Characterization of the High-Affinity Drug Ligand Binding Site of Mouse Recombinant TSPO


 , and
, and
Abstract
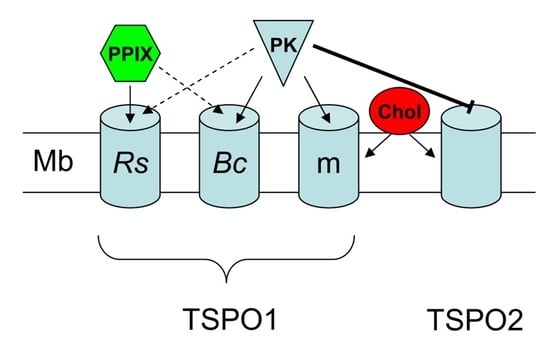
:
1. Introduction
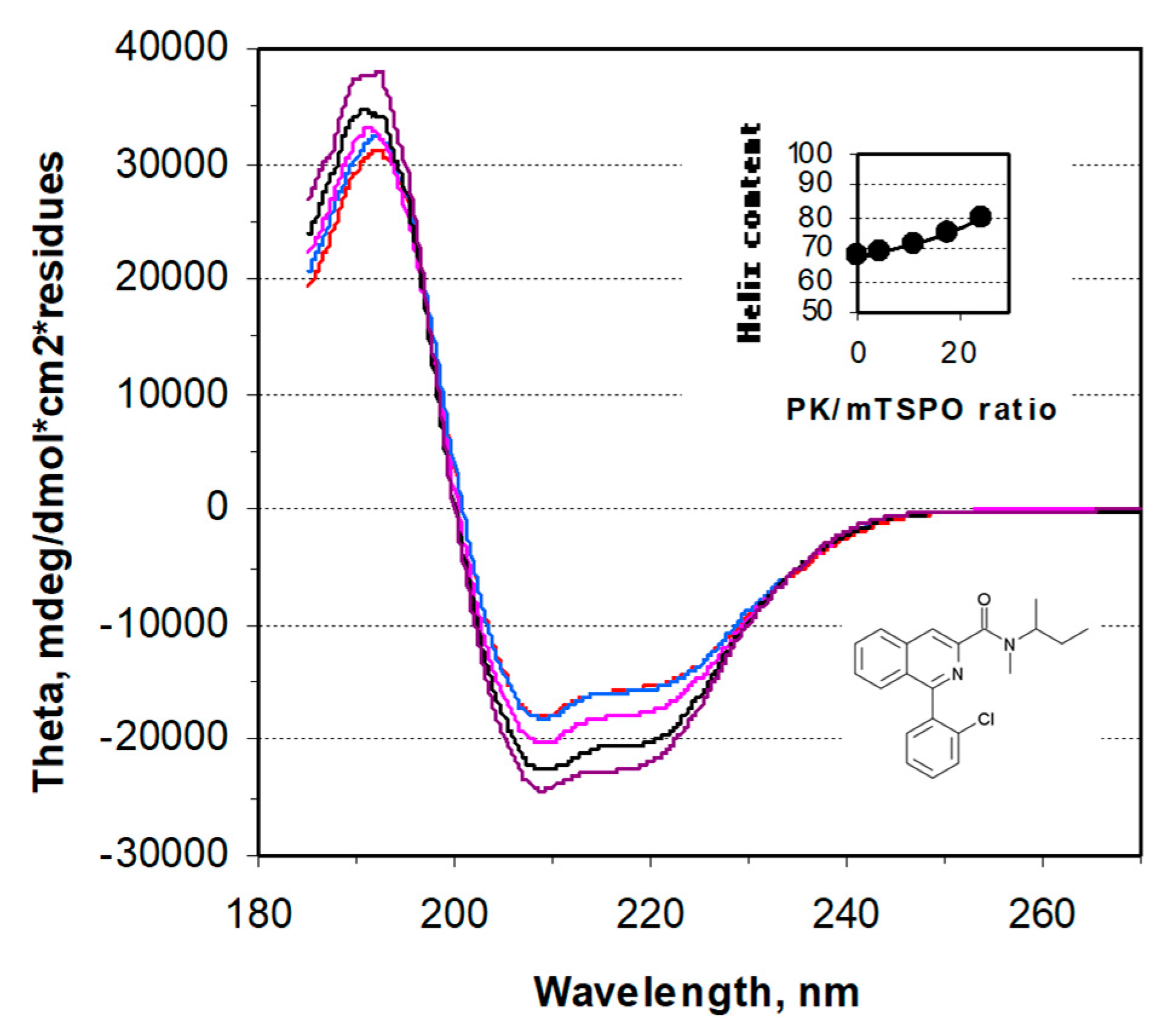
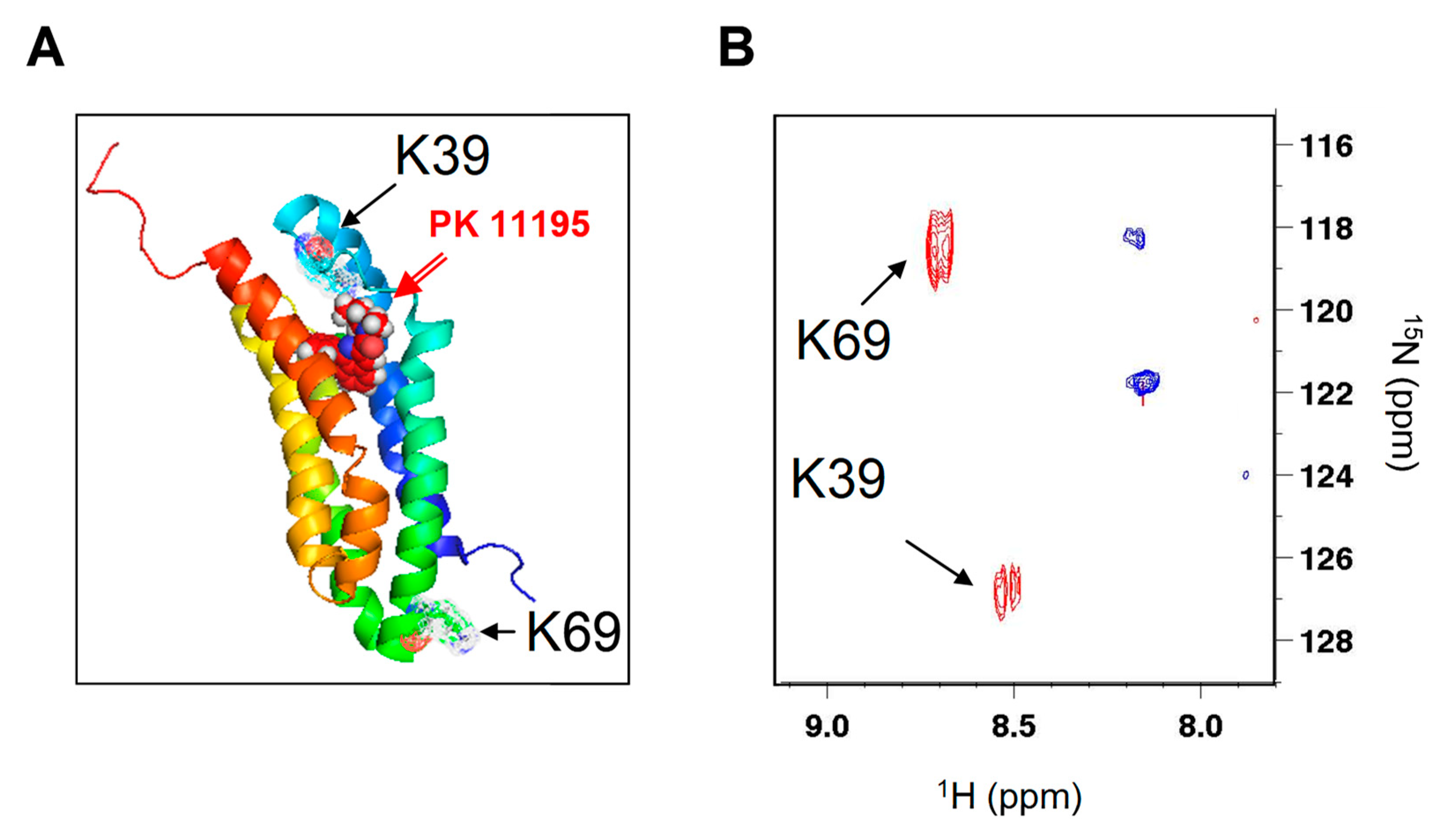
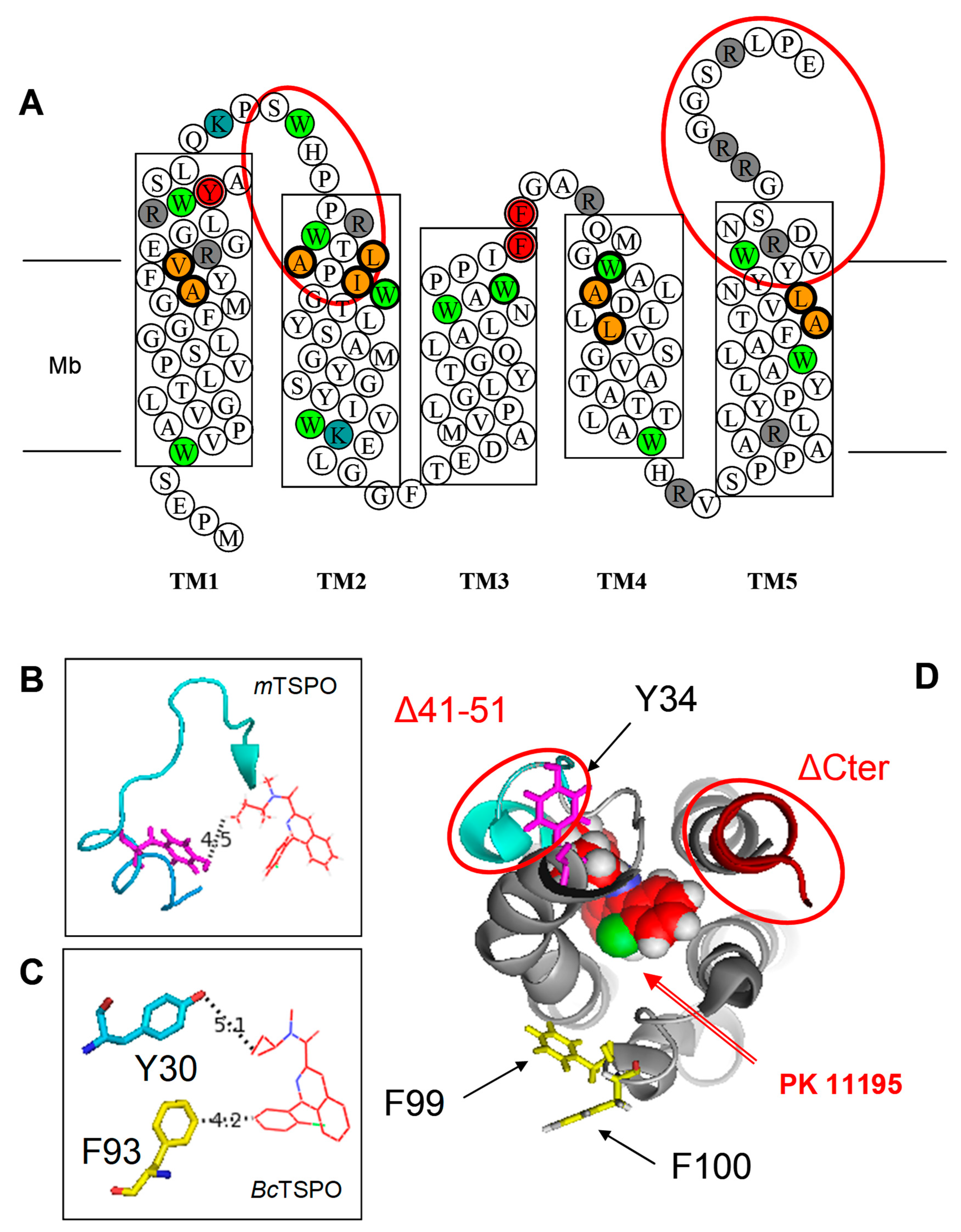
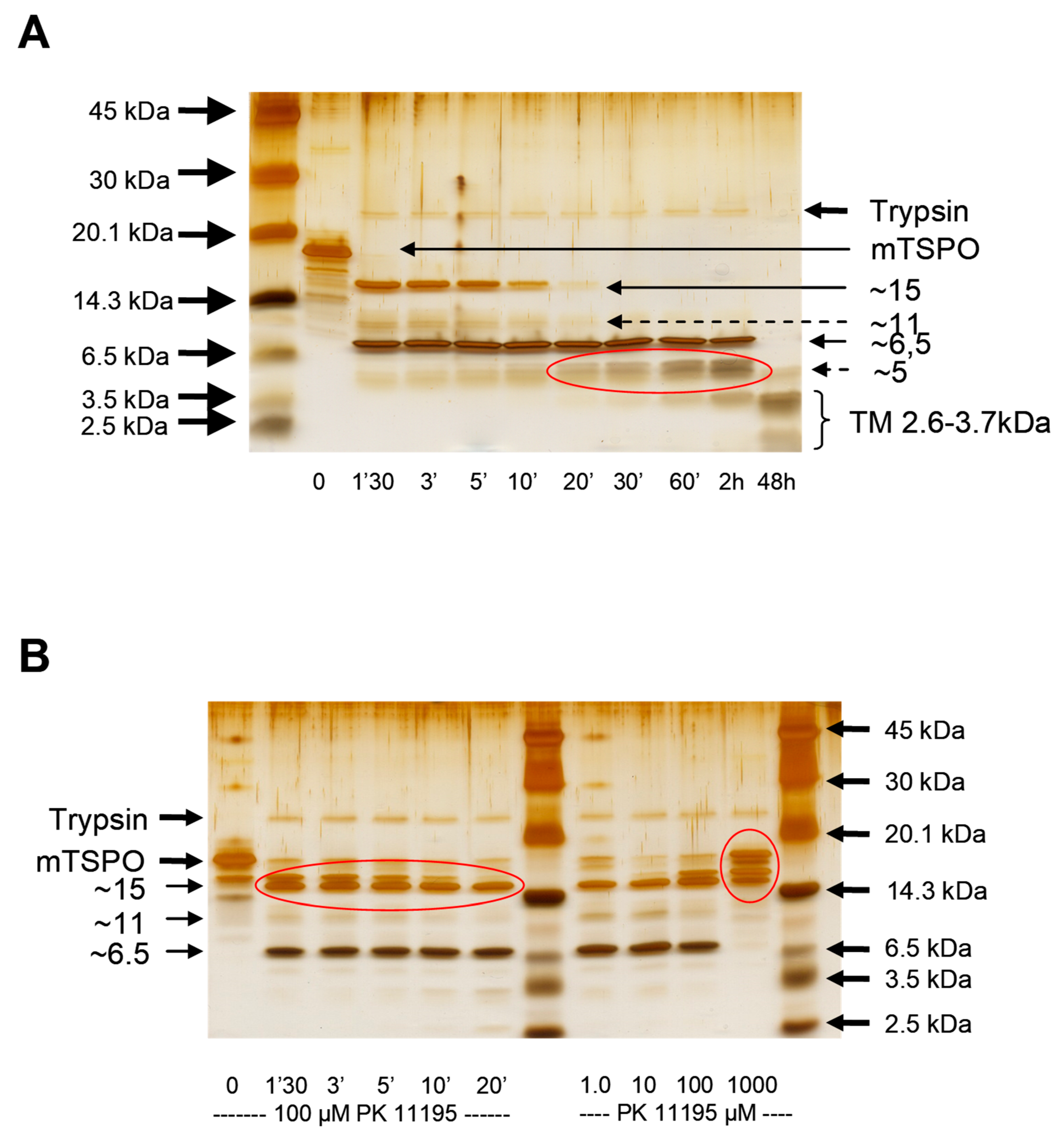
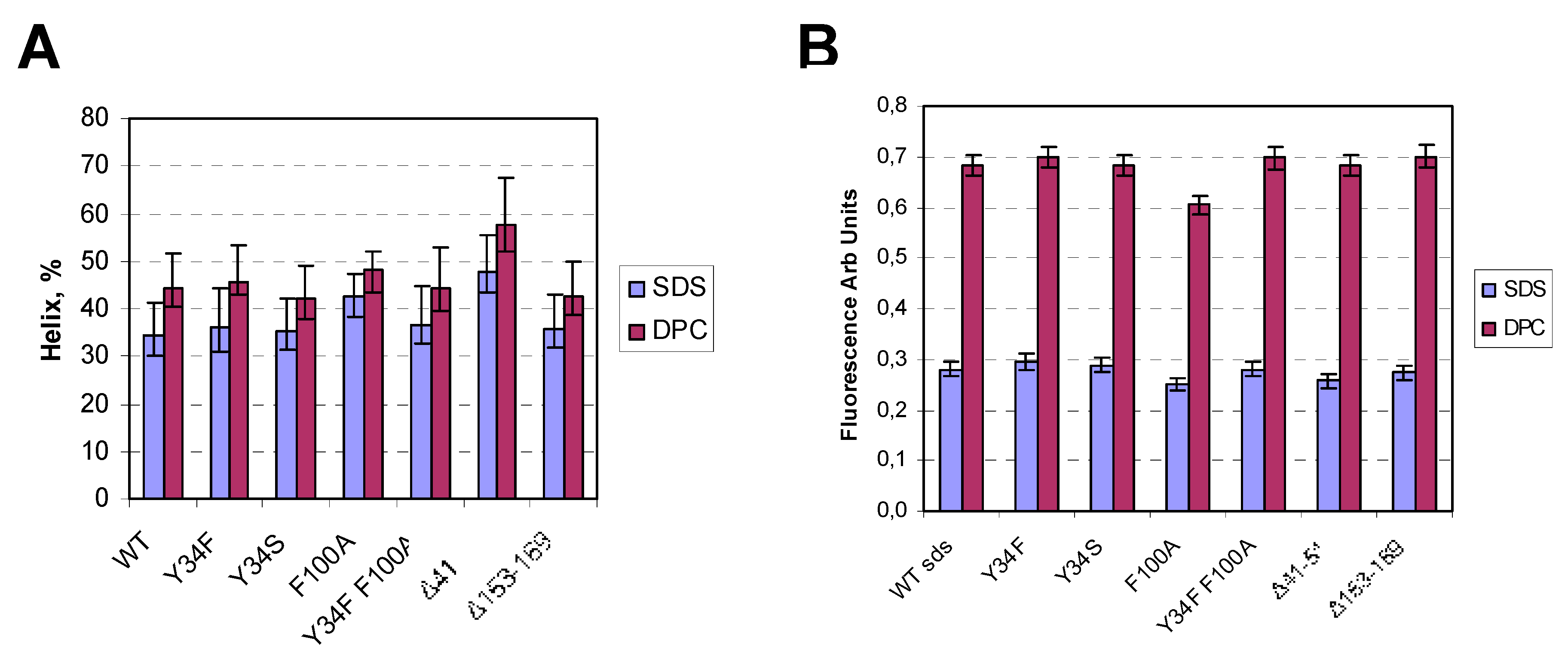
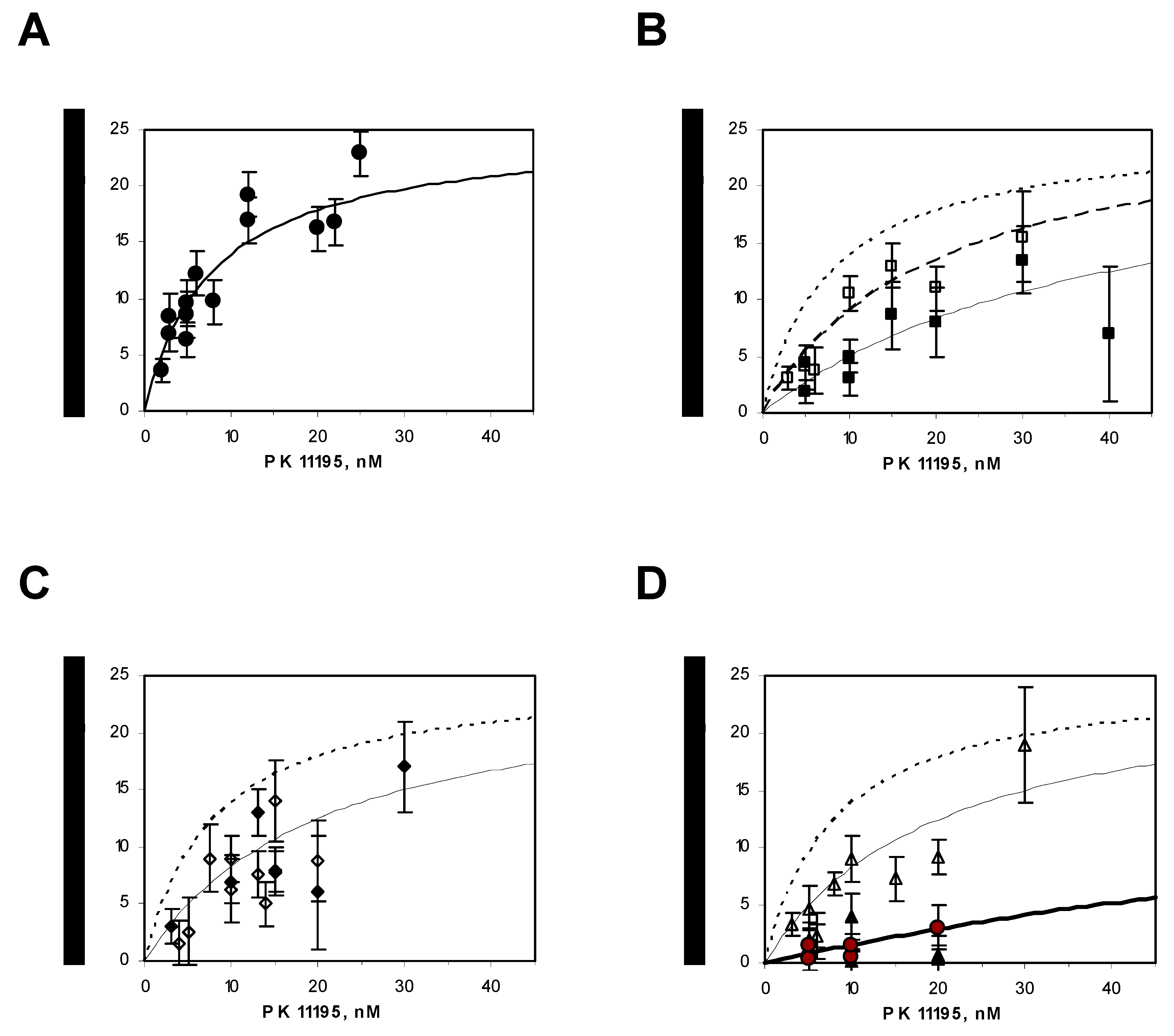
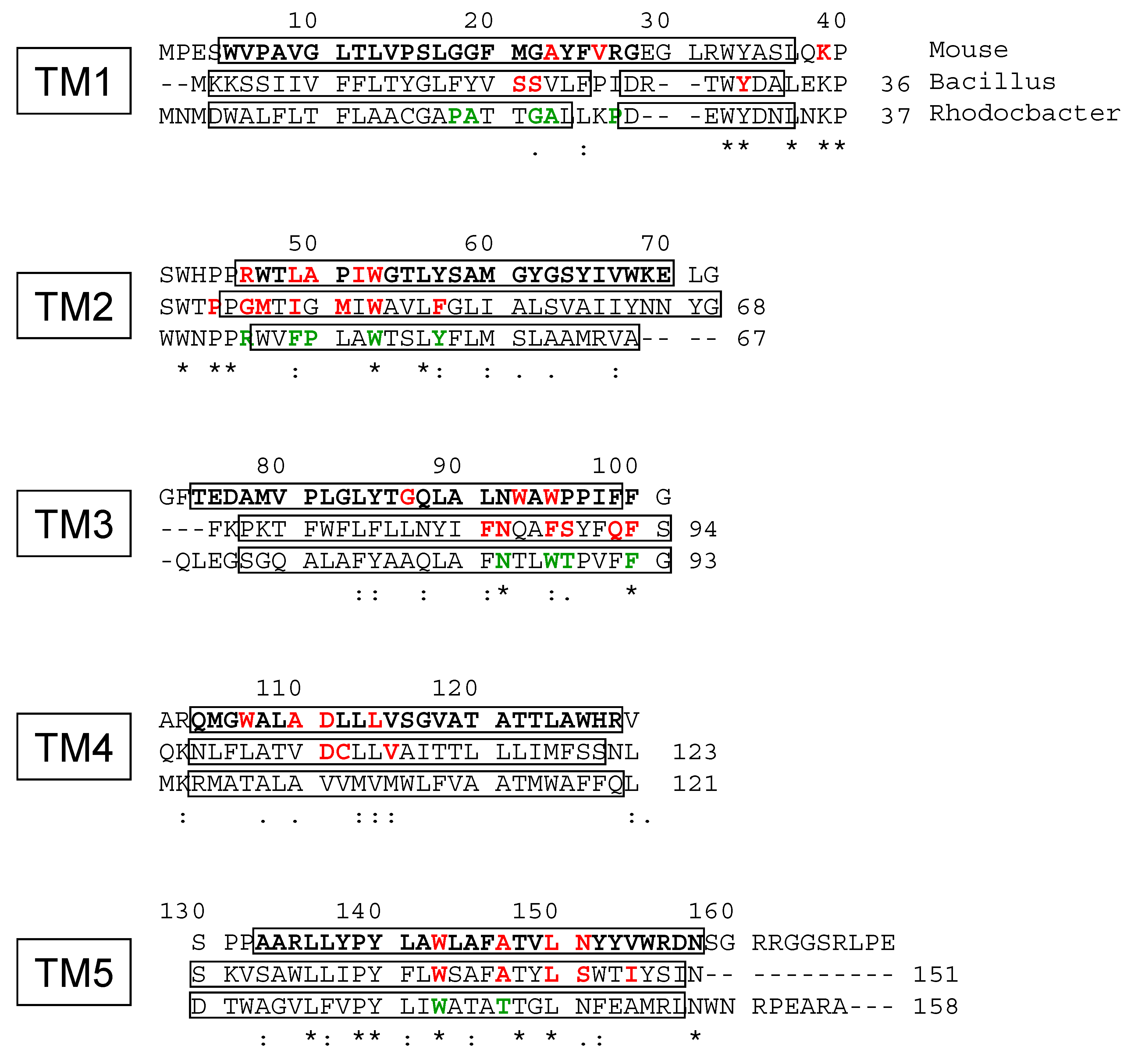
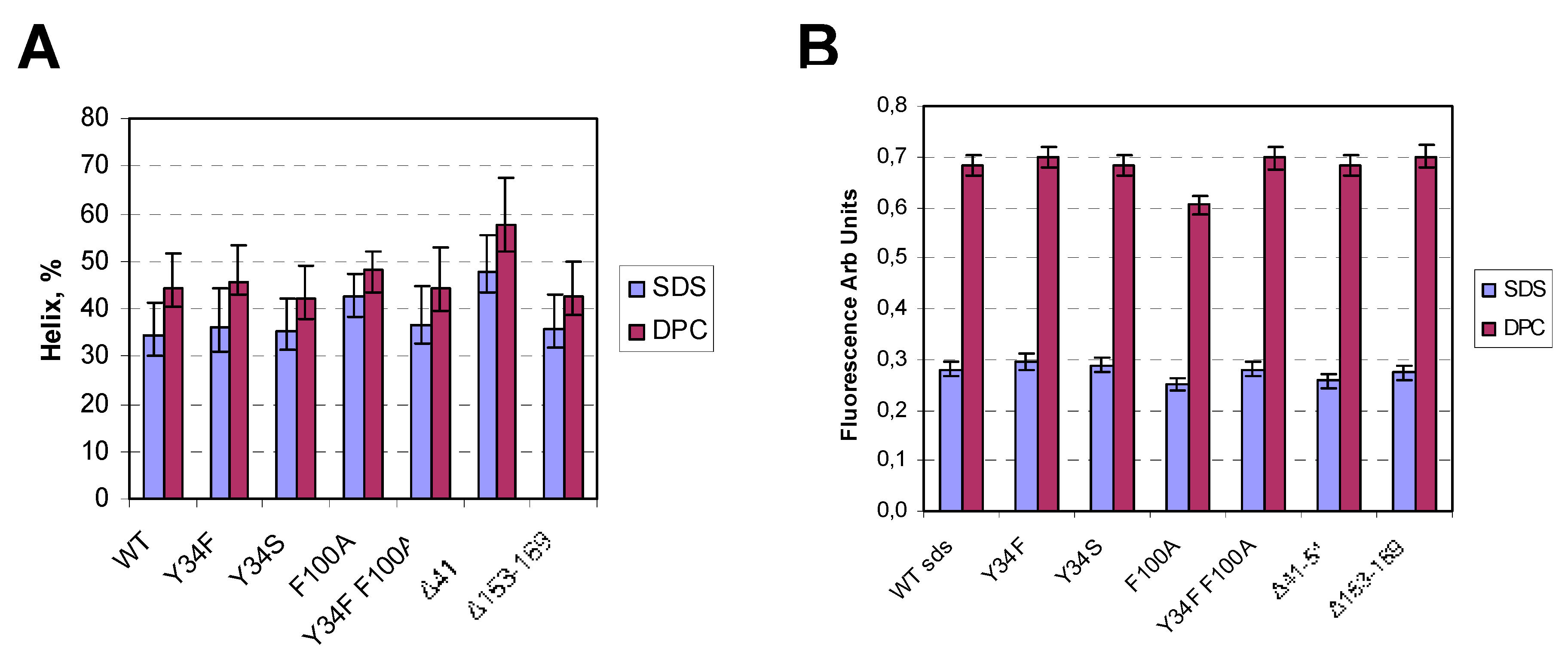
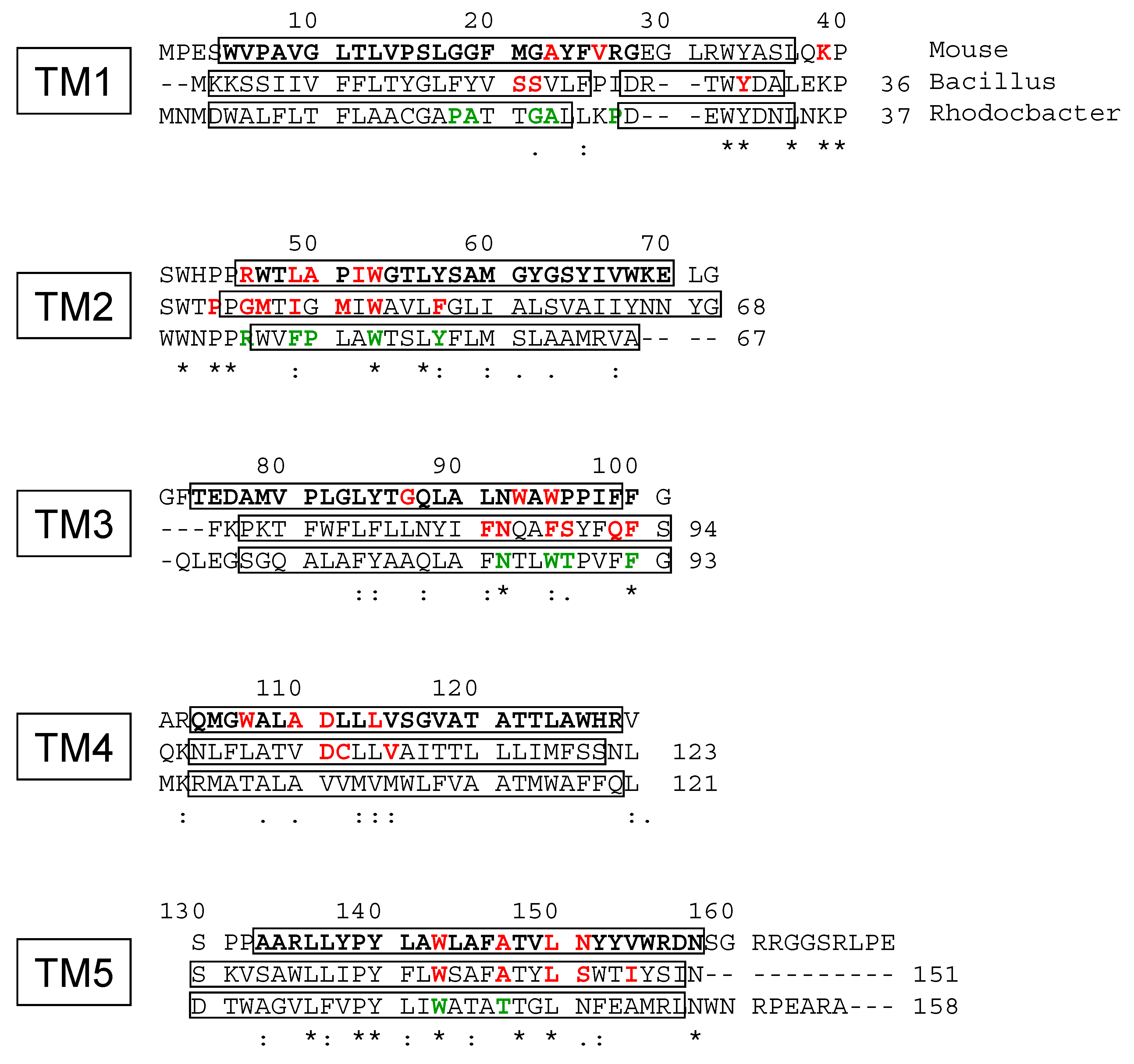
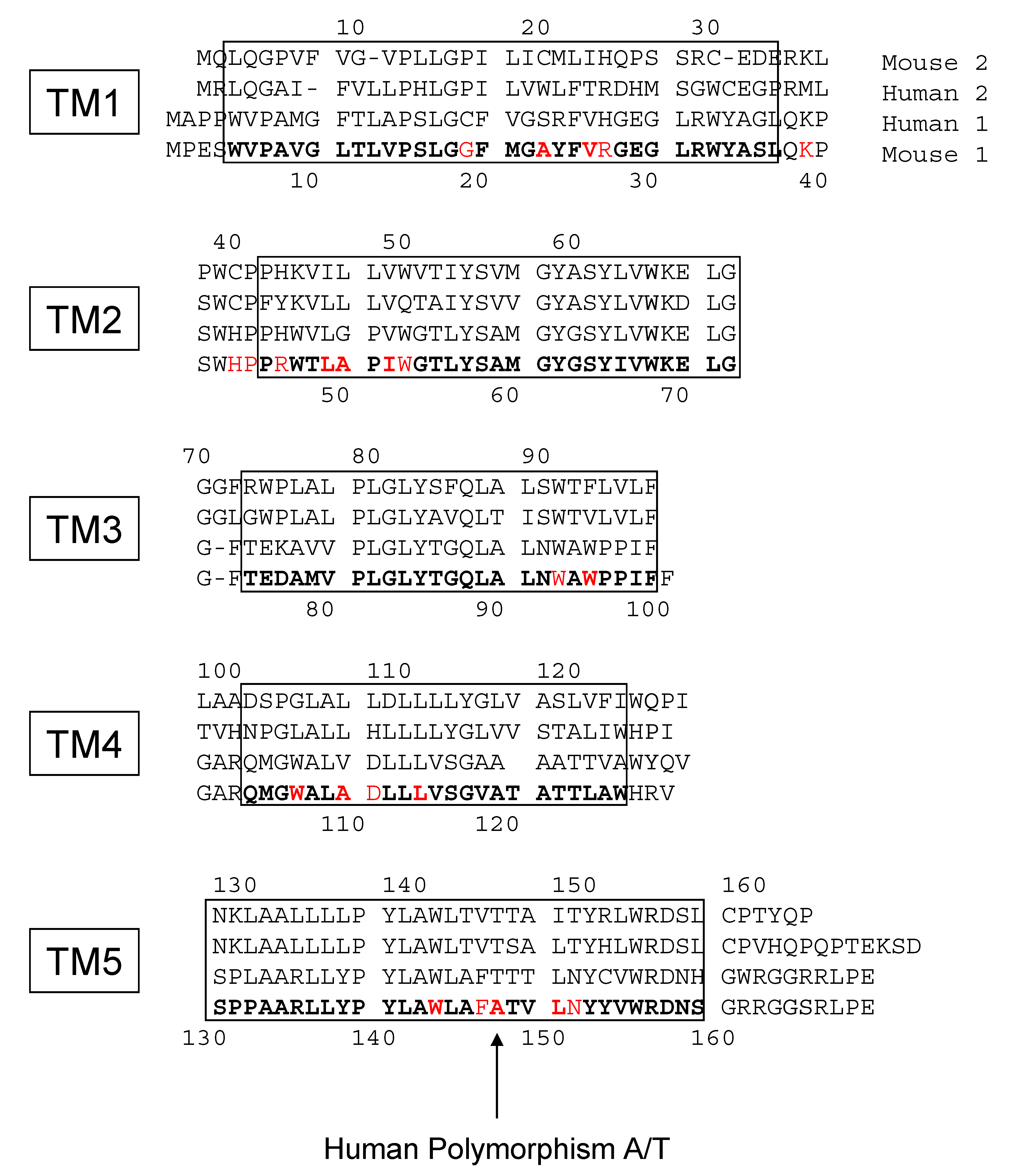
2. Results
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Recombinant Mouse TSPO
4.2. NMR Experiments
4.3. Trypsin Digestion
4.4. Site-Directed Mutagenesis
4.5. CD Spectroscopy
4.6. Fluorescence Spectroscopy
4.7. Reconstitution of TSPO in Liposomes
4.8. Radioligand-Binding Assays
4.9. Molecular Graphics and Distances Calculation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PK 11195 | N-butan-2-yl-1-(2-chlorophenyl)-N-methylisoquinoline-3-carboxamide |
| TSPO | Translocator protein |
| mTSPO | mouse TSPO |
| BcTSPO | Bacillus cereus TSPO |
| RsTSPO | Rhodobacter sphaeroides TSPO |
| AtTSPO | Arabidopsis thaliana TSPO |
| HSQC | Heteronuclear Single Quantum Coherence |
| PPIX | Protoporphyrin IX |
| NMR | Nuclear Magnetic Resonance |
| DPC | Dodecylphosphocholine |
| CD | Circular Dichroism |
| TM | Transmembrane |
| WT | Wild Type |
References
- Papadopoulos, V.; Baraldi, M.; Guilarte, T.R.; Knudsen, T.B.; Lacapère, J.-J.; Lindemann, P.; Norenberg, M.D.; Nutt, D.; Weizman, A.; Zhang, M.R.; et al. Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 2006, 27, 402–409. [Google Scholar] [CrossRef]
- Fan, J.; Lindemann, P.; Feuilloley, M.G.J.; Papadopoulos, V. Structural and functional evolution of the translocator protein (18kDa). Cur. Mol. Med. 2012, 12, 369–386. [Google Scholar]
- Lacapère, J.-J.; Papadopoulos, V. Peripheral-type benzodiazepine receptor: Structure and function of a cholesterol-binding protein in steroid and bile acid biosynthesis. Steroids 2003, 68, 569–585. [Google Scholar]
