Use of Multifactorial Treatments to Address the Challenge of Translating Experimental Myocardial Infarct Reduction Strategies


Abstract
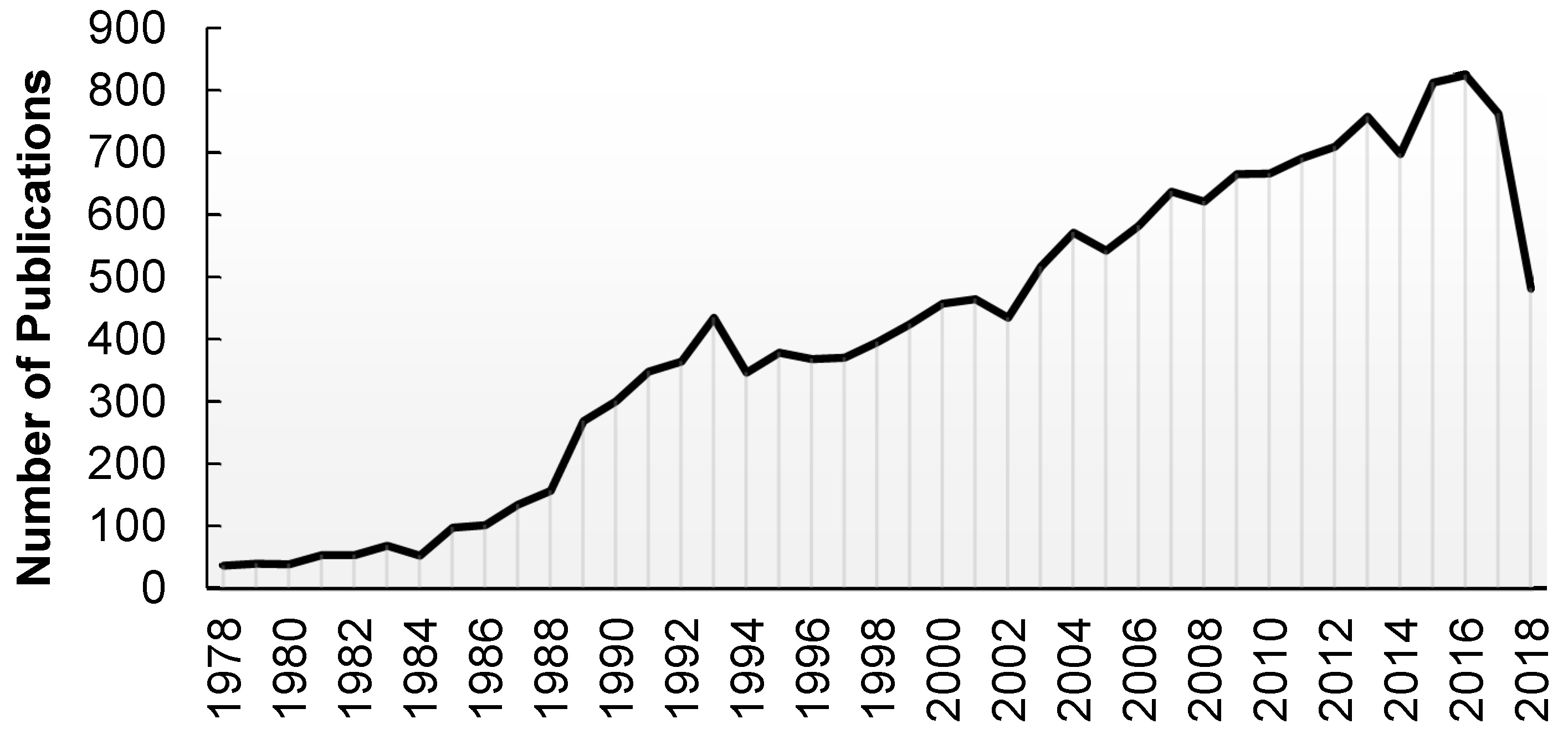
1. Introduction
2. Pathophysiology of MI
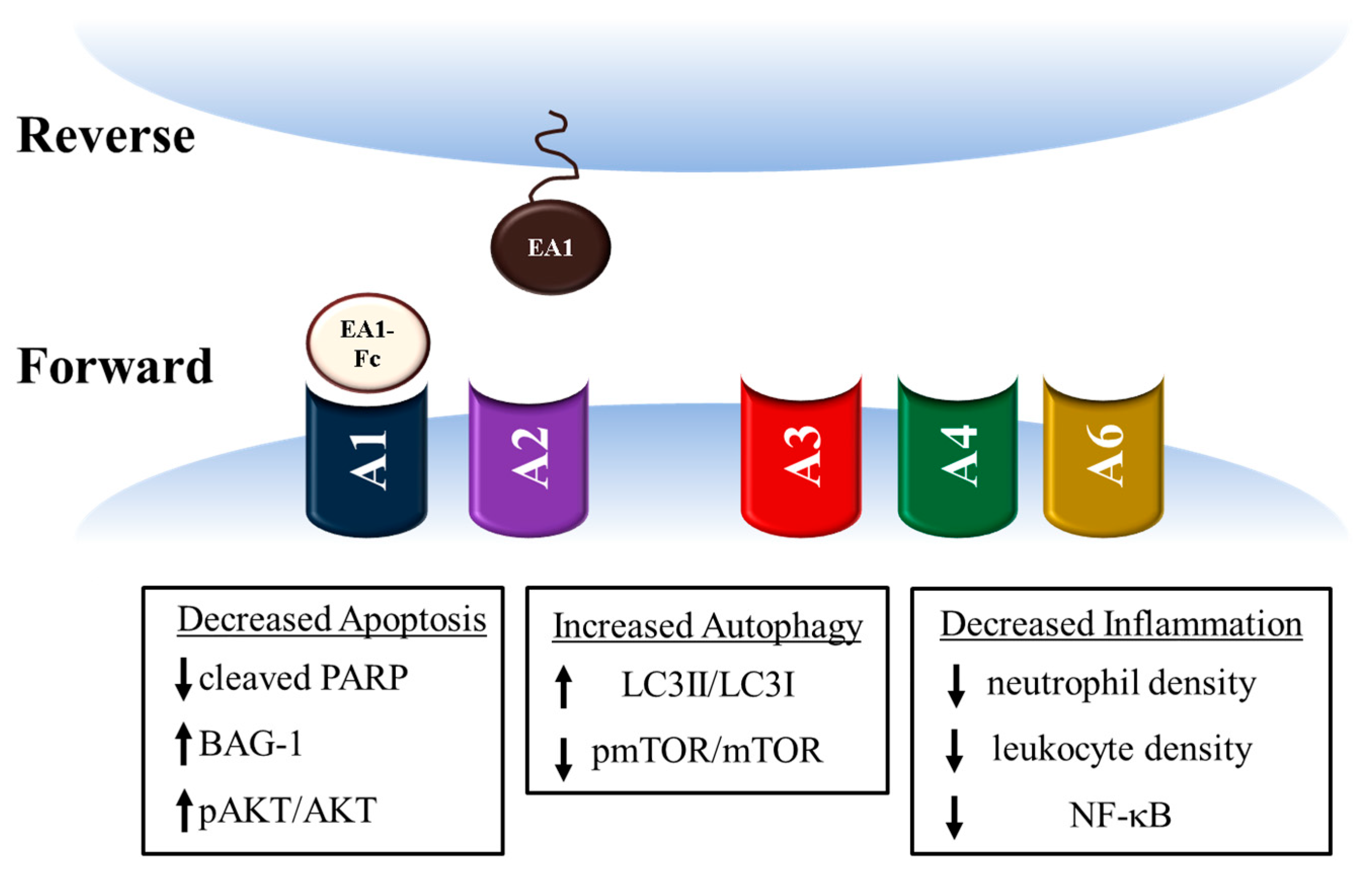
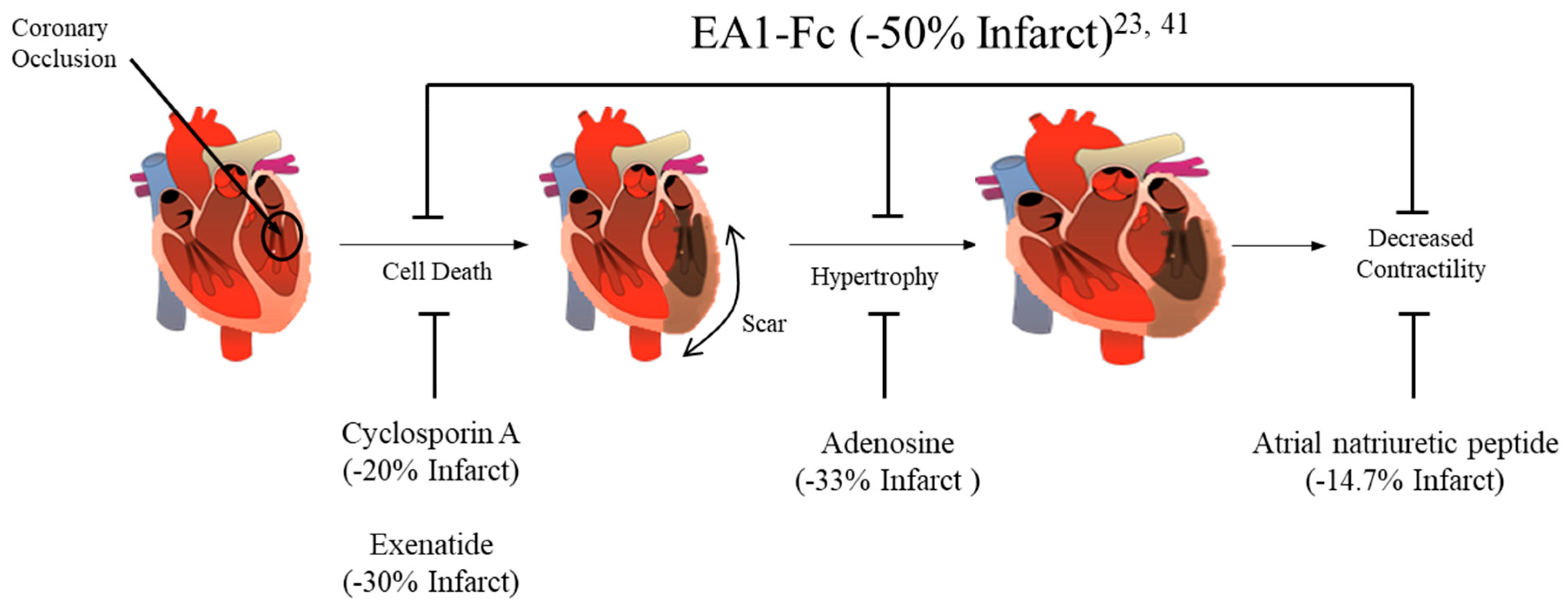
3. Ephrin A1 Ligand Intramyocardial Injection is Cardioprotective
4. Potential mechanisms for Ephrin A1-mediated cardioprotection
5. Conclusions
6. Patents
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular Disease |
| MI | Myocardial Infarction |
| EA1 | EphrinA1 |
| ATP | Adenosine Triphosphate |
| ROS | Reactive Oxygen Species |
| ECM | Extracellular Matrix |
| EF | Ejection Fraction |
| FS | Fractional Shortening |
References
- Centers for Disease Control. FastStat—Leading Causes of Death. Available online: https://www.cdc.gov/nchs/fastats/leading-causes-of-death.htm (accessed on 1 February 2019).
