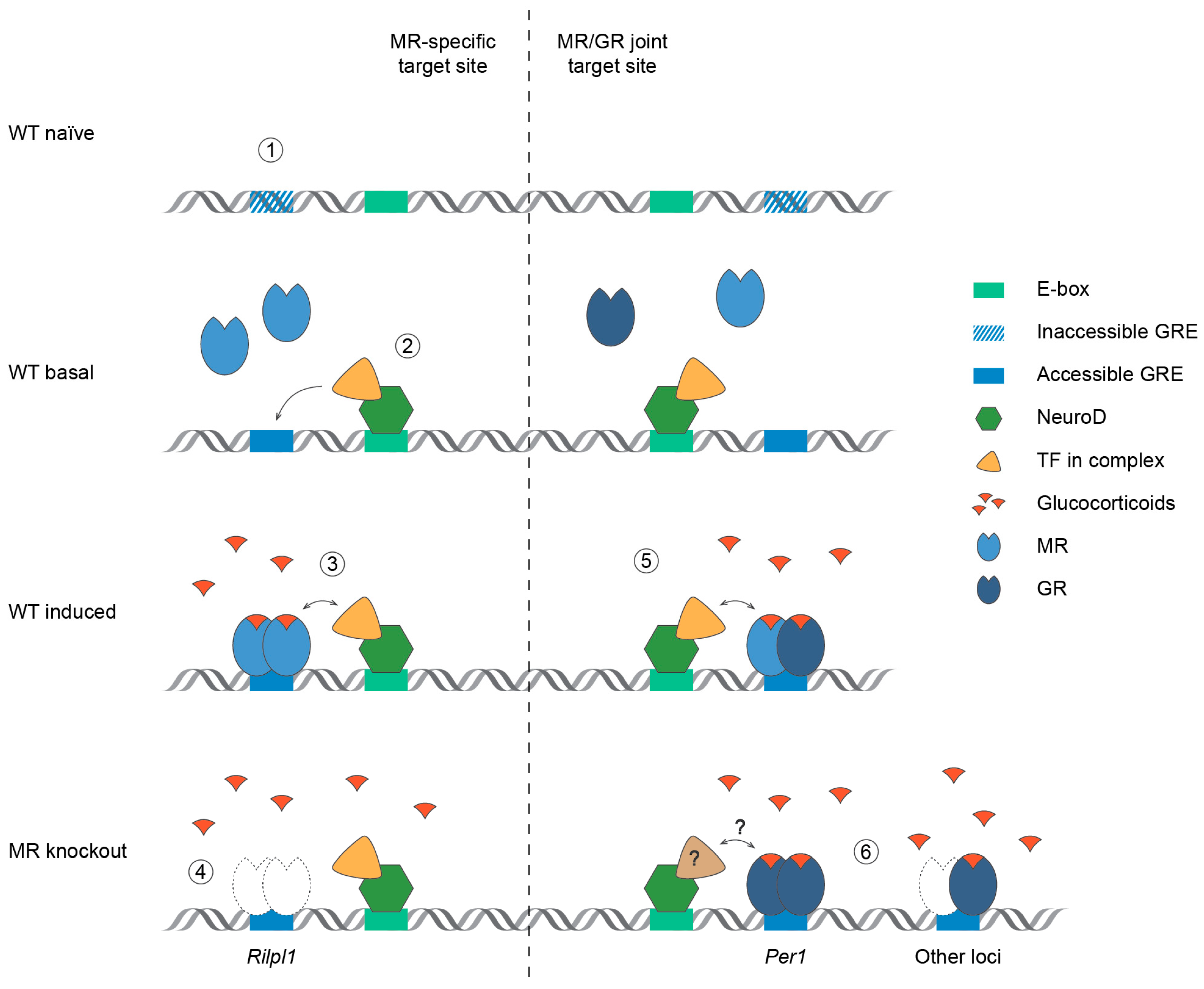
3. Discussion
3.1. Effects on DNA Binding
3.2. Mechanism of Glucocorticoid Signaling Potentiation
3.3. MR Selective Signaling and Future Implications
4. Materials and Methods
4.1. Animals
4.2. Plasma Corticosterone
4.3. ChIP-qPCR
4.4. Reporter Assays
4.5. Plasmids
4.6. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- De Kloet, E.R.; Otte, C.; Kumsta, R.; Kok, L.; Hillegers, M.H.; Hasselmann, H.; Kliegel, D.; Joels, M. Stress and Depression: A Crucial Role of the Mineralocorticoid Receptor. J. Neuroendocrinol. 2016, 28. [Google Scholar] [CrossRef] [PubMed]
- Klok, M.D.; Giltay, E.J.; Van der Does, A.J.; Geleijnse, J.M.; Antypa, N.; Penninx, B.W.; de Geus, E.J.; Willemsen, G.; Boomsma, D.I.; van Leeuwen, N.; et al. A common and functional mineralocorticoid receptor haplotype enhances optimism and protects against depression in females. Transl. Psychiatry 2011, 1, e62. [Google Scholar] [CrossRef]
- Klok, M.D.; Vreeburg, S.A.; Penninx, B.W.; Zitman, F.G.; de Kloet, E.R.; DeRijk, R.H. Common functional mineralocorticoid receptor polymorphisms modulate the cortisol awakening response: Interaction with SSRIs. Psychoneuroendocrinology 2011, 36, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Otte, C.; Hinkelmann, K.; Moritz, S.; Yassouridis, A.; Jahn, H.; Wiedemann, K.; Kellner, M. Modulation of the mineralocorticoid receptor as add-on treatment in depression: A randomized, double-blind, placebo-controlled proof-of-concept study. J. Psychiatr. Res. 2010, 44, 339–346. [Google Scholar] [CrossRef]
- Otte, C.; Wingenfeld, K.; Kuehl, L.K.; Kaczmarczyk, M.; Richter, S.; Quante, A.; Regen, F.; Bajbouj, M.; Zimmermann-Viehoff, F.; Wiedemann, K.; et al. Mineralocorticoid receptor stimulation improves cognitive function and decreases cortisol secretion in depressed patients and healthy individuals. Neuropsychopharmacology 2015, 40, 386–393. [Google Scholar] [CrossRef] [PubMed]
- Judd, L.L.; Schettler, P.J.; Brown, E.S.; Wolkowitz, O.M.; Sternberg, E.M.; Bender, B.G.; Bulloch, K.; Cidlowski, J.A.; de Kloet, E.R.; Fardet, L.; et al. Adverse consequences of glucocorticoid medication: Psychological, cognitive, and behavioral effects. Am. J. Psychiatry 2014, 171, 1045–1051. [Google Scholar] [CrossRef]
- DeBattista, C.; Belanoff, J.; Glass, S.; Khan, A.; Horne, R.L.; Blasey, C.; Carpenter, L.L.; Alva, G. Mifepristone versus placebo in the treatment of psychosis in patients with psychotic major depression. Biol. Psychiatry 2006, 60, 1343–1349. [Google Scholar] [CrossRef] [PubMed]