- Dupont, A.-C.; Largeau, B.; Santiago Ribeiro, M.J.; Guilloteau, D.; Tronel, C.; Arlicot, N. Translocator protein-18kDa (TSPO) positron emission tomography (PET) imaging and its clinical impact in neurodegenerative diseases. Int. J. Mol. Sci. 2017, 18, 785. [Google Scholar] [CrossRef] [PubMed]
- Denora, N.; Natile, G. An update view of the translocator protein (TSPO). Int. J. Mol. Sci. 2017, 18, 2640. [Google Scholar] [CrossRef]
- Zeng, J.; Guareschi, R.; Damre, M.; Cao, R.; Kless, A.; Neumaier, B.; Bauer, A.; Giorgetti, E.; Carloni, P.; Rosetti, G. Structural prediction of the dimeric form of the mammalian translocator membrane protein TSPO: A key target for brain diagnostics. Int. J. Mol. Sci. 2018, 19, 2588. [Google Scholar] [CrossRef]
- Owen, D.R.; Yeo, A.J.; Gunn, R.N.; Song, K.; Wadsworth, G.; Lewis, A.; Rhodes, C.; Pulford, D.J.; Bennacef, I.; Parker, C.A.; et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J. Cereb. Blood Flow Metab. 2012, 32, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Scarf, A.M.; Luus, C.; Da Pozzo, E.; Selleri, S.; Guarino, C.; Martini, C.; Ittner, L.M.; Kasiou, M. Evidence for complex binding profiles and species differences at translocator protein (TSPO) (18kDa). Curr. Mol. Med. 2012, 12, 488–493. [Google Scholar]
- Jaremko, L.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Structure of the mitochondrial translocator protein in complex with a diagnostic ligand. Science 2014, 343, 1363–1366. [Google Scholar] [CrossRef]
- Murail, S.; Robert, J.C.; Coïc, Y.M.; Neumann, J.M.; Ostuni, M.A.; Yao, Z.X.; Papadopoulos, V.; Jamin, N.; Lacapère, J.-J. Secondary and tertiary structures of the transmembrane domains of the translocator protein TSPO determined by NMR. Stabilization of the TSPO tertiary fold upon ligand binding. Biochim. Biophys. Acta 2008, 1778, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Farges, R.; Joseph-Liauzun, E.; Shire, D.; Caput, D.; Le Fur, G.; Loison, G.; Ferrara, P. Molecular basis for the different binding properties of benzodiazepines to human and bovine peripheral-type benzodiazepine receptor. FEBS 1993, 335, 305–308. [Google Scholar] [CrossRef]
- Farges, R.; Joseph-Liauzun, E.; Shire, D.; Caput, D.; Le Fur, G.; Ferrara, P. Site-directed mutagenesis of the peripheral benzodiazepine receptor: Identification of amino acids implicated in the binding site of Ro5-4864. Mol. Pharmacol. 1994, 46, 1160–1167. [Google Scholar]
