Differential Regulation of Type I and Type III Interferon Signaling


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
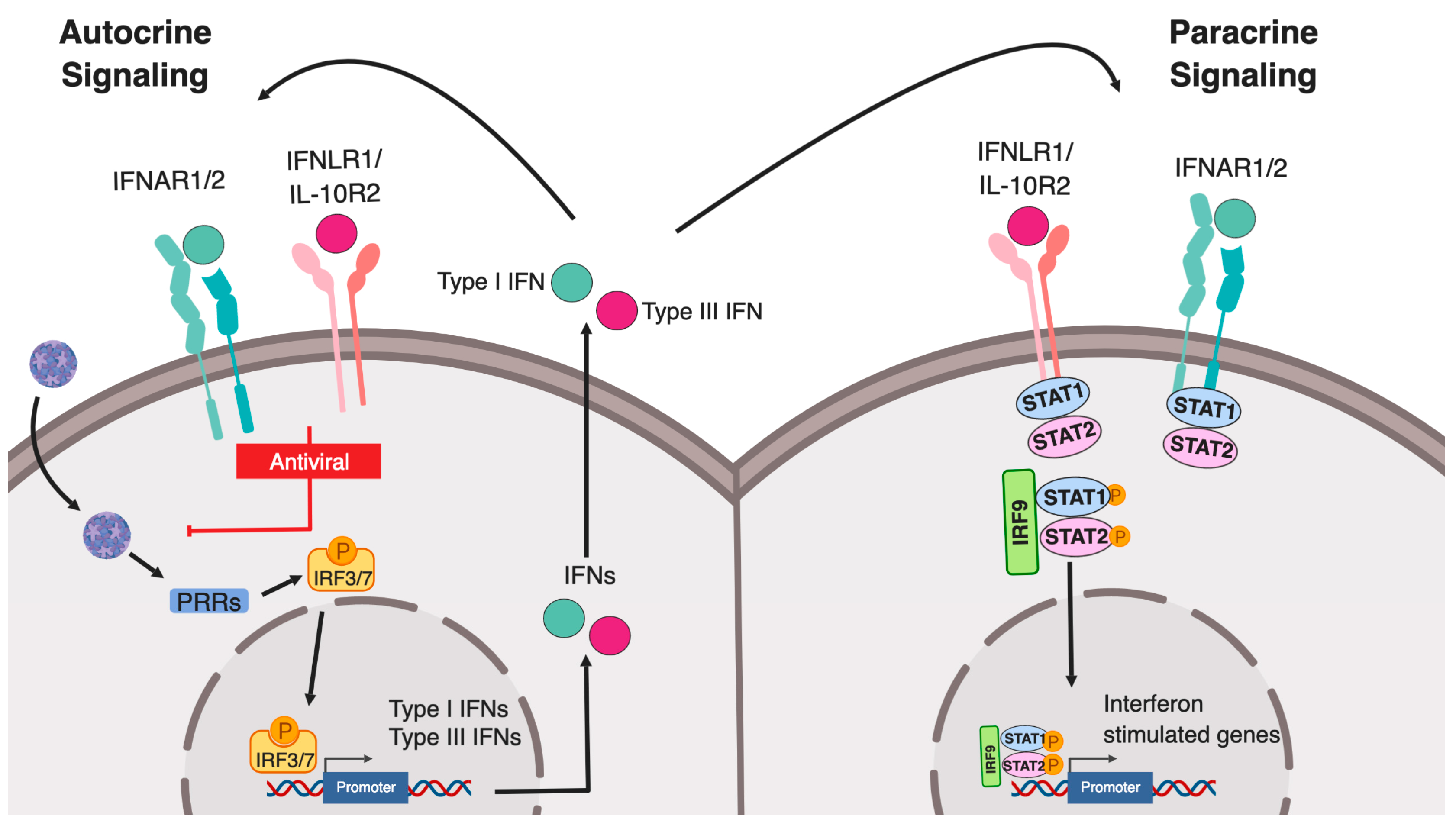
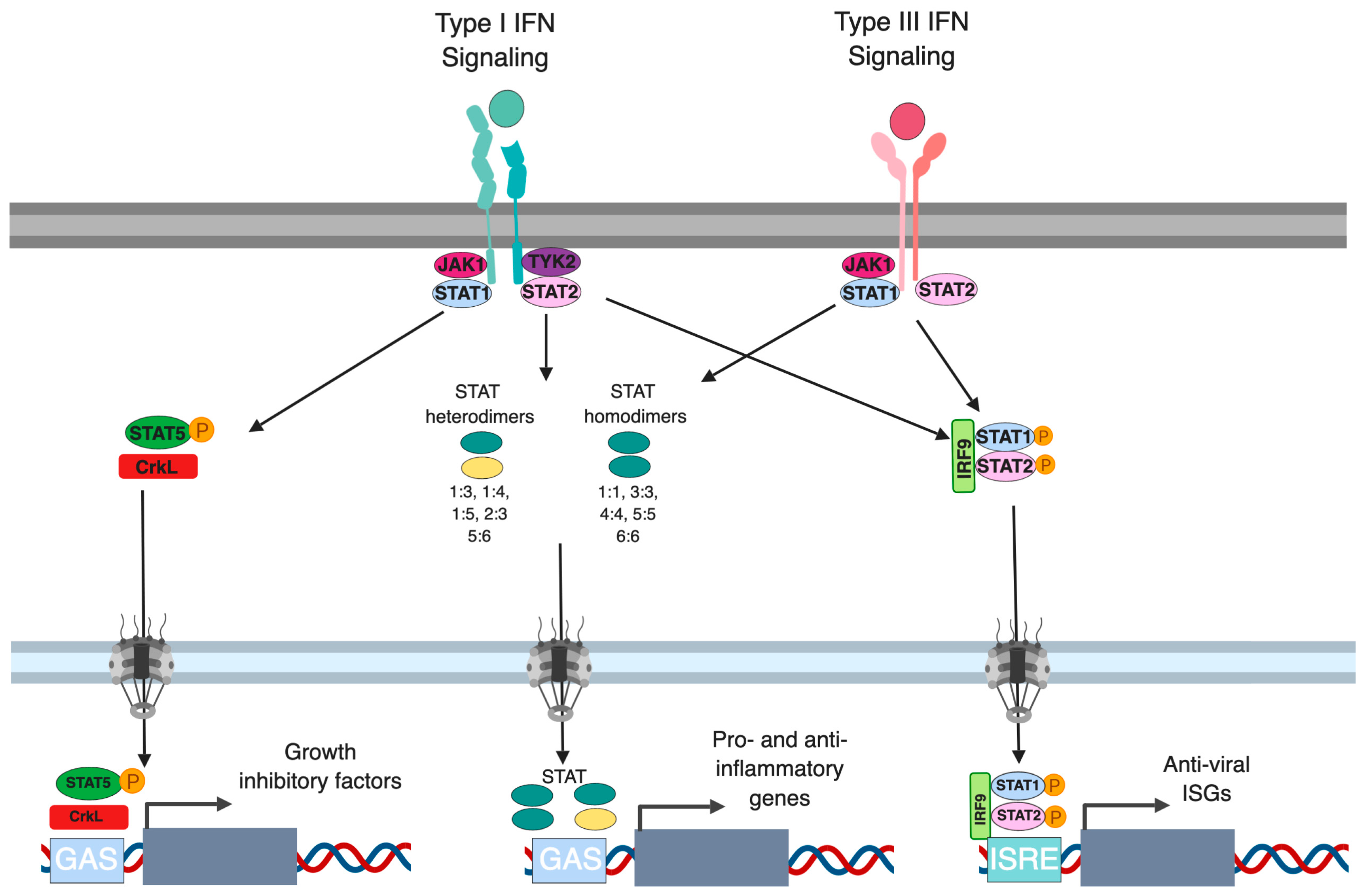
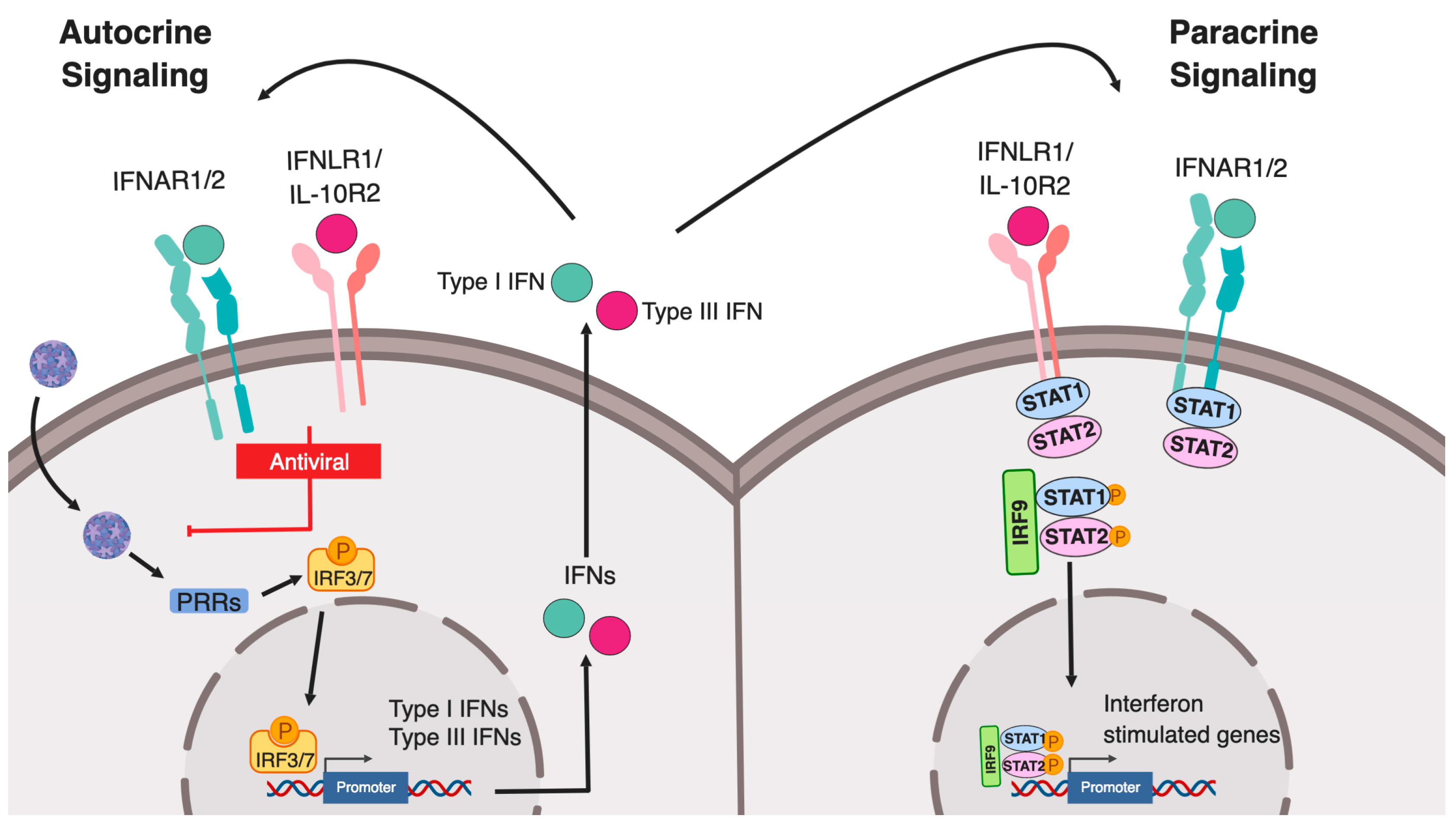
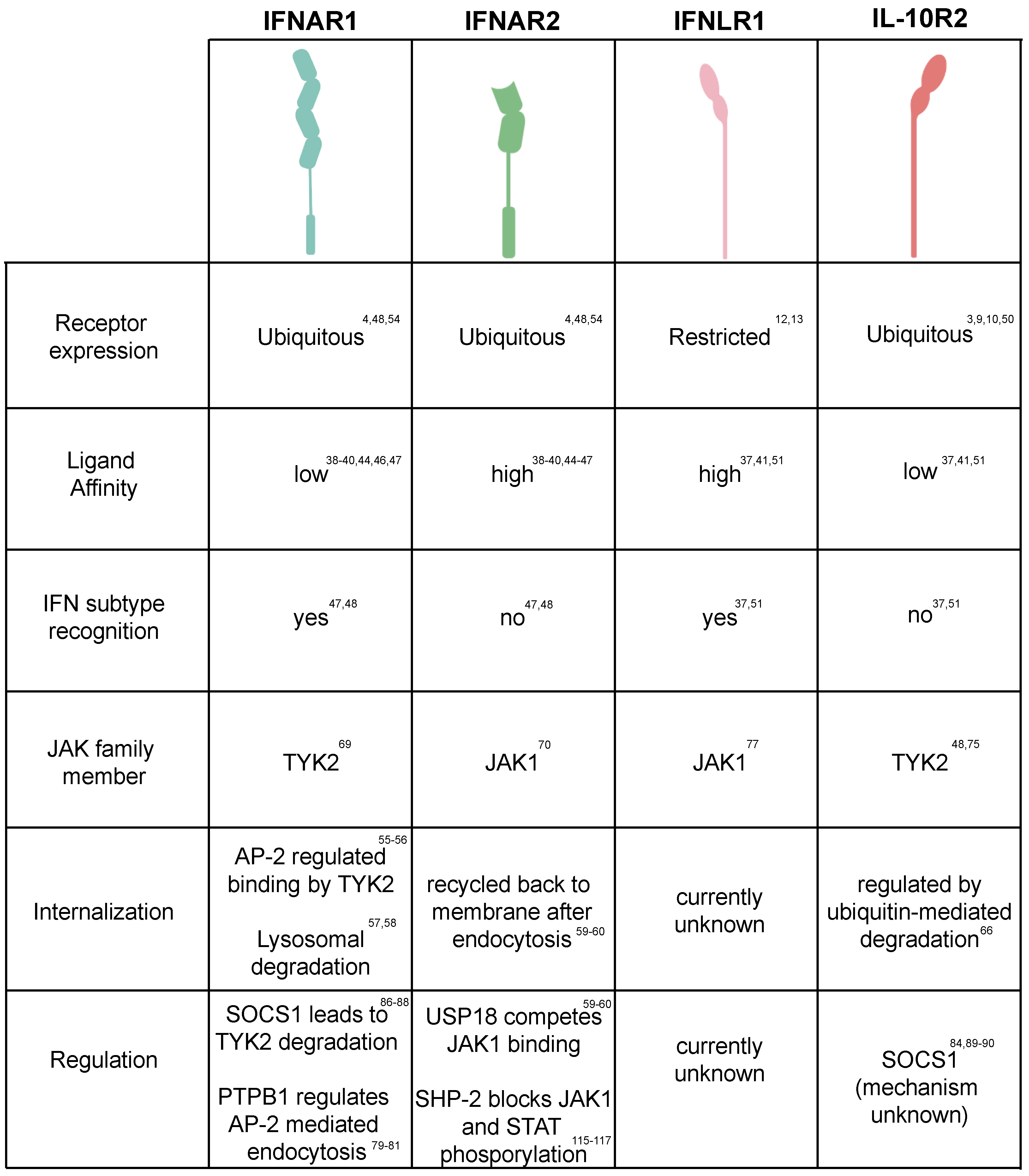
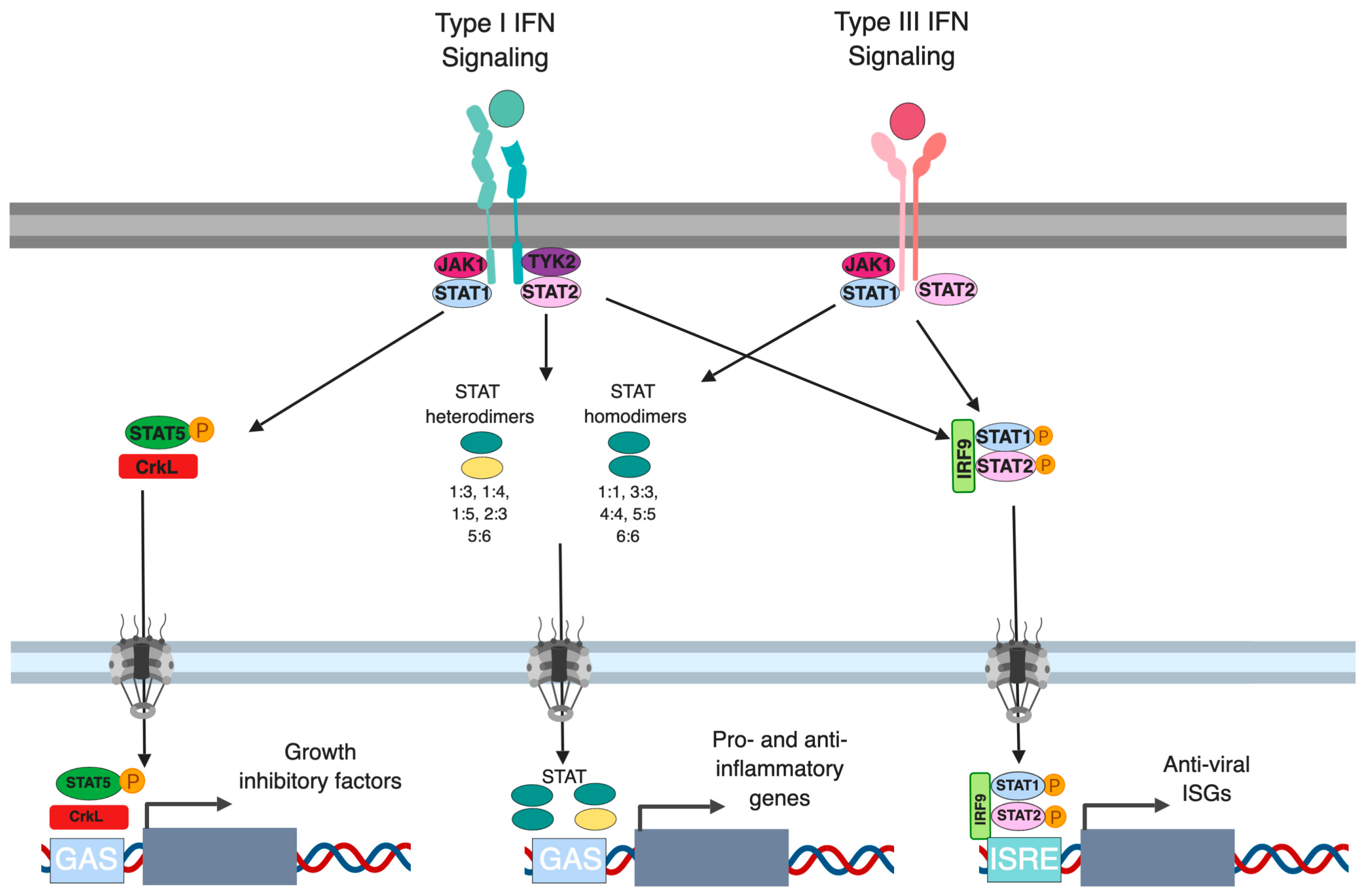
2. Canonical Type I and Type III IFN Signaling
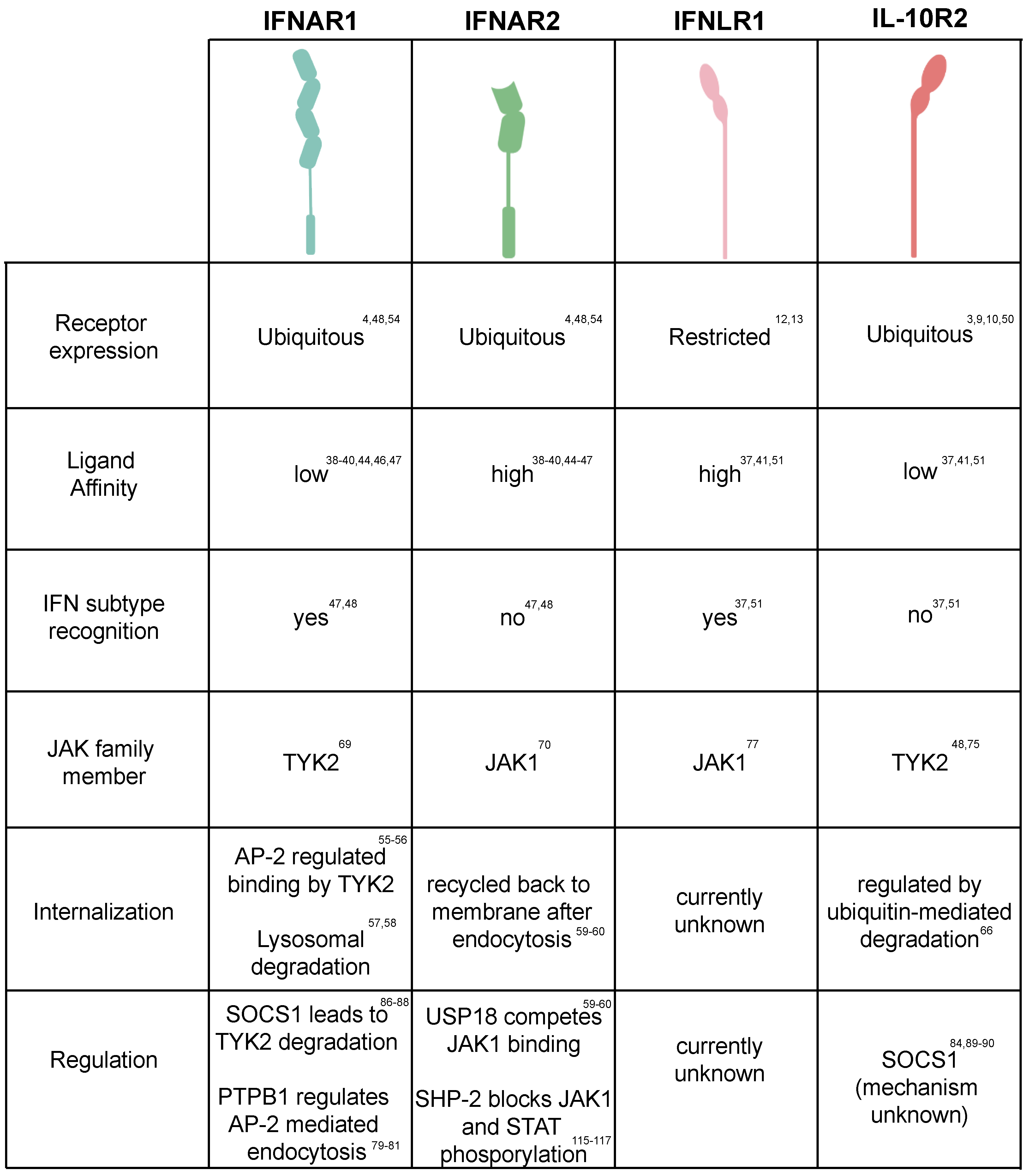
3. Regulation of JAK/STAT Activation
3.1. JAK Activation/Inactivation
3.2. Regulation of STATs
4. Alternative Signaling Pathways Downstream Type I and III IFNs
4.1. CrkL-RAP1 Pathway in Interferon Signaling
4.2. Phosphatidyl-Inositol 3-Kinase (PI3K)-Signaling Pathway
4.3. MAP Kinases in Type I and Type III IFN-Mediated Signaling
5. Modulation of ISG Expression
5.1. Role of Additional Transcription Factors in ISG Expression
5.2. Epigenetic Regulation of IFN and ISGs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Isaacs, A.; Lindenmann, J. Virus interference. I. The interferon. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1957, 147, 258–267. [Google Scholar]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef]
- Pestka, S.; Krause, C.D.; Sarkar, D.; Walter, M.R.; Shi, Y.; Fisher, P.B. Interleukin-10 and related cytokines and receptors. Annu. Rev. Immunol. 2004, 22, 929–979. [Google Scholar] [CrossRef] [PubMed]
- Langer, J.A.; Cutrone, E.C.; Kotenko, S. The Class II cytokine receptor (CRF2) family: Overview and patterns of receptor-ligand interactions. Cytokine Growth Factor Rev. 2004, 15, 33–48. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, D.W.; Nardelli, B.; Tsareva, T.; Mather, D.; Feng, P.; Semenuk, M.; Taylor, K.; Buergin, M.; Chinchilla, D.; Roshke, V.; et al. Interferon-κ, a Novel Type I Interferon Expressed in Human Keratinocytes. J. Biol. Chem. 2001, 276, 39765–39771. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Yanai, H. Interferon signalling network in innate defence. Cell Microbiol. 2006, 8, 907–922. [Google Scholar] [CrossRef] [PubMed]
- Gibbert, K.; Schlaak, J.F.; Yang, D.; Dittmer, U. IFN-α subtypes: Distinct biological activities in anti-viral therapy. Br. J. Pharmacol. 2013, 168, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. THE IFNγ RECEPTOR:A Paradigm for Cytokine Receptor Signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon- y: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [PubMed]
- Valente, G.; Ozmen, L.; Novelli, F.; Geuna, M. Distribution of interferon-γ receptor in human tissues. Eur. J. Immunol. 1992, 22, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda Interferon (IFN-), a Type III IFN, Is Induced by Viruses and IFNs and Displays Potent Antiviral Activity against Select Virus Infections In Vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef]
- Ank, N.; Iversen, M.B.; Bartholdy, C.; Staeheli, P.; Hartmann, R.; Jensen, U.B.; Dagnaes-Hansen, F.; Thomsen, A.R.; Chen, Z.; Haugen, H.; et al. An Important Role for Type III Interferon (IFN-/IL-28) in TLR-Induced Antiviral Activity. J. Immunol. 2008, 180, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, 1–12. [Google Scholar] [CrossRef]
- Mordstein, M.; Kochs, G.; Dumoutier, L.; Renauld, J.C.; Paludan, S.R.; Klucher, K.; Staeheli, P. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008, 4, 1–7. [Google Scholar] [CrossRef]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef]
- Pulverer, J.E.; Rand, U.; Lienenklaus, S.; Kugel, D.; Zietara, N.; Kochs, G.; Naumann, R.; Weiss, S.; Staeheli, P.; Hauser, H.; et al. Temporal and Spatial Resolution of Type I and III Interferon Responses In Vivo. J. Virol. 2010, 84, 8626–8638. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakõiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-lambda determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Mahlakõiv, T.; Hernandez, P.; Gronke, K.; Diefenbach, A.; Staeheli, P. Leukocyte-Derived IFN-α/β and Epithelial IFN-λ Constitute a Compartmentalized Mucosal Defense System that Restricts Enteric Virus Infections. PLoS Pathog. 2015, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jewell, N.A.; Cline, T.; Mertz, S.E.; Smirnov, S.V.; Flano, E.; Schindler, C.; Grieves, J.L.; Durbin, R.K.; Kotenko, S.V.; Durbin, J.E.; et al. Interferon-{lambda} is the predominant interferon induced by influenza A virus infection in vivo. J. Virol. 2010, 84, 11515–11522. [Google Scholar] [CrossRef] [PubMed]