- Warris, L.T.; van den Heuvel-Eibrink, M.M.; Aarsen, F.K.; Pluijm, S.M.; Bierings, M.B.; van den Bos, C.; Zwaan, C.M.; Thygesen, H.H.; Tissing, W.J.; Veening, M.A.; et al. Hydrocortisone as an Intervention for Dexamethasone-Induced Adverse Effects in Pediatric Patients with Acute Lymphoblastic Leukemia: Results of a Double-Blind, Randomized Controlled Trial. J. Clin. Oncol. 2016, 34, 2287–2293. [Google Scholar] [CrossRef]
- Van Weert, L.T.C.M.; Buurstede, J.C.; Mahfouz, A.; Braakhuis, P.S.M.; Polman, J.A.E.; Sips, H.C.M.; Roozendaal, B.; Balog, J.; de Kloet, E.R.; Datson, N.A.; et al. NeuroD Factors Discriminate Mineralocorticoid From Glucocorticoid Receptor DNA Binding in the Male Rat Brain. Endocrinology 2017, 158, 1511–1522. [Google Scholar] [CrossRef]
- Murre, C. Helix-loop-helix proteins and the advent of cellular diversity: 30 years of discovery. Genes Dev. 2019, 33, 6–25. [Google Scholar] [CrossRef] [PubMed]
- Fong, A.P.; Yao, Z.; Zhong, J.W.; Cao, Y.; Ruzzo, W.L.; Gentleman, R.C.; Tapscott, S.J. Genetic and epigenetic determinants of neurogenesis and myogenesis. Dev. Cell 2012, 22, 721–735. [Google Scholar] [CrossRef]
- Conerly, M.L.; Yao, Z.; Zhong, J.W.; Groudine, M.; Tapscott, S.J. Distinct Activities of Myf5 and MyoD Indicate Separate Roles in Skeletal Muscle Lineage Specification and Differentiation. Dev. Cell 2016, 36, 375–385. [Google Scholar] [CrossRef]
- Van Weert, L.T.C.M.; Buurstede, J.C.; Sips, H.C.M.; Vettorazzi, S.; Mol, I.M.; Hartmann, J.; Prekovic, S.; Zwart, W.; Schmidt, M.V.; Roozendaal, B.; et al. Identification of mineralocorticoid receptor target genes in the mouse hippocampus. J. Neuroendocrinol. 2019. submitted. [Google Scholar]
- Reul, J.M.; de Kloet, E.R. Two receptor systems for corticosterone in rat brain: Microdistribution and differential occupation. Endocrinology 1985, 117, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Mifsud, K.R.; Reul, J.M. Acute stress enhances heterodimerization and binding of corticosteroid receptors at glucocorticoid target genes in the hippocampus. Proc. Natl. Acad. Sci. USA 2016, 113, 11336–11341. [Google Scholar] [CrossRef] [PubMed]
- Polman, J.A.; Welten, J.E.; Bosch, D.S.; de Jonge, R.T.; Balog, J.; van der Maarel, S.M.; de Kloet, E.R.; Datson, N.A. A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci. 2012, 13, 118. [Google Scholar] [CrossRef] [PubMed]