- Thygesen, K.; Alpert, J.S.; Jaffe, A.S.; Chaitman, B.R.; Bax, J.J.; Morrow, D.A. Fourth Universal Definition of Myocardial Infarction. J. Am. Coll. Cardiol. 2018, 72, 2231–2264. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C. ECG Diagnosis: ST-Elevation Myocardial Infarction. Perm. J. 2014, 18, e133. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cardiovascular Disease-Prevention of Recurrences of Myocardial Infarction and Stroke Study. Available online: https://www.who.int/cardiovascular_diseases/priorities/secondary_prevention/country/en/index1.html (accessed on 8 March 2019).
- Global Burden of Disease Collaborative Network. Global Burden of Disease Study 2017 (GBD 2017) Results; Institute for Health Metrics and Evaluation (IHME): Seattle, WA, USA, 2018; Available online: http://ghdx.healthdata.org/gbd-results-tool (accessed on 8 March 2019).
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E.; Cushman, M.; Das, S.R.; Deo, R.; De Ferranti, S.D.; Floyd, J.; Fornage, M.; Gillespie, C.; et al. Heart Disease and Stroke Statistics’2017 Update: A Report from the American Heart Association. Circulation 2017. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report from the American Heart Association. Circulation 2018. [Google Scholar] [CrossRef]
- Burns, R.J.; Gibbons, R.J.; Yi, Q.; Roberts, R.S.; Miller, T.D.; Schaer, G.L.; Anderson, J.L.; Yusuf, S. CORE Study Investigators. The Relationships of Left Ventricular Ejection Fraction, End-Systolic Volume Index and Infarct Size to Six-month Mortality After Hospital Discharge Following Myocardial Infarction Treated by Thrombolysis. J. Am. Coll. Cardiol. 2002, 39, 30–36. [Google Scholar] [CrossRef]
- Roes, S.D.; Kelle, S.; Kaandorp, T.A.; Kokocinski, T.; Poldermans, D.; Lamb, H.J.; Boersma, E.; van der Wall, E.E.; Fleck, E.; de Roos, A.; et al. Comparison of Myocardial Infarct Size Assessed with Contrast-Enhanced Magnetic Resonance Imaging and Left Ventricular Function and Volumes to Predict Mortality in Patients with Healed Myocardial Infarction. Am. J. Cardiol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Miki, T. Limitation of Myocardial Infarct Size in the Clinical Setting: Current Status and Challenges in Translating Animal Experiments into Clinical Therapy. Basic Res. Cardiol. 2008. [Google Scholar] [CrossRef]
- Koruth, J.S.; Lala, A.; Pinney, S.; Reddy, V.Y.; Dukkipati, S.R. The Clinical Use of Ivabradine. J. Am. Coll. Cardiol. 2017. [Google Scholar] [CrossRef]
- Niccoli, G.; Borovac, J.A.; Vetrugno, V.; Camici, P.G.; Crea, F. Ivabradine in Acute Coronary Syndromes: Protection Beyond Heart Rate Lowering. Int. J. Cardiol. 2017. [Google Scholar] [CrossRef]
- Priti, K.; Ranwa, B.L.; Gokhroo, R.K.; Kishore, K.; Bisht, D.S.; Gupta, S. Ivabradine vs. Metoprolol in Patients with Acute Inferior Wall Myocardial Infarction-“Expanding Arena for Ivabradine”. Cardiovasc. Ther. 2017. [Google Scholar] [CrossRef]
- Zhang, R.L.; Christensen, L.P.; Tomanek, R.J. Chronic Heart Rate Reduction Facilitates Cardiomyocyte Survival After Myocardial Infarction. Anat. Rec. (Hoboken) 2010. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, D.M.; Smith, R.S.; Piras, B.A.; Beyers, R.J.; Lin, D.; Hossack, J.A.; French, B.A. Heart Rate Reduction with Ivabradine Protects Against Left Ventricular Remodeling by Attenuating Infarct Expansion and Preserving Remote-Zone Contractile Function and Synchrony in a Mouse Model of Reperfused Myocardial Infarction. J. Am. Heart Assoc. 2016. [Google Scholar] [CrossRef]