- Li, H.; Papadopoulos, V. Peripheral-type benzodiazepine receptor function in cholesterol transport. Identification of a putative cholesterol recognition/interaction amino acid sequence and consensus pattern. Endocrinology 1998, 139, 4991–4997. [Google Scholar] [CrossRef] [PubMed]
- Yeliseev, A.A.; Kaplan, S. TspO of Rhodobacter spaheroides. J. Biol. Chem. 2000, 275, 5657–5667. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liu, J.; Zheng, Y.; Garavito, M.; Ferguson-Miller, S. Crystal structures of translocator protein (TSPO) and mutant mimic of a human polymorphism. Science 2015, 347, 555–558. [Google Scholar] [CrossRef]
- Guo, Y.; Kalathur, R.C.; Liu, Q.; Kloss, B.; Bruni, R.; Ginter, C.; Kloppmann, E.; Rost, B.; Hendrickson, W.A. Structure and activity of tryptophan-rich TSPO proteins. Science 2015, 347, 551–555. [Google Scholar] [CrossRef]
- Fan, J.; Rone, M.B.; Papadopoulos, V. Translocator protein 2 is involved in cholesterol redistribution during erythropoiesis. J. Biol. Chem. 2009, 284, 30484–30497. [Google Scholar] [CrossRef]
- Jaremko, M.; Jaremko, Ł.; Giller, K.; Becker, S.; Zweckstetter, M. Structural integrity of the A147T polymorph of mammalian TSPO. Chem. Bio. Chem. 2015, 17, 1598–1601. [Google Scholar] [CrossRef]
- Lacapere, J.-J.; Iatmanen-Harbi, S.; Senicourt, L.; Lequin, O.; Tekely, P.; Purusottam, R.N.; Hellwig, P.; Kriegel, S.; Ravaud, S.; Juillan-Binard, C.; et al. Structural studies of TSPO, a mitochondrial membrane protein. In Membrane Proteins Production for Structural Studies; Muss-Veteau, I., Ed.; Springer: New York, NY, USA; Heidelberg/Berlin, Germany; Dordrecht, The Netherlands; London, UK, 2014; pp. 393–421. ISBN 978-1-4939-0661-1. [Google Scholar]
- Woogler, N.; Bournazos, A.; Sophocleous, R.A.; Evesson, F.J.; Lek, A.; Driemer, N.; Sutton, R.B.; Cooper, S.T. Limited proteolysis as a tool to probe the tertiary conformation of dysferlin and structural consequences of patient missense variant L344P. J. Biol. Chem. 2017, 292, 18577–18591. [Google Scholar]