- Hernández, P.P.; Mahlakõiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hölscher, C.; Dumoutier, L.; et al. Interferon-λ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698. [Google Scholar] [CrossRef]
- Lin, J.D.; Feng, N.; Sen, A.; Balan, M.; Tseng, H.C.; McElrath, C.; Smirnov, S.V.; Peng, J.; Yasukawa, L.L.; Durbin, R.K.; et al. Distinct Roles of Type I and Type III Interferons in Intestinal Immunity to Homologous and Heterologous Rotavirus Infections. PLoS Pathog. 2016, 12, 1–29. [Google Scholar]
- Saxena, K.; Simon, L.M.; Zeng, X.-L.; Blutt, S.E.; Crawford, S.E.; Sastri, N.P.; Karandikar, U.C.; Ajami, N.J.; Zachos, N.C.; Kovbasnjuk, O.; et al. A paradox of transcriptional and functional innate interferon responses of human intestinal enteroids to enteric virus infection. Proc Natl Acad Sci. USA 2017, 114, E570–E579. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef]
- Pervolaraki, K.; Stanifer, M.L.; Münchau, S.; Renn, L.A.; Albrecht, D.; Kurzhals, S.; Senís, E.; Grimm, D.; Schröder-Braunstein, J.; Rabin, R.L.; et al. Type I and type III interferons display different dependency on mitogen-activated protein kinases to mount an antiviral state in the human gut. Front. Immunol. 2017, 8, 1–16. [Google Scholar] [CrossRef]
- Pervolaraki, K.; Rastgou Talemi, S.; Albrecht, D.; Bormann, F.; Bamford, C.; Mendoza, J.L.; Garcia, K.C.; McLauchlan, J.; Hofer, T.; Stanifer, M.L.; et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 2018, 14, e1007420. [Google Scholar] [CrossRef]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III Interferon (IFN) Induces a Type I IFN-Like Response in a Restricted Subset of Cells through Signaling Pathways Involving both the Jak-STAT Pathway and the Mitogen-Activated Protein Kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef]
- Marcello, T.; Grakoui, A.; Barba-Spaeth, G.; Machlin, E.S.; Kotenko, S.V.; Macdonald, M.R.; Rice, C.M. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006, 131, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Bolen, C.R.; Ding, S.; Robek, M.D.; Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Jilg, N.; Lin, W.; Hong, J.; Schaefer, E.A.; Wolski, D.; Meixong, J.; Goto, K.; Brisac, C.; Chusri, P.; Fusco, D.N.; et al. Kinetic differences in the induction of interferon stimulated genes by interferon-α and interleukin 28B are altered by infection with hepatitis C virus. Hepatology 2014, 59, 1250–1261. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-lambda determine persistence of enteric murine norovirus infection_supplemental data. Science 2014, 347, 266–269. [Google Scholar]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef]
- Levy, D.E.; Marié, I.J.; Durbin, J.E. Induction and function of type i and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Odendall, C.; Kagan, J.C. The unique regulation and functions of type III interferons in antiviral immunity. Curr. Opin. Virol. 2015, 12, 47–52. [Google Scholar] [CrossRef]
- Mendoza, J.L.; Schneider, W.M.; Hoffmann, H.; Jong, Y.P.; De Rice, C.M.; Garcia, K.C.; Mendoza, J.L.; Schneider, W.M.; Hoffmann, H.; Vercauteren, K.; et al. The IFN-l-IFN-l R1-IL-10R b Complex Reveals Structural Features Underlying Type III IFN Functional Plasticity Article The IFN-l -IFN-l R1-IL-10R b Complex Reveals Structural Features Underlying. Immunity 2017, 46, 379–392. [Google Scholar] [CrossRef]
- Lavoie, T.B.; Kalie, E.; Crisafulli-cabatu, S.; Abramovich, R.; Digioia, G.; Moolchan, K.; Pestka, S.; Schreiber, G. Cytokine Binding and activity of all human alpha interferon subtypes. Cytokine 2011, 56, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Jaks, E.; Gavutis, M.; Uzé, G.; Martal, J.; Piehler, J. Differential Receptor Subunit Affinities of Type I Interferons Govern Differential Signal Activation. J. Mol. Biol. 2007, 366, 525–539. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Roisman, L.C.; Jaks, E.; Gavutis, M.; Piehler, J.; Van der Heyden, J.; Uze, G.; Schreiber, G. Inquiring into the Differential Action of Interferons (IFNs): An IFN-2 Mutant with Enhanced Affinity to IFNAR1 Is Functionally Similar to IFN-. Mol. Cell. Biol. 2006, 26, 1888–1897. [Google Scholar] [CrossRef]