- Pataskar, A.; Jung, J.; Smialowski, P.; Noack, F.; Calegari, F.; Straub, T.; Tiwari, V.K. NeuroD1 reprograms chromatin and transcription factor landscapes to induce the neuronal program. EMBO J. 2016, 35, 24–45. [Google Scholar] [CrossRef]
- Berger, S.; Wolfer, D.P.; Selbach, O.; Alter, H.; Erdmann, G.; Reichardt, H.M.; Chepkova, A.N.; Welzl, H.; Haas, H.L.; Lipp, H.P.; et al. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc. Natl. Acad. Sci. USA 2006, 103, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.C.; Schiltz, R.L.; Sung, M.H.; Yen, P.M.; Stamatoyannopoulos, J.A.; Biddie, S.C.; Johnson, T.A.; Miranda, T.B.; John, S.; Hager, G.L. Dynamic exchange at regulatory elements during chromatin remodeling underlies assisted loading mechanism. Cell 2011, 146, 544–554. [Google Scholar] [CrossRef] [PubMed]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef]
- Hodge, R.D.; Bakken, T.E.; Miller, J.A.; Smith, K.A.; Barkan, E.R.; Graybuck, L.T.; Close, J.L.; Long, B.; Penn, O.; Yao, Z.; et al. Conserved cell types with divergent features between human and mouse cortex. bioRxiv 2018, 384826. [Google Scholar] [CrossRef]
- Pooley, J.R.; Flynn, B.P.; Grontved, L.; Baek, S.; Guertin, M.J.; Kershaw, Y.M.; Birnie, M.T.; Pellatt, A.; Rivers, C.A.; Schiltz, R.L.; et al. Genome-Wide Identification of Basic Helix-Loop-Helix and NF-1 Motifs Underlying GR Binding Sites in Male Rat Hippocampus. Endocrinology 2017, 158, 1486–1501. [Google Scholar] [CrossRef]
- Fong, A.P.; Yao, Z.; Zhong, J.W.; Johnson, N.M.; Farr, G.H., 3rd; Maves, L.; Tapscott, S.J. Conversion of MyoD to a neurogenic factor: Binding site specificity determines lineage. Cell Rep. 2015, 10, 1937–1946. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.; Noshiro, M.; Choi, M.; Morita, K.; Kawamoto, T.; Fujimoto, K.; Kato, Y.; Makishima, M. The basic helix-loop-helix proteins differentiated embryo chondrocyte (DEC) 1 and DEC2 function as corepressors of retinoid X receptors. Mol. Pharmacol. 2009, 76, 1360–1369. [Google Scholar] [CrossRef]
- Hemmer, M.C.; Wierer, M.; Schachtrup, K.; Downes, M.; Hubner, N.; Evans, R.M.; Uhlenhaut, N.H. E47 modulates hepatic glucocorticoid action. Nat. Commun. 2019, 10, 306. [Google Scholar] [CrossRef]