- Ponniah, G.; Nowak, C.; Kita, A.; Cheng, G.; Kori, Y.; Liu, H. Conformational changes of recombinant monoclonal antibodies by limited proteolytic digestion, stable isotope labeling, and liquid chromatography-mass spectroscopy. Anal. Biochem. 2016, 497, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jaremko, L.; Jaremko, M.; Giller, K.; Becker, S.; Zweckstetter, M. Conformational flexibility in the transmembrane protein TSPO. Chem. Eur. J. 2015, 21, 16555–16563. [Google Scholar] [CrossRef]
- Lacapère, J.-J.; Delavoie, F.; Li, H.; Péranzi, G.; Maccario, J.; Papadopoulos, V.; Vidic, B. Structural and functional study of reconstituted peripheral benzodiazepine receptor. Biochem. Biophys. Res. Commun. 2001, 284, 536–541. [Google Scholar] [CrossRef]
- Robert, J.-C.; Lacapere, J.-J. Bacterial overexpressed membrane proteins: An example, the TSPO. Methods Mol. Biol. 2010, 654, 29–45. [Google Scholar]
- Jamin, N.; Neumann, J.-M.; Ostuni, M.A.; Vu, T.K.; Yao, Z.X.; Murail, S.; Robert, J.-C.; Giatzakis, C.; Papadopoulos, V.; Lacapere, J.-J. Characterization of the cholesterol recognition amino acid consensus sequence of the peripheral-type benzodiazepine receptor. Mol. Endocrinol. 2005, 19, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Korkhov, V.M.; Sachse, C.; Short, J.M.; Tate, C.G. Three-dimensional structure of TspO by electron cryomicroscopy of helical crystals. Structure 2010, 18, 677–687. [Google Scholar] [CrossRef]
- Li, F.; Xia, Y.; Meiler, J.; Ferguson-Miller, S. Characterization and modeling of the oligomeric state and ligand binding behavior of purified translocator protein 18kDa from Rhodobacter sphaeroides. Biochemistry 2013, 52, 5884–5899. [Google Scholar] [CrossRef] [PubMed]
- Ostuni, M.A.; Harbi-Iatmamen, S.; Teboul, D.; Robert, J.-C.; Lacapere, J.-J. Characterization of membrane protein preparations: Measurement of detergent content and ligand binding after proteoliposomes reconstitution. Methods Mol. Biol. 2010, 654, 3–18. [Google Scholar] [PubMed]
- Rigaud, J.-L.; Levy, D. Reconstitution of membrane proteins into liposomes. Methods Enzymol. 2003, 372, 65–86. [Google Scholar]