- D’Amario, D.; Leone, A.M.; Borovac, J.A.; Cannata, F.; Siracusano, A.; Niccoli, G.; Crea, F. Granulocyte Colony-Stimulating Factor for the Treatment of Cardiovascular Diseases: An Update with a Critical Appraisal. Pharmacol. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Lv, H.; Yao, K.; Ge, L.; Ye, Z.; Ding, H.; Zhang, Y.; Lu, H.; Huang, Z.; Zhang, S.; et al. Effects of Different Doses of Granulocyte Colony-Stimulating Factor Mobilization Therapy on Ischemic Cardiomyopathy. Sci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pourtaji, A.; Jahani, V.; Moallem, S.M.H.; Karimani, A.; Mohammadpour, A.H. Application of G-CSF in congestive heart failure treatment. Curr. Cardiol. Rev. 2018. [Google Scholar] [CrossRef]
- Ibanez, B.; Macaya, C.; Sánchez-Brunete, V.; Pizarro, G.; Fernández-Friera, L.; Mateos, A.; Fernández-Ortiz, A.; García-Ruiz, J.M.; García-Álvarez, A.; Iñiguez, A.; et al. Effect of Early Metoprolol on Infarct Size in ST-segment-elevation Myocardial Infarction Patients Undergoing Primary Percutaneous Coronary Intervention: The Effect of Metoprolol in Cardioprotection During an Acute Myocardial Infarction (METOCARD-CNIC) Trial. Circulation 2013. [Google Scholar] [CrossRef]
- García-Prieto, J.; Villena-Gutiérrez, R.; Gómez, M.; Bernardo, E.; Pun-García, A.; García-Lunar, I.; Crainiciuc, G.; Fernández-Jiménez, R.; Sreeramkumar, V.; Bourio-Martínez, R.; et al. Neutrophil Stunning by Metoprolol Reduces Infarct Size. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Riedle, N.; Dickhaus, H.; Erbacher, M.; Steen, H.; Andrassy, M.; Lossnitzer, D.; Hardt, S.; Rottbauer, W.; Zugck, C.; Giannitsis, E.; et al. Early Assessment of Infarct Size and Prediction of Functional Recovery by Quantitative Myocardial Blush Grade in Patients with Acute Coronary Syndromes Treated According to Current Guidelines. Catheter. Cardiovasc. Interv. 2010. [Google Scholar] [CrossRef]
- Ibanez, B.; James, S.; Agewall, S.; Antunes, M.J.; Bucciarelli-Ducci, C.; Bueno, H.; Caforio, A.L.P.; Crea, F.; Goudevenos, J.A.; Halvorsen, S.; et al. 2017 ESC Guidelines for the Management of Acute Myocardial Infarction in Patients Presenting with ST-segment Elevation: The Task Force for the Management of Acute Myocardial Infarction in Patients Presenting with ST-segment Elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2017. [Google Scholar] [CrossRef]
- DuSablon, A.; Parks, J.; Whitehurst, K.; Estes, H.; Chase, R.; Vlahos, E.; Sharma, U.; Wert, D.; Virag, J. EphrinA1-Fc Attenuates Myocardial Ischemia/Reperfusion Injury in Mice. PLoS ONE 2017, 12, e0189307. [Google Scholar] [CrossRef]
- Baron, T.; Hambraeus, K.; Sundström, J.; Erlinge, D.; Jernberg, T.; Lindahl, B. Type 2 Myocardial Infarction in Clinical Practice. Heart 2015. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Ferdinandy, P.; Andreadou, I.; Bøtker, H.E.; Heusch, G.; Ibáñez, B.; Ovize, M.; Schulz, R.; Yellon, D.M.; Hausenloy, D.J.; et al. Multitarget Strategies to Reduce Myocardial Ischemia/Reperfusion Injury: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 73, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Gersh, B.J. The Pathophysiology of Acute Myocardial Infarction and Strategies of Protection beyond Reperfusion: A Continual Challenge. Eur. Heart J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandy, P.; Hausenloy, D.J.; Heusch, G.; Baxter, G.F.; Schulz, R. Interaction of Risk Factors, Comorbidities, and Comedications with Ischemia/Reperfusion Injury and Cardioprotection by Preconditioning, Postconditioning, and Remote Conditioning. Pharmacol. Rev. 2014, 66, 1142–1174. [Google Scholar] [CrossRef]
- Fanaroff, A.C.; Morrow, V.; Krucoff, M.W.; Seltzer, J.H.; Perin, E.C.; Taylor, D.A.; Miller, L.W.; Zeiher, A.M.; Fernández-Avilés, F.; Losordo, D.W.; et al. A Path Forward for Regenerative Medicine. Circ. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017. [Google Scholar] [CrossRef]
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing Myocardial Infarct Size: Challenges and Future Opportunities. Heart 2016. [Google Scholar] [CrossRef]