- François-Newton, V.; de Freitas Almeida, G.M.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uzé, G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE 2011, 6, e22200. [Google Scholar] [CrossRef] [PubMed]
- Renauld, J.-C. Class II cytokine receptors and their ligands: Key antiviral and inflammatory modulators. Nat. Rev. Immunol. 2003, 3, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.F. Structural design and molecular evolution of a cytokine receptor superfamily. Proc. Natl. Acad. Sci. USA 1990, 87, 6934–6938. [Google Scholar] [CrossRef] [PubMed]
- Cohen, B.; Novick, D.; Barak, S. Ligand-Induced Association of the Type I Interferon Receptor Components. Mol. Cell. Biol. 1995, 15, 4208–4214. [Google Scholar] [CrossRef] [PubMed]
- Piehler, J.; Schreiber, G. Mutational and structural analysis of the binding interface between type I interferons and their receptor ifnar2. J. Mol. Biol. 1999, 294, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Lamken, P.; Lata, S.; Gavutis, M.; Piehler, J.; Wolfgang, J. Ligand-induced Assembling of the Type I Interferon Receptor on Supported Lipid Bilayers. J. Mol. Biol. 2004, 341, 303–318. [Google Scholar] [CrossRef] [PubMed]
- de Weerd, N.A.; Samarajiwa, S.A.; Hertzog, P.J. Type I interferon receptors: Biochemistry and biological functions. J. Biol. Chem. 2007, 282, 20053–20057. [Google Scholar] [CrossRef]
- De Weerd, N.A.; Nguyen, T. The interferons and their receptors—Distribution and regulation. Immunol Cell Biol. 2012, 90, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Makowska, Z.; Duong, F.H.T.; Trincucci, G.; Tough, D.F.; Heim, M.H. Interferon-β and interferon-λ signaling is not affected by interferon-induced refractoriness to interferon-α in vivo. Hepatology 2011, 53, 1171–1180. [Google Scholar] [CrossRef] [PubMed]
- Gad, H.H.; Dellgren, C.; Hamming, O.J.; Vends, S.; Paludan, S.R.; Hartmann, R. Interferon-λ is functionally an interferon but structurally related to the interleukin-10 family. J. Biol. Chem. 2009, 284, 20869–20875. [Google Scholar] [CrossRef] [PubMed]
- Miknis, Z.J.; Magracheva, E.; Li, W.; Zdanov, A.; Kotenko, S.V.; Wlodawer, A. Crystal Structure of Human Interferon-γ1 in Complex with Its High-Affinity Receptor Interferon-γR1. J. Mol. Biol. 2010, 404, 650–664. [Google Scholar] [CrossRef] [PubMed]
- Jordan, W.J.; Eskdale, J.; Boniotto, M.; Rodia, M.; Kellner, D.; Gallagher, G. Modulation of the human cytokine response by interferon lambda-1 (IFN-lambda1/IL-29). Genes Immunity 2007, 8, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G.; et al. Article Dendritic Cells in Response to Bacterial Flagellin Enhances Mucosal Innate Immune Defense. Immunity 2011, 36, 276–287. [Google Scholar] [CrossRef]
- Fuchs, S.Y. Hope and Fear for Interferon: The Receptor-Centric Outlook on the Future of Interferon Therapy. J. Interferon Cytokine Res. 2013, 33, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Suresh Kumar, K.G.; Barriere, H.; Carbone, C.J.; Liu, J.; Swaminathan, G.; Xu, P.; Li, Y.; Baker, D.P.; Peng, J.; Lukacs, G.L. Site-specific ubiquitination exposes a linear motif to promote interferon-α receptor endocytosis. J. Cell. Biol. 2007, 179, 935–950. [Google Scholar] [CrossRef]
- Kumar, K.G.S.; Varghese, B.; Banerjee, A.; Baker, D.P.; Constantinescu, S.N.; Pellegrini, S.; Fuchs, S.Y. Basal ubiquitin-independent internalization of interferon alpha receptor is prevented by Tyk2-mediated masking of a linear endocytic motif. J. Biol. Chem. 2008, 283, 18566–18572. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.G.S.; Tang, W.; Ravindranath, A.K.; Clark, W.A.; Croze, E.; Fuchs, S.Y. SCF HOS ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-a receptor. EMBO J. 2003, 22, 5480–5490. [Google Scholar] [CrossRef]
- Marijanovic, Z.; Ragimbeau, J.; Kumar, K.G.S.; Fuchs, S.Y.; Pellegrini, S. TYK2 activity promotes ligand-induced IFNAR1 proteolysis. Biochem. J. 2006, 397, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Marijanovic, Z.; Ragimbeau, J.; van der Heyden, J.; Uze, G.; Pellegrini, S. Comparable potency of IFNalpha2 and IFNbeta on immediate JAK/STAT activation but differential down-regulation of IFNAR2. Biochem. J. 2007, 407, 141–151. [Google Scholar] [CrossRef]