- Owen, D.R.; Fan, J.; Campioli, E.; Venugopal, S.; Midzak, A.; Daly, E.; Harlay, A.; Issop, L.; Libri, V.; Kalogiannopoulou, D.; et al. TSPO mutations in rats and human polymorphism impair the rate of steroid synthesis. Biochem. J. 2017, 474, 3985–3999. [Google Scholar] [CrossRef]
- Milenkovic, V.M.; Bader, S.; Sudria-Lopez, D.; Siebert, R.; Brandl, C.; Nothdurfter, C.; Weber, B.H.F.; Rupprecht, R.; Wetzel, C.H. Effects of genetic variants in the TSPO gene on protein structure and stability. PLoS ONE 2018, 13, e0195627. [Google Scholar] [CrossRef]
- Lindemann, P.; Koch, A.; Degenhardt, B.; Haus, G.; Grimm, B.; Papadopouilos, V. A novel Arabidopsis thaliana protein is a functional peripheral-type benzodiazepine receptor. Plant Cell Physiol. 2004, 45, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Kuraukas, V.; Hessel, A.; Ma, P.; Lunetti, P.; Weinhäupl, K.; Imbert, L.; Brutscher, B.; King, M.S.; Sounier, R.; Dolce, V.; et al. How Detergent Impacts Membrane Proteins: Atomic-Level Views of Mitochondrial Carriers in Dodecylphosphocholine. J. Phys. Chem. Lett. 2018, 9, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Pebay-Peyroula, E.; Dahout-Gonzalez, C.; Kahn, R.; Trézéguet, V.; Lauquin, G.J.; Brandolin, G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003, 426, 39–44. [Google Scholar] [CrossRef]
- Toyoshima, C.; Nakasako, M.; Nomura, H.; Ogawa, H. Crystal structure of the calcium pump of sarcoplasmic reticulum at 2.6 A resolution. Nature 2000, 405, 647–655. [Google Scholar] [CrossRef]
- Stokes, D.L.; Delavoie, F.; Rice, W.J.; Champeil, P.; McIntosh, D.B.; Lacapère, J.-J. Structural studies of a stabilized phosphoenezyme intermediate of Ca-ATPase. J. Biol. Chem. 2005, 280, 18063–18072. [Google Scholar] [CrossRef] [PubMed]
- Ruppecht, R.; Papdopoulos, V.; Rammes, G.; Baghai, T.C.; Fan, J.; Akula, N.; Groyer, G.; Adams, D.; Schumacher, M. Translocator protein (18kDa) (TSPO) as a therapeutic target for neurological and psychiatric disorders. Nat. Rev. Drug. Discov. 2010, 9, 971–988. [Google Scholar] [CrossRef]
- Jaipura, G.; Leonov, A.; Giller, K.; Vasa, S.K.; Jaremko, T.; Jaremko, M.; Linser, R.; Becker, S.; Zweckstetter, M. Cholesterol-mediated allosteric regulation of the mitochondrial translocator protein structure. Nat. Commun. 2017, 8, 14893. [Google Scholar] [CrossRef]