- Hong, C.Y.; Gong, E.Y.; Kim, K.; Suh, J.H.; Ko, H.M.; Lee, H.J.; Choi, H.S.; Lee, K. Modulation of the expression and transactivation of androgen receptor by the basic helix-loop-helix transcription factor Pod-1 through recruitment of histone deacetylase 1. Mol. Endocrinol. 2005, 19, 2245–2257. [Google Scholar] [CrossRef]
- Tetel, M.J.; Auger, A.P.; Charlier, T.D. Who’s in charge? Nuclear receptor coactivator and corepressor function in brain and behavior. Front. Neuroendocrinol. 2009, 30, 328–342. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.D.; Nitiyanandan, R.; Meraji, S.; Daer, R.; Godeshala, S.; Goklany, S.; Haynes, K.; Rege, K. An inhibitor screen identifies histone-modifying enzymes as mediators of polymer-mediated transgene expression from plasmid DNA. J. Control. Release 2018, 286, 210–223. [Google Scholar] [CrossRef]
- Ochiai, H.; Fujimuro, M.; Yokosawa, H.; Harashima, H.; Kamiya, H. Transient activation of transgene expression by hydrodynamics-based injection may cause rapid decrease in plasmid DNA expression. Gene Ther. 2007, 14, 1152–1159. [Google Scholar] [CrossRef]
- Oakley, R.H.; Busillo, J.M.; Cidlowski, J.A. Cross-talk between the glucocorticoid receptor and MyoD family inhibitor domain-containing protein provides a new mechanism for generating tissue-specific responses to glucocorticoids. J. Biol. Chem. 2017, 292, 5825–5844. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, M.R.; Girardot, C.; Sigismondo, G.; Krijgsveld, J. Expanding the Circuitry of Pluripotency by Selective Isolation of Chromatin-Associated Proteins. Mol. Cell 2016, 64, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Labonte, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.; Tan, C.; Scarpa, J.R.; Moy, G.; Loh, Y.E.; Cahill, M.; et al. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Bagot, R.C.; Cates, H.M.; Purushothaman, I.; Lorsch, Z.S.; Walker, D.M.; Wang, J.; Huang, X.; Schluter, O.M.; Maze, I.; Pena, C.J.; et al. Circuit-wide Transcriptional Profiling Reveals Brain Region-Specific Gene Networks Regulating Depression Susceptibility. Neuron 2016, 90, 969–983. [Google Scholar] [CrossRef] [PubMed]
- Boulle, F.; Massart, R.; Stragier, E.; Paizanis, E.; Zaidan, L.; Marday, S.; Gabriel, C.; Mocaer, E.; Mongeau, R.; Lanfumey, L. Hippocampal and behavioral dysfunctions in a mouse model of environmental stress: Normalization by agomelatine. Transl. Psychiatry 2014, 4, e485. [Google Scholar] [CrossRef] [PubMed]