- Chmiest, D.; Sharma, N.; Zanin, N.; Viaris De Lesegno, C.; Shafaq-Zadah, M.; Sibut, V.; Dingli, F.; Hupé, P.; Wilmes, S.; Piehler, J.; et al. Spatiotemporal control of interferon-induced JAK/STAT signalling and gene transcription by the retromer complex. Nat Commun. 2016, 7, 13476. [Google Scholar] [CrossRef] [PubMed]
- Ragimbeau, J.; Dondi, E.; Alcover, Â.; Eid, P.; Uze, G.; Pellegrini, S. The tyrosine kinase Tyk2 controls IFNAR1 cell surface expression. EMBO J. 2003, 22, 537–547. [Google Scholar] [CrossRef]
- Liu, J.; Huangfu, W.; Kumar, K.G.S.; Qian, J.; Casey, J.P.; Hamanaka, R.B.; Grigoriadou, C.; Aldabe, R.; Diehl, J.A.; Fuchs, S.Y. Article Virus-Induced Unfolded Protein Response Attenuates Antiviral Defenses via Phosphorylation-Dependent Degradation of the Type I Interferon Receptor. Cell Host Microbe 2009, 5, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Wallace, E.; Docke, W.-D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Parks, O.B.; Pociask, D.A.; Hodzic, Z.; Kolls, J.K.; Good, M. Interleukin-22 Signaling in the Regulation of Intestinal Health and Disease. Front. Cell Dev. Biol. 2015, 3, 85. [Google Scholar] [CrossRef] [PubMed]
- Weathington, N.M.; Snavely, C.A.; Chen, B.B.; Zhao, J.; Zhao, Y.; Mallampalli, R.K. Glycogen synthase kinase-3beta stabilizes the interleukin (IL)-22 receptor from proteasomal degradation in murine lung epithelia. J. Biol. Chem. 2014, 289, 17610–17619. [Google Scholar] [CrossRef]
- Jiang, H.; Lu, Y.; Yuan, L.; Liu, J. Regulation of interleukin-10 receptor ubiquitination and stability by beta-TrCP-containing ubiquitin E3 ligase. PLoS ONE 2011, 6, e27464. [Google Scholar] [CrossRef]
- Du, Z.; Kelly, E.; Mecklenbrauker, I.; Agle, L.; Herrero, C.; Paik, P.; Ivashkiv, L.B. Selective regulation of IL-10 signaling and function by zymosan. J. Immunol. 2006, 176, 4785–4792. [Google Scholar] [CrossRef]
- Sen, A.; Sharma, A.; Greenberg, H.B. Rotavirus degrades multiple type interferon receptors to inhibit IFN signaling and protects against mortality from endotoxin in suckling mice. J. Virol. 2017, 92, e01394-e17. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Piazza, F.; Krishnan, K.; Pine, R.; Krolewski, J.J. Definition of the interferon-alpha receptor-binding domain on the TYK2 kinase. J. Biol. Chem. 1998, 273, 4046–4051. [Google Scholar] [CrossRef]
- Usacheva, A.; Sandoval, R.; Domanski, P.; Kotenko, S.V.; Nelms, K.; Goldsmith, M.A.; Colamonici, O.R. Contribution of the Box 1 and Box 2 motifs of cytokine receptors to Jak1 association and activation. J. Biol. Chem. 2002, 277, 48220–48226. [Google Scholar] [CrossRef]
- Wagner, T.C.; Velichko, S.; Vogel, D.; Rani, M.R.S.; Leung, S.; Ransohoff, R.M.; Stark, G.R.; Perez, H.D.; Croze, E. Interferon Signaling Is Dependent on Specific Tyrosines Located within the Intracellular Domain of IFNAR2c. J. Biol. Chem. 2002, 277, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Krishnan, K.; Greenlund, A.C.; Gupta, S.; Lim, J.T.E.; Schreiber, R.D.; Schindler, C.W.; Krolewskil, J.J. Phosphorylated interferon-a receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. EMBO J. 1996, 15, 1064–1074. [Google Scholar] [CrossRef]
- Gibbs, V.C.; Takahashi, M.; Aguet, M.; Chuntharapai, A. A negative regulatory region in the intracellular domain of the human interferon-alpha receptor. J. Biol. Chem. 1996, 271, 28710–28716. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Lee, C.; Piganis, R.; Plumlee, C.; de Weerd, N.; Hertzog, P.J.; Schindler, C.A. A conserved IFN-alpha receptor tyrosine motif directs the biological response to type I IFNs. J. Immunol. 2008, 180, 5483–5489. [Google Scholar] [CrossRef]
- Fuchs, S.; Kaiser-Labusch, P.; Bank, J.; Ammann, S.; Kolb-Kokocinski, A.; Edelbusch, C.; Omran, H.; Ehl, S. Tyrosine kinase 2 is not limiting human antiviral type III interferon responses. Eur. J. Immunol. 2016, 46, 2639–2649. [Google Scholar] [CrossRef]
- Kreins, A.Y.; Ciancanelli, M.J.; Okada, S.; Kong, X.-F.; Ramirez-Alejo, N.; Kilic, S.S.; El Baghdadi, J.; Nonoyama, S.; Boisson-Dupuis, S. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J. Exp. Med. 2015, 212, 1641–1662. [Google Scholar] [CrossRef]
- Dumoutier, L.; Tounsi, A.; Michiels, T.; Sommereyns, C.; Kotenko, S.V.; Renauld, J.-C. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and antiproliferative activity of IL-29/interferon-lambda 1: Similarities with type I interferon signaling. J. Biol. Chem. 2004, 279, 32269–32274. [Google Scholar] [CrossRef]
- Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 2013, 255, 25–39. [Google Scholar] [CrossRef]
- Carbone, C.J.; Zheng, H.; Bhattacharya, S.; Lewis, J.R.; Reiter, A.M.; Henthorn, P.; Zhang, Z.Y.; Baker, D.P.; Ukkiramapandian, R.; Bence, K.K.; et al. Protein tyrosine phosphatase 1B is a key regulator of IFNAR1 endocytosis and a target for antiviral therapies. Proc. Natl. Acad. Sci. USA 2012, 109, 19226–19231. [Google Scholar] [CrossRef]
- Myers, M.P.; Andersen, J.N.; Cheng, A.; Tremblay, M.L.; Horvath, C.M.; Parisien, J.P.; Salmeen, A.; Barford, D.; Tonks, N.K. TYK2 and JAK2 Are Substrates of Protein-tyrosine Phosphatase, 1B. J. Biol. Chem. 2001, 276, 47771–47774. [Google Scholar] [CrossRef]
- Simoncic, P.D.; Lee-loy, A.; Barber, D.L.; Tremblay, M.L.; Mcglade, C.J. The T Cell Protein Tyrosine Phosphatase Is a Negative Regulator of Janus Family Kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef]
- Irie-Sasaki, J.; Sasaki, T.; Matsumoto, W.; Opavsky, A.; Cheng, M.; Welstead, G.; Griffiths, E.; Krawczyk, C.; Richardson, C.D.; Aitken, K.; et al. CD45 is a JAK phosphatase and negatively regulates cytokine receptor signalling. Nature 2001, 409, 349–354. [Google Scholar] [CrossRef]
- Malakhova, O.A.; Kim KIl Luo, J.; Zou, W.; Suresh, K.G.; Fuchs, S.Y.; Shuai, K.; Zhang, D. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef]
- Blumer, T.; Coto-Llerena, M.; Duong, F.H.T.; Heim, M.H. SOCS1 is an inducible negative regulator of interferon λ (IFN-λ)-induced gene expression in vivo. J. Biol. Chem. 2017, 292, 17928–17938. [Google Scholar] [CrossRef]
- Burkart, C.; Arimoto, K.; Tang, T.; Cong, X.; Xiao, N.; Liu, Y.-C.; Kotenko, S.V.; Ellies, L.G.; Zhang, D.E. Usp18 deficient mammary epithelial cells create an antitumour environment driven by hypersensitivity to IFN-λ and elevated secretion of Cxcl10. EMBO Mol. Med. 2013, 5, 1035–1050. [Google Scholar] [CrossRef]
- Song, M.M.; Shuai, K. The suppressor of cytokine signaling (SOCS) 1 and SOCS3 but not SOCS2 proteins inhibit interferon-mediated antiviral and antiproliferative activities. J. Biol. Chem. 1998, 273, 35056–35062. [Google Scholar] [CrossRef] [PubMed]
- Piganis, R.A.R.; De Weerd, N.A.; Gould, J.A.; Schindler, C.W.; Mansell, A.; Nicholson, S.E.; Hertzog, P.J. Suppressor of Cytokine Signaling (SOCS) 1 inhibits type I interferon (IFN) signaling via the interferon α receptor (IFNAR1)-associated tyrosine kinase tyk2. J. Biol. Chem. 2011, 286, 33811–33818. [Google Scholar] [CrossRef]
- Fenner, J.E.; Starr, R.; Cornish, A.L.; Zhang, J.; Metcalf, D.; Schreiber, R.D.; Hilton, D.J.; Alexander, W.S.; Hertzog, P.J. Suppressor of cytokine signaling 1 regulates the immune response to infection by a unique inhibition of type I interferon activity. Nat. Immunol. 2006, 7, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Beigel, F.; Olszak, T.; Zitzmann, K.; Eichhorst, S.T.; Otte, J.-M.; Diebold, J.; Diepolder, H.; Adler, B.; Auernhammer, C.J. IL-28A and IL-29 mediate antiproliferative and antiviral signals in intestinal epithelial cells and murine CMV infection increases colonic IL-28A expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 289, G960–G968. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, S.; Guan, Y.; Chen, L. Type III Interferon Induces Distinct SOCS1 Expression Pattern that Contributes to Delayed but Prolonged Activation of Jak/STAT Signaling Pathway: Implications for Treatment Non-Response in HCV Patients. PLoS ONE 2015, 10, e0133800. [Google Scholar] [CrossRef] [PubMed]
- Sarasin-Filipowicz, M.; Wang, X.; Yan, M.; Duong, F.H.T.; Poli, V.; Hilton, D.J.; Zhang, D.-E.; Heim, M.H. Alpha Interferon Induces Long-Lasting Refractoriness of JAK-STAT Signaling in the Mouse Liver through Induction of USP18/UBP43. Mol. Cell. Biol. 2009, 29, 4841–4851. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Messina, N.L.; Hii, L.; Gould, J.A.; Sabapathy, K.; Robertson, A.P.S.; Trapani, J.A.; Levy, D.E.; Hertzog, P.J.; Clarke, C.J.P.; et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 2010, 8, e1000361. [Google Scholar] [CrossRef]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Antenus, M.; Williams, K.L.; Erikson, J.; Wherry, E.J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Lew, D.J.; Decker, T.; Kessler, D.S.; Darnell, J.E. Synergistic interaction between interferon-oa and interferon-γ through induced synthesis of one subunit of the transcription factor ISGF3. EMBO J. 1990, 9, 1105–1111. [Google Scholar] [CrossRef]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; Deken XDe Brochiero, E.; Miot, F.; Grandvaux, N. IFN β/TNF α synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH Oxidase-mediated airway antiviral response. Cell Res. 2013, 23, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Herrero, C.; Li, W.; Antoniv, T.T.; Falck-pedersen, E.; Koch, A.E.; Woods, J.M.; Iii, G.K.H.; Ivashkiv, L.B. Sensitization of IFN-γ Jak-STAT signaling during macrophage activation. Nat. Immunol. 2002, 3, 859–866. [Google Scholar] [CrossRef]
- Darnell, J.E.; Kerr, I.M.; Stark, G.R. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 1994, 264, 1415–1421. [Google Scholar] [CrossRef]
- Meinke, A.; Barahmand-Pour, F.; Wohrl, S.; Stoiber, D.; Decker, T. Activation of different Stat5 isoforms contributes to cell-type-restricted signaling in response to interferons. Mol. Cell. Biol. 1996, 16, 6937–6944. [Google Scholar] [CrossRef]
- Fasler-Kan, E.; Pansky, A.; Wiederkehr, M.; Battegay, M.; Heim, M.H. Interferon-alpha activates signal transducers and activators of transcription 5 and 6 in Daudi cells. Eur. J. Biochem. 1998, 254, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.D.; Smith, J.D.; Murphy, T.L.; Murphy, K.M. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2. J. Biol. Chem. 2000, 275, 2693–2697. [Google Scholar] [CrossRef] [PubMed]
- Uddin, S.; Lekmine, F.; Sassano, A.; Rui, H.; Fish, E.N.; Platanias, L.C. Role of Stat5 in type I interferon-signaling and transcriptional regulation. Biochem. Biophys. Res. Commun. 2003, 308, 325–330. [Google Scholar] [CrossRef]
- Kotenko, S.V. IFN-λs. Curr. Opin. Immunol. 2011, 23, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C.; Fish, E.N. Signaling pathways activated by interferons. Exp. Hematol. 1999, 27, 1583–1592. [Google Scholar] [CrossRef]
- Platanias, L.C. Mechanisms of type-I-and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Darnell, J.E. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; Darnell, J.E.J. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Wen, Z.; Darnell, J.E.J. Mapping of Stat3 serine phosphorylation to a single residue (727) and evidence that serine phosphorylation has no influence on DNA binding of Stat1 and Stat3. Nucleic Acids Res. 1997, 25, 2062–2067. [Google Scholar] [CrossRef]
- Uddin, S.; Sassano, A.; Deb, D.K.; Verma, A.; Majchrzak, B.; Rahman, A.; Malik, A.B.; Fish, E.N.; Platanias, L.C. Protein kinase C-δ (PKC-δ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002, 277, 14408–14416. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, G.G. A linear signal transduction pathway involving phosphatidylinositol 3-kinase, protein kinase Cε, and MAPK in mesangial cells regulates interferon-γ-induced STAT1α transcriptional activation. J. Biol. Chem. 2004, 279, 27399–27409. [Google Scholar] [CrossRef]
- Staab, J.; Herrmann-Lingen, C.; Meyer, T. CDK8 as the STAT1 serine 727 kinase? JAK-STAT 2013, 2, e24275. [Google Scholar] [CrossRef] [PubMed]
- Bancerek, J.; Poss, Z.C.; Steinparzer, I.; Sedlyarov, V.; Pfaffenwimmer, T.; Mikulic, I.; Dolken, L.; Strobl, B.; Muller, M.; Taatjes, D.J.; et al. CDK8 kinase phosphorylates transcription factor STAT1 to selectively regulate the interferon response. Immunity 2013, 38, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Ramsauer, K.; Sadzak, I.; Porras, A.; Pilz, A.; Nebreda, A.R.; Decker, T.; Kovarik, P. p38 MAPK enhances STAT1-dependent transcription independently of Ser-727 phosphorylation. Proc. Natl. Acad. Sci. USA 2002, 99, 12859–12864. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-L.; Friedman, B.A.; Schmid, S.; Gertz, J.; Myers, R.M.; Tenoever, B.R.; Maniatis, T. IkappaB kinase epsilon (IKK(epsilon)) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. USA 2011, 108, 21170–21175. [Google Scholar] [CrossRef] [PubMed]
- Tenoever, B.R.; Ng, S.-L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef]
- Xu, D.; Qu, C.-K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- You, M.; Yu, D.H.; Feng, G.S. Shp-2 tyrosine phosphatase functions as a negative regulator of the interferon-stimulated Jak/STAT pathway. Mol. Cell. Biol. 1999, 19, 2416–2424. [Google Scholar] [CrossRef]