- Jang, W.J.; Yang, J.H.; Song, Y.; Hahn, J.-Y.; Chun, W.J.; Oh, J.H.; Kim, W.S.; Lee, Y.T.; Yu, C.W.; Lee, H.J.; et al. Second-Generation Drug-Eluting Stenting versus Coronary Artery Bypass Grafting for Treatment of Coronary Chronic Total Occlusion. J. Cardiol. 2019. [Google Scholar] [CrossRef]
- Mohr, F.W.; Morice, M.C.; Kappetein, A.P.; Feldman, T.E.; Ståhle, E.; Colombo, A.; Mack, M.J.; Holmes, D.R., Jr.; Morel, M.A.; Van Dyck, N.; et al. Coronary Artery Bypass Graft Surgery Versus Percutaneous Coronary Intervention in Patients with Three-Vessel Disease and Left Main Coronary Disease: 5-year Follow-up of the Randomised, Clinical SYNTAX Trial. Lancet 2013. [Google Scholar] [CrossRef]
- Kim, Y.G.; Park, D.W.; Lee, W.S.; Park, G.M.; Sun, B.J.; Lee, C.H.; Hwang, K.W.; Cho, S.W.; Kim, Y.R.; Song, H.G.; et al. Influence of Diabetes Mellitus on Long-Term (Five-Year) Outcomes of Drug-Eluting Stents and Coronary Artery Bypass Grafting for Multivessel Coronary Revascularization. Am. J. Cardiol. 2012. [Google Scholar] [CrossRef]
- LaNoue, K.; Nicklas, W.J.; Williamson, J.R. Control of Citric Acid Cycle Activity in Rat Heart Mitochondria. J. Biol. Chem. 1970, 245, 102–111. [Google Scholar] [PubMed]
- Taegtmeyer, H.; Overturf, M.L. Effects of Moderate Hypertension on Cardiac Function and Metabolism in the Rabbit. Hypertension 1988. [Google Scholar] [CrossRef]
- Sack, M.N.; Rader, T.A.; Park, S.; Bastin, J.; McCune, S.A.; Kelly, D.P. Fatty Acid Oxidation Enzyme Gene Expression Is Downregulated in the Failing Heart. Circulation 1996. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The Failing Heart Utilizes 3-Hydroxybutyrate as a Metabolic Stress Defense. JCI Insight 2019. [Google Scholar] [CrossRef]
- MacLellan, W.R.; Schneider, M.D. Death by Design: Programmed Cell Death in Cardiovascular Biology and Disease. Circ. Res. 1997. [Google Scholar] [CrossRef]
- Freude, B.; Masters, T.N.; Kostin, S.; Robicsek, F.; Schaper, J. Cardiomyocyte Apoptosis in Acute and Chronic Conditions. Basic Res. Cardiol. 1998. [Google Scholar] [CrossRef]
- Dries, J.L.; Kent, S.D.; Virag, J.A.I. Intramyocardial Administration of Chimeric EphrinA1-Fc Promotes Tissue Salvage Following Myocardial Infarction in Mice. J. Physiol. 2011. [Google Scholar] [CrossRef]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The Role of Autophagy in Cardiomyocytes in the Basal State and in Response to Hemodynamic Stress. Nat. Med. 2007. [Google Scholar] [CrossRef]
- Dorn, G.W.; Diwan, A. The Rationale for Cardiomyocyte Resuscitation in Myocardial Salvage. J. Mol. Med. 2008. [Google Scholar] [CrossRef]
- Porrello, E.R.; Delbridge, L.M.D. Cardiomyocyte Autophagy Is Regulated by Angiotensin II Type 1 and Type 2 Receptors. Autophagy 2009. [Google Scholar] [CrossRef]
- Fishbein, M.C.; Maclean, D.; Maroko, P.R. Experimental Myocardial Infarction in the Rat: Qualitative and Quantitative Changes during Pathologic Evolution. Am. J. Pathol. 1978. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Smith, C.W.; Entman, M.L. The Inflammatory Response in Myocardial Infarction. Cardiovasc. Res. 2002. [Google Scholar] [CrossRef]
- Virag, J.I.; Murry, C.E. Myofibroblast and Endothelial Cell Proliferation during Murine Myocardial Infarct Repair. Am. J. Pathol. 2003. [Google Scholar] [CrossRef]
- Dorn, G.W., II. Pharmacogenetic Profiling in the Treatment of Heart Disease. Transl. Res. 2009, 154, 295–302. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. The Immune System and Cardiac Repair. Pharmacol. Res. 2008. [Google Scholar] [CrossRef]
- Lambert, J.M.; Lopez, E.F.; Lindsey, M.L. Macrophage Roles Following Myocardial Infarction. Int. J. Cardiol. 2008. [Google Scholar] [CrossRef]
- Nah, D.Y.; Rhee, M.Y. The Inflammatory Response and Cardiac Repair after Myocardial Infarction. Korean Circ. J. 2009. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Fibroblasts and the Extracellular Matrix in Right Ventricular Disease. Cardiovasc. Res. 2017. [Google Scholar] [CrossRef]