- Geffroy, B.; Sadoul, B.; Bouchareb, A.; Prigent, S.; Bourdineaud, J.P.; Gonzalez-Rey, M.; Morais, R.N.; Mela, M.; Nobre Carvalho, L.; Bessa, E. Nature-Based Tourism Elicits a Phenotypic Shift in the Coping Abilities of Fish. Front. Physiol. 2018, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Yamamoto, K.R. Mineralocorticoid and glucocorticoid receptor activities distinguished by nonreceptor factors at a composite response element. Science 1993, 259, 1161–1165. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Full Name | Forward & Reverse (5′ > 3′) | Product Length (bp) |
|---|---|---|---|
| Fkbp5 | FK506 binding protein 5 | TGCCAGCCACATTCAGAACA TCAAGTGAGTCTGGTCACTGC | 122 |
| Kif1c | Kinesin family member 1C | GCTGGGGTGTACACAGATGG TGACTAGCCAGAGCAGTATGTC | 156 |
| Klf9 | Kruppel-like factor 9 | ATCTAGGGCAGTTTGTTCAA GGCAGGTTCATCTGAGGACA | 96 |
| Per1 | Period circadian clock 1 | GGAGGCGCCAAGGCTGAGTG CGGCCAGCGCACTAGGGAAC | 73 |
| Rilpl1 | Rab interacting lysosomal protein-like 1 | CAGGCAGATGCCAGGCT CCCATGCCTGTTCCTCTAGT | 106 |
| Zfp219 | Zinc finger protein 219 | AGTCCATCACATTCTGTTGCTTTC TAGTCAGCTATGACCATGCAGT | 131 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Weert, L.T.C.M.; Buurstede, J.C.; Sips, H.C.M.; Mol, I.M.; Puri, T.; Damsteegt, R.; Roozendaal, B.; Sarabdjitsingh, R.A.; Meijer, O.C. Mechanistic Insights in NeuroD Potentiation of Mineralocorticoid Receptor Signaling. Int. J. Mol. Sci. 2019, 20, 1575. https://doi.org/10.3390/ijms20071575
van Weert LTCM, Buurstede JC, Sips HCM, Mol IM, Puri T, Damsteegt R, Roozendaal B, Sarabdjitsingh RA, Meijer OC. Mechanistic Insights in NeuroD Potentiation of Mineralocorticoid Receptor Signaling. International Journal of Molecular Sciences. 2019; 20(7):1575. https://doi.org/10.3390/ijms20071575
Chicago/Turabian Stylevan Weert, Lisa T. C. M., Jacobus C. Buurstede, Hetty C. M. Sips, Isabel M. Mol, Tanvi Puri, Ruth Damsteegt, Benno Roozendaal, R. Angela Sarabdjitsingh, and Onno C. Meijer. 2019. "Mechanistic Insights in NeuroD Potentiation of Mineralocorticoid Receptor Signaling" International Journal of Molecular Sciences 20, no. 7: 1575. https://doi.org/10.3390/ijms20071575
APA Stylevan Weert, L. T. C. M., Buurstede, J. C., Sips, H. C. M., Mol, I. M., Puri, T., Damsteegt, R., Roozendaal, B., Sarabdjitsingh, R. A., & Meijer, O. C. (2019). Mechanistic Insights in NeuroD Potentiation of Mineralocorticoid Receptor Signaling. International Journal of Molecular Sciences, 20(7), 1575. https://doi.org/10.3390/ijms20071575