- Wu, T.R.; Hong, Y.K.; Wang, X.-D.; Ling, M.Y.; Dragoi, A.M.; Chung, A.S.; Campbell, A.G.; Han, Z.-Y.; Feng, G.-S.; Chin, Y.E. SHP-2 is a dual-specificity phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. J. Biol. Chem. 2002, 277, 47572–47580. [Google Scholar] [CrossRef]
- Steen, H.C.; Nogusa, S.; Thapa, R.J.; Basagoudanavar, S.H.; Gill, A.L.; Merali, S.; Barrero, C.A.; Balachandran, S.; Gamero, A.M. Identification of STAT2 serine 287 as a novel regulatory phosphorylation site in type I interferon-induced cellular responses. J. Biol. Chem. 2013, 288, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nan, J.; Willard, B.; Wang, X.; Yang, J.; Stark, G.R. Negative regulation of type I IFN signaling by phosphorylation of STAT2 on T387. EMBO J. 2017, 36, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Vanhatupa, S.; Gronholm, J.; Palvimo, J.J.; Silvennoinen, O. SUMO-1 conjugation selectively modulates STAT1-mediated gene responses. Blood 2005, 106, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Bhattacharya, S.; Yunus, A.A.; Lima, C.D.; Schindler, C. Stat1 and SUMO modification. Blood 2006, 108, 3237–3244. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef]
- Wieczorek, M.; Ginter, T.; Brand, P.; Heinzel, T.; Kramer, O.H. Acetylation modulates the STAT signaling code. Cytokine Growth Factor Rev. 2012, 23, 293–305. [Google Scholar] [CrossRef]
- Zhuang, S. Regulation of STAT signaling by acetylation. Cell Signal. 2013, 25, 1924–1931. [Google Scholar] [CrossRef]
- Sung, P.S.; Cheon, H.; Cho, C.H.; Hong, S.-H.; Park, D.Y.; Seo, H.-I.; Park, S.H.; Yoon, S.K.; Stark, G.R.; Shin, E.C. Roles of unphosphorylated ISGF3 in HCV infection and interferon responsiveness. Proc. Natl. Acad. Sci. USA 2015, 112, 10443–10448. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Stark, G.R. Unphosphorylated STAT1 prolongs the expression of interferon-induced immune regulatory genes. Proc. Natl. Acad. Sci. USA 2009, 106, 9373–9378. [Google Scholar] [CrossRef] [PubMed]
- Cheon, H.; Holvey-Bates, E.G.; Schoggins, J.W.; Forster, S.; Hertzog, P.; Imanaka, N.; Rice, C.M.; Jackson, M.W.; Junk, D.J.; Stark, G.R. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J. 2013, 32, 2751–2763. [Google Scholar] [CrossRef] [PubMed]
- Shresta, S.; Sharar, K.L.; Prigozhin, D.M.; Snider, H.M.; Beatty, P.R.; Harris, E. Critical roles for both STAT1-dependent and STAT1-independent pathways in the control of primary dengue virus infection in mice. J. Immunol. 2005, 175, 3946–3954. [Google Scholar] [CrossRef]
- Brierley, M.M.; Marchington, K.L.; Jurisica, I.; Fish, E.N. Identification of GAS-dependent interferon-sensitive target genes whose transcription is STAT2-dependent but ISGF3-independent. FEBS J. 2006, 273, 1569–1581. [Google Scholar] [CrossRef]
- Perry, S.T.; Buck, M.D.; Lada, S.M.; Schindler, C.; Shresta, S. STAT2 mediates innate immunity to Dengue virus in the absence of STAT1 via the type I interferon receptor. PLoS Pathog. 2011, 7, e1001297. [Google Scholar] [CrossRef]
- Fink, K.; Grandvaux, N. STAT2 and IRF9: Beyond ISGF3. JAK-STAT 2013, 2, e27521. [Google Scholar] [CrossRef]
- Boudinot, P.; Langevin, C.; Secombes, C.J.; Levraud, J.P. The peculiar characteristics of fish type I interferons. Viruses 2016, 8, 298. [Google Scholar] [CrossRef]
- Grumbach, I.M.; Mayer, I.A.; Uddin, S.; Lekmine, F.; Majchrzak, B.; Yamauchi, H.; Fujita, S.; Druker, B.; Fish, E.N.; Platanias, L.C. Engagement of the CrkL adaptor in interferon alpha signalling in BCR-ABL-expressing cells. Br. J. Haematol. 2001, 112, 327–336. [Google Scholar] [CrossRef]
- Lekmine, F.; Sassano, A.; Uddin, S.; Majchrzak, B.; Miura, O.; Druker, B.J.; Fish, E.N.; Imamoto, A.; Platanias, L.C. The CrkL Adapter Protein Is Required for Type I Interferon-Dependent Gene Transcription and Activation of the Small G-Protein Rap1. Biochem. Biophys. Res. Commun. 2002, 750, 744–750. [Google Scholar] [CrossRef]
- Fish, E.N.; Uddin, S.; Korkmaz, M.; Majchrzak, B.; Druker, B.J.; Platanias, L.C. Activation of a CrkL-stat5 signaling complex by type I interferons. J. Biol. Chem. 1999, 274, 571–573. [Google Scholar] [CrossRef]