- Wells, J.M.; Gaggar, A.; Blalock, J.E. MMP Generated Matrikines. Matrix Biol. 2015. [Google Scholar] [CrossRef]
- Turner, N.A. Inflammatory and Fibrotic Responses of Cardiac Fibroblasts to Myocardial Damage Associated Molecular Patterns (DAMPs). J. Mol. Cell Cardiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial Infarction. Circ. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shinde, A.V.; Humeres, C.; Frangogiannis, N.G. The Role of α-Smooth Muscle Actin in Fibroblast-Mediated Matrix Contraction and Remodeling. Biochim. Biophys. Acta Mol. Basis Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Katwadi, K.; Kwek, X.Y.; Ismail, N.I.; Chinda, K.; Ong, S.G.; Hausenloy, D.J. Non-Coding RNAs as Therapeutic Targets for Preventing Myocardial Ischemia-Reperfusion Injury. Expert Opin. Ther. Targets. 2018. [Google Scholar] [CrossRef] [PubMed]
- French, B.A.; Holmes, J.W. Implications of Scar Structure and Mechanics for Post-Infarction Cardiac Repair and Regeneration. Exp. Cell Res. 2019. [Google Scholar] [CrossRef]
- Wenk, J.F.; Klepach, D.; Lee, L.C.; Zhang, Z.; Ge, L.; Tseng, E.E.; Martin, A.; Kozerke, S.; Gorman, J.H.; Gorman, R.C.; et al. First Evidence of Depressed Contractility in the Border Zone of a Human Myocardial Infarction. Ann. Thorac. Surg. 2012. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Braunwald, E. Ventricular Enlargement Following Infarction Is a Modifiable Process. Am. J. Cardiol. 1991. [Google Scholar] [CrossRef]
- Gaudron, P.; Eilles, C.; Kugler, I.; Ertl, G. Progressive Left Ventricular Dysfunction and Remodeling after Myocardial Infarction. Potential Mechanisms and Early Predictors. Circulation 1993. [Google Scholar] [CrossRef]
- Goldstein, S.; Ali, A.S.; Sabbah, H. Ventricular Remodeling. Mechanisms and Prevention. Cardiol. Clin. 1998, 16, 623–632. [Google Scholar] [CrossRef]
- Holmes, J.W.; Borg, T.K.; Covell, J.W. Structure and Mechanics of Healing Myocardial Infarcts. Annu. Rev. Biomed. Eng. 2005. [Google Scholar] [CrossRef]
- Cirillo, M.; Arpesella, G. Rewind the Heart: A Novel Technique to Reset Heart Fibers’ Orientation in Surgery for Ischemic Cardiomyopathy. Med. Hypotheses 2008. [Google Scholar] [CrossRef] [PubMed]
- Bodor, G.S.; Porterfield, D.; Voss, E.M.; Smith, S.; Apple, F.S. Cardiac Troponin-I Is Not Expressed in Fetal and Healthy or Diseased Adult Human Skeletal Muscle Tissue. Clin. Chem. 1995, 41, 1710–1715. [Google Scholar] [PubMed]
- Chapelle, J.-P. Cardiac Troponin I and Troponin T: Recent Players in the Field of Myocardial Markers. Clin. Chem. Lab. Med. 1999, 37, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Braunwald, E.; Antman, E.M.; Beasley, J.W.; Califf, R.M.; Cheitlin, M.D.; Hochman, J.S.; Jones, R.H.; Kereiakes, D.; Kupersmith, J.; Levin, T.N.; et al. ACC/AHA Guideline Update for the Management of Patients with Unstable Angina and Non-ST-Segment Elevation Myocardial Infarction—2002: Summary Article: A Report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients with Unstable Angina). Circulation 2002, 106, 1893–1900. [Google Scholar]
- Nageh, T.; Sherwood, R.A.; Harris, B.M.; Byrne, J.A.; Thomas, M.R. Cardiac Troponin T and I and Creatine Kinase-MB as Markers of Myocardial Injury and Predictors of Outcome Following Percutaneous Coronary Intervention. Int. J. Cardiol. 2003, 92, 285–293. [Google Scholar] [CrossRef]
- Oyama, M.A.; Sisson, D.D. Cardiac Troponin-I Concentration in Dogs with Cardiac Disease. J. Vet. Intern. Med. 2004. [Google Scholar] [CrossRef]
- Jaffe, A.S. Use of Biomarkers in the Emergency Department and Chest Pain Unit. Cardiol. Clin. 2005. [Google Scholar] [CrossRef]
- Ranya, N.; Sweis, A.J. Acute Myocardial Infarction (MI). Available online: https://www.merckmanuals.com/professional/cardiovascular-disorders/coronary-artery-disease/acute-myocardial-infarction-mi#v27852674 (accessed on 3 February 2019).