- Jaipura, G.; Giller, K.; Leonov, A.; Becker, S.; Zweckstetter, M. Insight into cholesterol/membrane intercations using paramagnetic solid-state NMR. Chem. Eur. J. 2018, 24, 1–7. [Google Scholar]
- Senicourt, L.; Iatmanen-Harbi, S.; Hattab, C.; Ostuni, M.A.; Giraud, M.-F.; Lacapere, J.-J. Recombinant overexpression of mammalian TSPO isoforms 1 and 2. Methods Mol. Biol. 2017, 1635, 1–25. [Google Scholar] [PubMed]
- Landrieu, I.; Hassan, S.; Sauty, M.; Dewitte, F.; Wieruszeski, J.-M.; Inzé, D.; De Veylder, L.; Lippens, G. Characterization of the Arabidopsis thaliana Arath;CDC25 dual-specificity tyrosine phosphatase. Biochem. Biophys. Res. Commun. 2004, 322, 734–739. [Google Scholar] [CrossRef]
- O’Grady, C.; Rempel, B.L.; Sokaribo, A.; Nokhrin, S.; Dmitriev, O.Y. One-step amino acid selective isotope labeling of proteins in prototrophic Escherichia coli strains. Anal. Biochem. 2012, 426, 126–128. [Google Scholar] [CrossRef]
- Switzer, R.C.; Merril, C.R.; Shifrin, S. A highly sensitive silver stain for detecting proteins and peptides in polyacrylamide gels. Anal. Biochem. 1979, 98, 231–237. [Google Scholar] [CrossRef]
- Schägger, H.; von Jagow, G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef]
- Jamin, N.; Lacapère, J.-J. Circular dichroism as a tool for controlling membrane protein folding or structural modifications. In Biophysical Analysis of Membrane Proteins. Investigating Structure and Function; Pebey-Peyroula, E., Ed.; Wiley-VCH Press: Weinheim, Germany, 2007; pp. 243–258. [Google Scholar]
- Sreerama, N.; Woody, R.W. Estimation of protein secondary structure from circular dichroism spectra: Comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal. Biochem. 2000, 287, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Teboul, D.; Beaufils, S.; Taveau, J.-C.; Iatmanen-Harbi, S.; Renault, A.; Venien-Bryan, C.; Vie, V.; Lacapere, J.-J. Mouse TSPO in a lipid environment interacting with a functionalized monolayer. Biochim. Biophys. Acta 2012, 654, 3–18. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMol Molecular Graphics System; DeLano Scientific LLC: Palo Alto, CA, USA, 2008; Available online: http://www.pymol.org (accessed on 20 January 2019).