- Månsson-Broberg, A.; Siddiqui, A.J.; Genander, M.; Grinnemo, K.H.; Hao, X.; Andersson, A.B.; Wärdell, E.; Sylvén, C.; Corbascio, M. Modulation of EphrinB2 Leads to Increased Angiogenesis in Ischemic Myocardium and Endothelial Cell Proliferation. Biochem. Biophys. Res. Commun. 2008. [Google Scholar] [CrossRef]
- Hirai, H.; Maru, Y.; Hagiwara, K.; Nishida, J.; Takaku, F. A Novel Putative Tyrosine Kinase Receptor Encoded by the Eph Gene. Science 1987. [Google Scholar] [CrossRef]
- Brückner, K.; Pasquale, E.B.; Klein, R. Tyrosine Phosphorylation of Transmembrane Ligands for Eph Receptors. Science 1997. [Google Scholar] [CrossRef]
- Mellitzer, G.; Xu, Q.; Wilkinson, D.G. Eph Receptors and Ephrins Restrict Cell Intermingling and Communication. Nature 1999. [Google Scholar] [CrossRef] [PubMed]
- Klein, R. Excitatory Eph Receptors and Adhesive Ephrin Ligands. Curr. Opin. Cell Biol. 2001. [Google Scholar] [CrossRef]
- Kullander, K.; Klein, R. Mechanisms and Functions of Eph and Ephrin Signalling. Nat. Rev. Mol. Cell Biol. 2002. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph Receptors and Ephrins in Cancer: Bidirectional Signalling and Beyond. Nat. Rev. Cancer 2010. [Google Scholar] [CrossRef]
- Zhou, R. The Eph Family Receptors and Ligands. Pharmacol. Ther. 1998. [Google Scholar] [CrossRef]
- Arvanitis, D.; Davy, A. Eph/Ephrin Signaling: Networks. Genes Dev. 2008. [Google Scholar] [CrossRef]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and Inhibition of the ICE/CED-3 Protease Necessary for Mammalian Apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef]
- Tewari, M.; Quan, L.T.; O’Rourke, K.; Desnoyers, S.; Zeng, Z.; Beidler, D.R.; Poirier, G.G.; Salvesen, G.S.; Dixit, V.M. Yama/CPP32 Beta, a Mammalian Homolog of CED-3, Is a CrmA-Inhibitable Protease That Cleaves the Death Substrate Poly(ADP-Ribose) Polymerase. Cell 1995, 81, 801–809. [Google Scholar] [CrossRef]
- Oliver, F.J.; de la Rubia, G.; Rolli, V.; Ruiz-Ruiz, M.C.; de Murcia, G.; Murcia, J.M. Importance of Poly(ADP-Ribose) Polymerase and Its Cleavage in Apoptosis. Lesson from an Uncleavable Mutant. J. Biol. Chem. 1998, 273, 33533–33539. [Google Scholar] [CrossRef]
- Doong, H.; Vrailas, A.; Kohn, E.C. What’s in the “BAG”?—A Functional Domain Analysis of the BAG-Family Proteins. Cancer Lett. 2002, 188, 25–32. [Google Scholar] [CrossRef]
- Townsend, P.A.; Cutress, R.I.; Carroll, C.J.; Lawrence, K.M.; Scarabelli, T.M.; Packham, G.; Stephanou, A.; Latchman, D.S. BAG-1 Proteins Protect Cardiac Myocytes from Simulated Ischemia/Reperfusion-Induced Apoptosis via an Alternate Mechanism of Cell Survival Independent of the Proteasome. J. Biol. Chem. 2004, 279, 20723–20728. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Rosenzweig, A. Convergent Signal Transduction Pathways Controlling Cardiomyocyte Survival and Function: The Role of PI 3-Kinase and Akt. J. Mol. Cell. Cardiol. 2005, 38, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Latronico, M.V.G.; Costinean, S.; Lavitrano, M.L.; Peschle, C.; Condorelli, G. Regulation of Cell Size and Contractile Function by AKT in Cardiomyocytes. Ann. N. Y. Acad. Sci. 2004, 1015, 250–260. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.; Yellon, D. Survival Kinases in Ischemic Preconditioning and Postconditioning. Cardiovasc. Res. 2006, 70, 240–253. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Murphy, A.N.; Brown, J.H. Akt Mediated Mitochondrial Protection in the Heart: Metabolic and Survival Pathways to the Rescue. J. Bioenerg. Biomembr. 2009, 41, 169–180. [Google Scholar] [CrossRef]
- Przyklenk, K.; Undyala, V.V.R.; Wider, J.; Sala-Mercado, J.A.; Gottlieb, R.A.; Mentzer, R.M. Acute Induction of Autophagy as a Novel Strategy for Cardioprotection: Getting to the Heart of the Matter. Autophagy 2011, 7, 432–433. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.N.; Kyoi, S.; Hariharan, N.; Takagi, H.; Sadoshima, J.; Gottlieb, R.A. Chapter 16 Novel Methods for Measuring Cardiac Autophagy In Vivo. Methods Enzymol. 2009, 453, 325–342. [Google Scholar] [CrossRef]
- Gustafsson, Å.B.; Gottlieb, R.A. Recycle or Die: The Role of Autophagy in Cardioprotection. J. Mol. Cell. Cardiol. 2008, 44, 654–661. [Google Scholar] [CrossRef]
- Wang, L.; Cheng, X.; Huang, C.; Huang, B.; Liang, Q. Rapamycin Protects Cardiomyocytes against Anoxia/Reoxygenation Injury by Inducing Autophagy through the PI3k/Akt Pathway. J. Huazhong Univ. Sci. Technol. Med. Sci. 2015, 35, 10–15. [Google Scholar] [CrossRef]
- Kadowaki, M.; Karim, M.R. Chapter 13 Cytosolic LC3 Ratio as a Quantitative Index of Macroautophagy. Methods Enzymol. 2009, 452, 199–213. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.; Shao, H.; Marks, R.M.; Polverini, P.J.; Dixit, V.M. Role of B61, the Ligand for the Eck Receptor Tyrosine Kinase, in TNF-α-Induced Angiogenesis. Science 1995. [Google Scholar] [CrossRef]
- Cheng, N.; Brantley, D.M.; Chen, J. The Ephrins and Eph Receptors in Angiogenesis. Cytokine Growth Factor Rev. 2002. [Google Scholar] [CrossRef]
- Moon, J.J.; Lee, S.H.; West, J.L. Synthetic Biomimetic Hydrogels Incorporated with Ephrin-A1 for Therapeutic Angiogenesis. Biomacromolecules 2007. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Pasqualini, R.; Lindberg, R.A.; Kain, R.; Freeman, A.L.; Pasquale, E.B. The Ephrin-A1 Ligand and Its Receptor, EphA2, Are Expressed during Tumor Neovascularization. Oncogene 2000. [Google Scholar] [CrossRef] [PubMed]
- Brantley-Sieders, D.M.; Fang, W.B.; Hwang, Y.; Hicks, D.; Chen, J. Ephrin-A1 Facilitates Mammary Tumor Metastasis through an Angiogenesis-Dependent Mechanism Mediated by EphA Receptor and Vascular Endothelial Growth Factor in Mice. Cancer Res. 2006. [Google Scholar] [CrossRef]
- Wykosky, J.; Palma, E.; Gibo, D.M.; Ringler, S.; Turner, C.P.; Debinski, W. Soluble Monomeric EphrinA1 Is Released from Tumor Cells and Is a Functional Ligand for the EphA2 Receptor. Oncogene 2008. [Google Scholar] [CrossRef]
- Shaut, C.A.; Saneyoshi, C.; Morgan, E.A.; Knosp, W.M.; Sexton, D.R.; Stadler, H.S. HOXA13 Directly Regulates EphA6 and EphA7 Expression in the Genital Tubercle Vascular Endothelia. Dev. Dyn. 2007. [Google Scholar] [CrossRef]
- Ivanov, A.; Romanovsky, A. Putative Dual Role of Ephrin-Eph Receptor Interactions in Inflammation. IUBMB Life 2006. [Google Scholar] [CrossRef]
- Lefer, D.J.; Bolli, R. Development of an NIH Consortium for PreclinicAl AssESsment of CARdioprotective Therapies (CAESAR): A Paradigm Shift in Studies of Infarct Size Limitation. J. Cardiovasc. Pharmacol. Ther. 2011. [Google Scholar] [CrossRef]
- O’Neal, W.T.; Griffin, W.F.; Dries-Devlin, J.L.; Kent, S.D.; Chen, J.; Willis, M.S.; Virag, J.A.I. Ephrin–Eph Signaling as a Potential Therapeutic Target for the Treatment of Myocardial Infarction. Med. Hypotheses 2013, 80, 738–744. [Google Scholar] [CrossRef] [PubMed]
- Spath, N.B.; Mills, N.L.; Cruden, N.L. Novel Cardioprotective and Regenerative Therapies in Acute Myocardial Infarction: A Review of Recent and Ongoing Clinical Trials. Future Cardiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kloner, R.A. Current State of Clinical Translation of Cardioprotective Agents for Acute Myocardial Infarction. Circ. Res. 2013. [Google Scholar] [CrossRef] [PubMed]
- Hale, S.L.; Kloner, R.A. Combination Therapy for Maximal Myocardial Infarct Size Reduction. Heart Dis. 2010, 3, 351–356. [Google Scholar] [CrossRef]
- Marzilli, M.; Luis, I.; Gowdak, H.W.; Hausenloy, D.J.; Lopaschuk, G.D.; Marber, C.M.; Laboratoires, L.; Brigitte, S.; Chevret, O. Reducing Myocardial Infarct Size: Myth or Reality? Heart Metab. 2016, 70, 2–3. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Grouping | % of All Deaths |
|---|---|
| Global | 15.96266586 |
| Low SDI | 8.554976163 |
| Middle SDI | 16.63194299 |
| High SDI | 16.59093766 |
| Mexico | 14.2476225 |
| United States | 18.65732133 |
| Southeast Asia | 13.05553801 |
| South Asia | 15.47558271 |
| Central Asia | 33.26288153 |
| Indonesia | 14.29116844 |
| Oceania | 13.89558582 |
| Australasia | 16.54619353 |
| Latin America and Caribbean | 14.05392127 |
| Tropical Latin America | 13.07831696 |
| Southern Latin America | 14.71951694 |
| Eastern Europe | 34.96210128 |
| Central Europe | 26.35190592 |
| Western Europe | 15.93160107 |
| United Kingdom | 14.30616833 |
| Sweden | 19.92329938 |
| Central Sub-Saharan Africa | 5.589027664 |
| Southern Sub-Saharan Africa | 6.811094508 |
| Sub-Saharan Africa | 4.942820494 |
| North Africa and Middle East | 24.78925909 |
| Western Sub-Saharan Africa | 4.443263763 |
| Eastern Sub-Saharan Africa | 4.873600872 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horton, J.L.; Virag, J. Use of Multifactorial Treatments to Address the Challenge of Translating Experimental Myocardial Infarct Reduction Strategies. Int. J. Mol. Sci. 2019, 20, 1449. https://doi.org/10.3390/ijms20061449
Horton JL, Virag J. Use of Multifactorial Treatments to Address the Challenge of Translating Experimental Myocardial Infarct Reduction Strategies. International Journal of Molecular Sciences. 2019; 20(6):1449. https://doi.org/10.3390/ijms20061449
Chicago/Turabian StyleHorton, Julie L., and Jitka Virag. 2019. "Use of Multifactorial Treatments to Address the Challenge of Translating Experimental Myocardial Infarct Reduction Strategies" International Journal of Molecular Sciences 20, no. 6: 1449. https://doi.org/10.3390/ijms20061449
APA StyleHorton, J. L., & Virag, J. (2019). Use of Multifactorial Treatments to Address the Challenge of Translating Experimental Myocardial Infarct Reduction Strategies. International Journal of Molecular Sciences, 20(6), 1449. https://doi.org/10.3390/ijms20061449