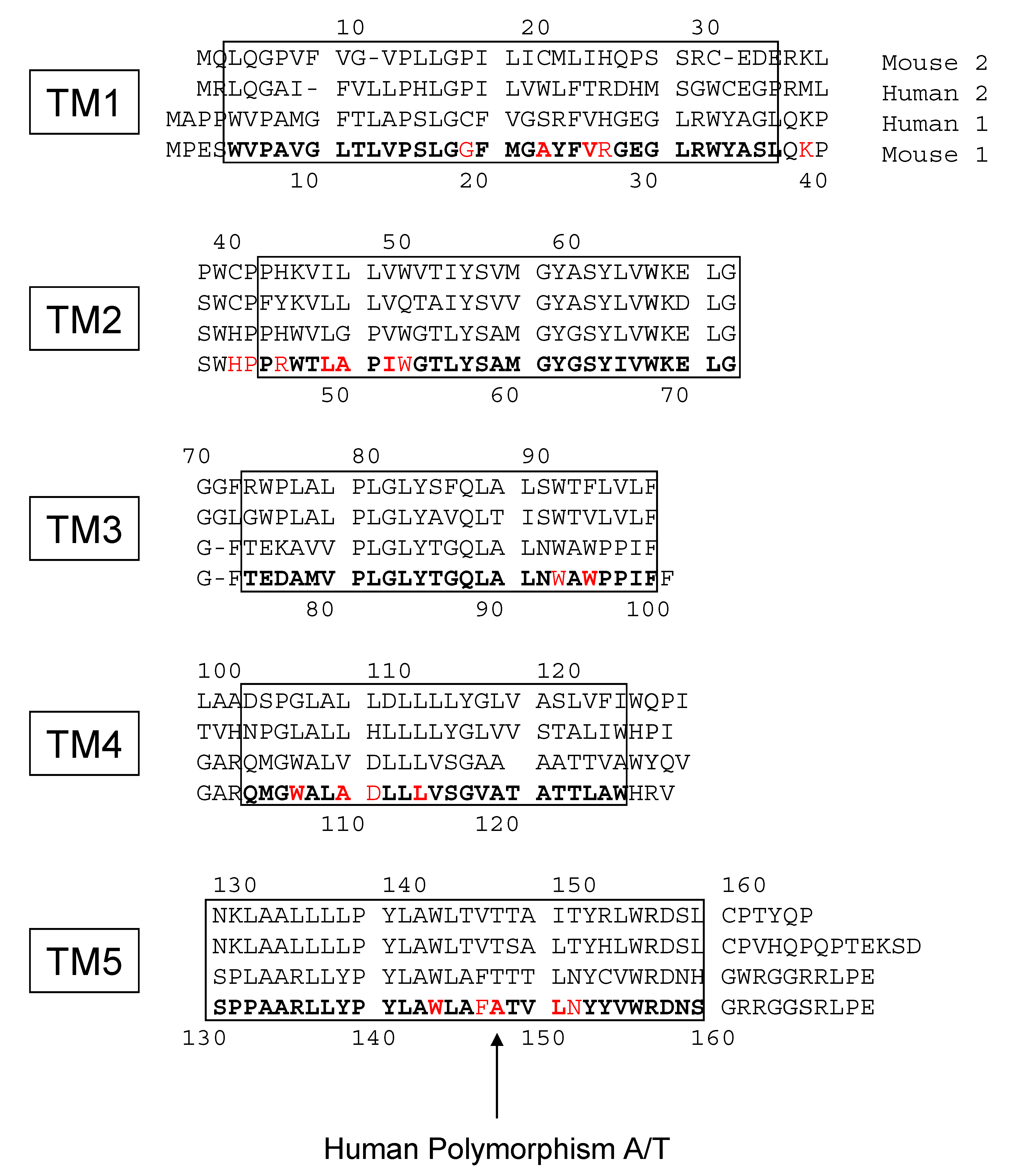

{kind=link}
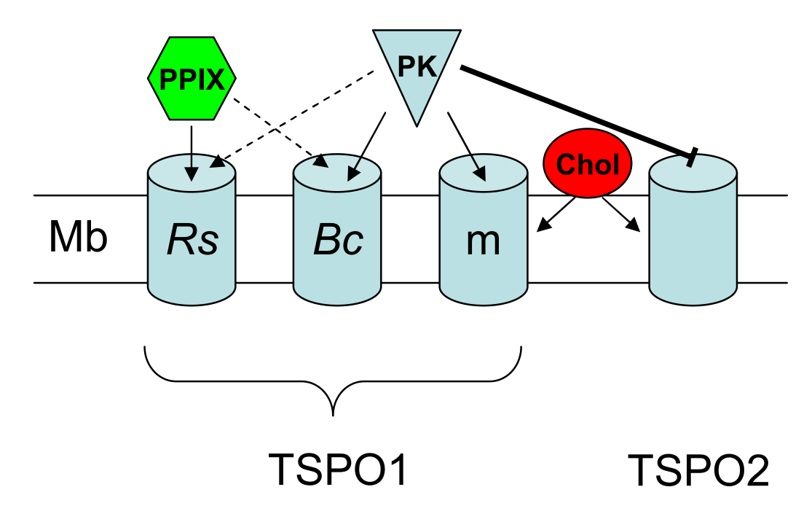
{kind=link}
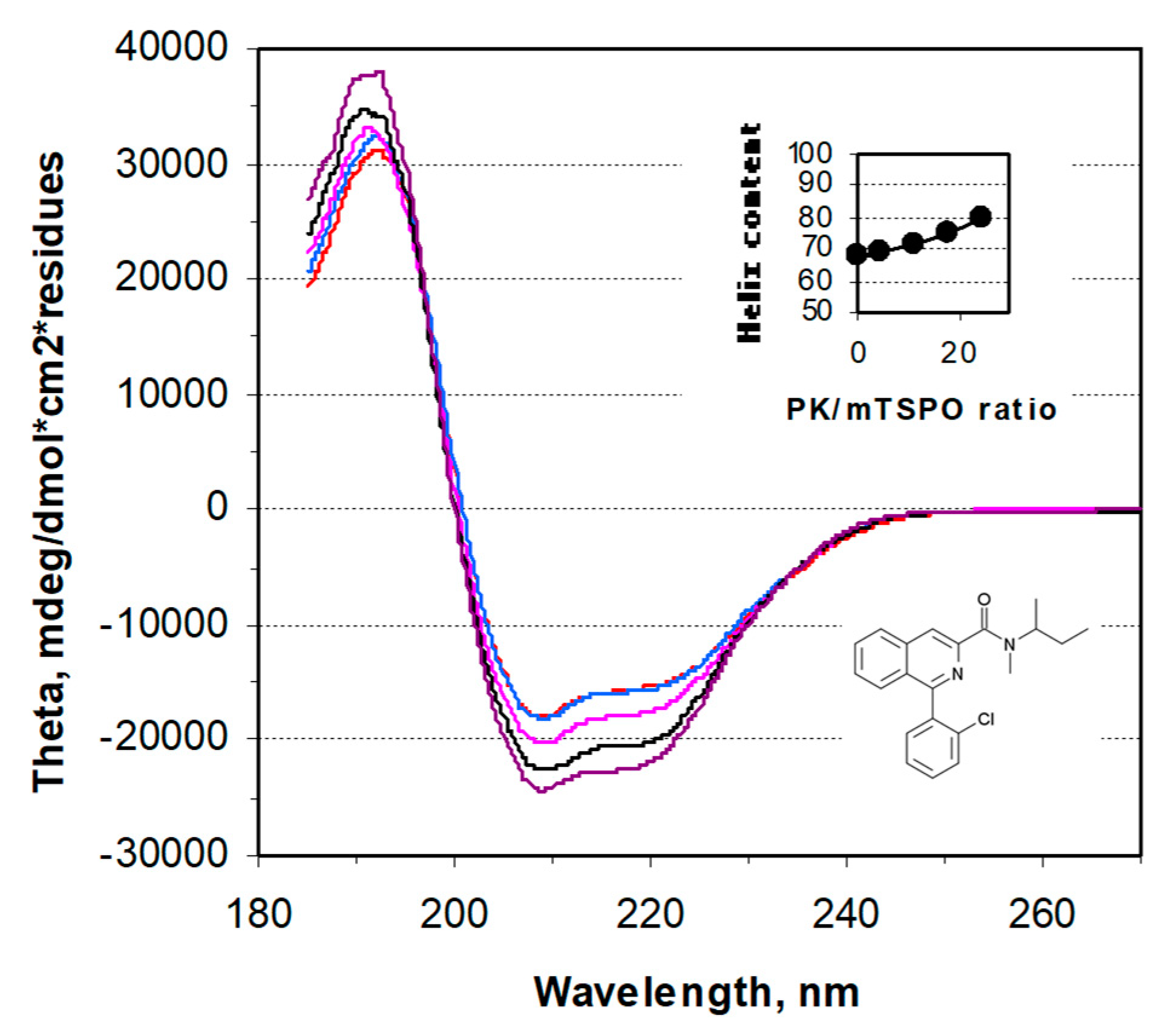
{kind=link}
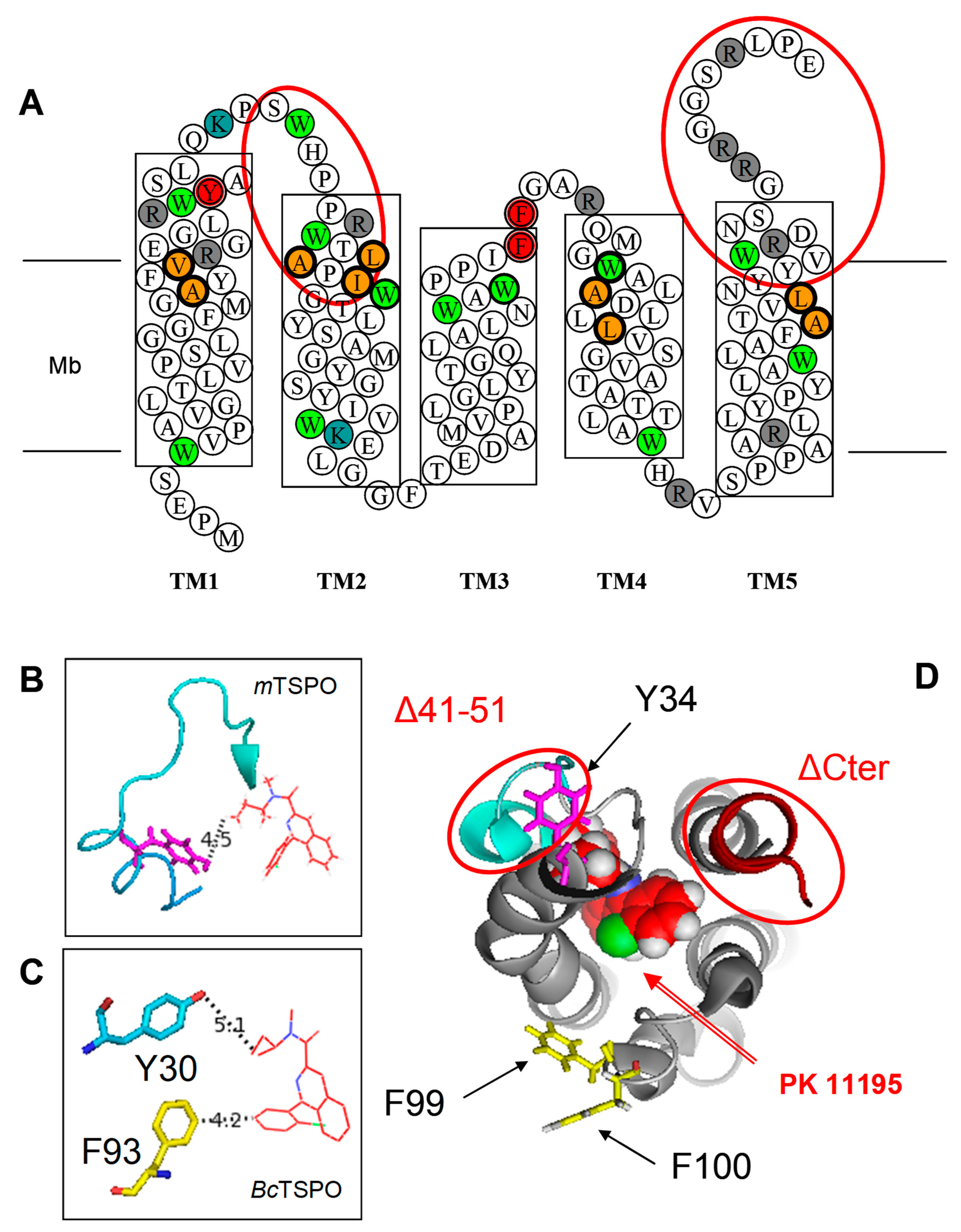{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Kd (nM) | Bmax (nmol/mg) |
|---|---|---|
| WT | 8 ± 3 | 25 ± 3 |
| Y34S | 40 ± 7 | 25 ± 5 |
| Y34F | 20 ± 3 | 27 ± 3 |
| Δ41–50 | 20 ± 8 | 25 ± 8 |
| ΔC-ter (153–169) | 20 ± 5 | 30 ± 10 |
| F100A | 20 ± 5 | 25 ± 7 |
| Y34F/F100A | 150 ± 20 | 25 ± 10 |
| Y34F/F99A | 150 ± 20 | 25 ± 10 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iatmanen-Harbi, S.; Senicourt, l.; Papadopoulos, V.; Lequin, O.; Lacapere, J.-J. Characterization of the High-Affinity Drug Ligand Binding Site of Mouse Recombinant TSPO. Int. J. Mol. Sci. 2019, 20, 1444. https://doi.org/10.3390/ijms20061444
Iatmanen-Harbi S, Senicourt l, Papadopoulos V, Lequin O, Lacapere J-J. Characterization of the High-Affinity Drug Ligand Binding Site of Mouse Recombinant TSPO. International Journal of Molecular Sciences. 2019; 20(6):1444. https://doi.org/10.3390/ijms20061444
Chicago/Turabian StyleIatmanen-Harbi, Soria, lucile Senicourt, Vassilios Papadopoulos, Olivier Lequin, and Jean-Jacques Lacapere. 2019. "Characterization of the High-Affinity Drug Ligand Binding Site of Mouse Recombinant TSPO" International Journal of Molecular Sciences 20, no. 6: 1444. https://doi.org/10.3390/ijms20061444
APA StyleIatmanen-Harbi, S., Senicourt, l., Papadopoulos, V., Lequin, O., & Lacapere, J.-J. (2019). Characterization of the High-Affinity Drug Ligand Binding Site of Mouse Recombinant TSPO. International Journal of Molecular Sciences, 20(6), 1444. https://doi.org/10.3390/ijms20061444