- Kaur, S.; Uddin, S.; Platanias, L.C. The PI3’ kinase pathway in interferon signaling. J. Interferon Cytokine Res. 2005, 25, 780–787. [Google Scholar] [CrossRef]
- Uddin, S.; Majchrzak, B.; Woodson, J.; Arunkumar, P.; Alsayed, Y.; Pine, R.; Young, P.R.; Fish, E.N.; Platanias, L.C. Activation of the p38 mitogen-activated protein kinase by type I interferons. J. Biol. Chem. 1999, 274, 30127–30131. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Li, Y.; Mears, H.; Platanias, L.C. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J. Interferon Cytokine Res. 2005, 25, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-J.; Hua, X.; He, S.-F.; Ren, H.; Qi, Z.-T. Interferon alpha regulates MAPK and STAT1 pathways in human hepatoma cells. Virol. J. 2011, 8, 157. [Google Scholar] [CrossRef]
- Ahmad, S.; Alsayed, Y.M.; Druker, B.J.; Platanias, L.C. The type I interferon receptor mediates tyrosine phosphorylation of the CrkL adaptor protein. J. Biol. Chem. 1997, 272, 29991–29994. [Google Scholar] [CrossRef]
- Uddin, S.; Yenush, L.; Sun, X.J.; Sweet, M.E.; White, M.F.; Platanias, L.C. Interferon-alpha engages the insulin receptor substrate-1 to associate with the phosphatidylinositol 3’-kinase. J. Biol. Chem. 1995, 270, 15938–15941. [Google Scholar] [CrossRef]
- Uddin, S.; Fish, E.N.; Sher, D.A.; Gardziola, C.; White, M.F.; Platanias, L.C. Activation of the phosphatidylinositol 3-kinase serine kinase by IFN-alpha. J. Immunol. 1997, 158, 2390–2397. [Google Scholar]
- Kaur, S.; Sassano, A.; Joseph, A.M.; Majchrzak-Kita, B.; Eklund, E.A.; Verma, A.; Brachmann, S.M.; Fish, E.N.; Platanias, L.C. Dual regulatory roles of phosphatidylinositol 3-kinase in IFN signaling. J. Immunol. 2008, 181, 7316–7323. [Google Scholar] [CrossRef]
- Goh, K.C.; Haque, S.J.; Williams, B.R. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999, 18, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Verma, A.; Grumbach, I.M.; Uddin, S.; Lekmine, F.; Ravandi, F.; Majchrzak, B.; Fujita, S.; Fish, E.N.; Platanias, L.C. The p38 MAPK pathway mediates the growth inhibitory effects of interferon-alpha in BCR-ABL-expressing cells. J. Biol. Chem. 2001, 276, 28570–28577. [Google Scholar] [CrossRef]
- Ishida, H.; Ohkawa, K.; Hosui, A.; Hiramatsu, N.; Kanto, T.; Ueda, K.; Takehara, T.; Hayashi, N. Involvement of p38 signaling pathway in interferon-alpha-mediated antiviral activity toward hepatitis C virus. Biochem. Biophys. Res. Commun. 2004, 321, 722–727. [Google Scholar] [CrossRef]
- Börgeling, Y.; Schmolke, M.; Viemann, D.; Nordhoff, C.; Roth, J.; Ludwig, S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J. Biol. Chem. 2014, 289, 13–27. [Google Scholar] [CrossRef]
- Platanias, L.C.; Sweet, M.E. Interferon alpha induces rapid tyrosine phosphorylation of the vav proto-oncogene product in hematopoietic cells. J. Biol. Chem. 1994, 269, 3143–3146. [Google Scholar]
- Uddin, S.; Lekmine, F.; Sharma, N.; Majchrzak, B.; Mayer, I.; Young, P.R.; Bokoch, G.M.; Fish, E.N.; Platanias, L.C. The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J. Biol. Chem. 2000, 275, 27634–27640. [Google Scholar] [PubMed]
- Micouin, A.; Wietzerbin, J.; Steunou, V.; Martyre, M.C. p95(vav) associates with the type I interferon (IFN) receptor and contributes to the antiproliferative effect of IFN-alpha in megakaryocytic cell lines. Oncogene 2000, 19, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Batra, S.; Sassano, A.; Majchrzak, B.; Levy, D.E.; Gaestel, M.; Fish, E.N.; Davis, R.J.; Platanias, L.C. Activation of mitogen-activated protein kinase kinase (MKK) 3 and MKK6 by type I interferons. J. Biol. Chem. 2005, 280, 10001–10010. [Google Scholar] [CrossRef]
- Wang, F.; Ma, Y.; Barrett, J.W.; Gao, X.; Loh, J.; Barton, E.; Virgin, H.W.; McFadden, G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat. Immunol. 2004, 5, 1266–1274. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-J.; Wang, W.; Wang, W.-B.; Ren, H.; Qi, Z.-T. Involvement of ERK pathway in interferon alpha-mediated antiviral activity against hepatitis C virus. Cytokine 2015, 72, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xiang, Y.; Sabapathy, K.; Silverman, R.H. An apoptotic signaling pathway in the interferon antiviral response mediated by RNase L and c-Jun NH2-terminal kinase. J. Biol. Chem. 2004, 279, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Alase, A.A.; El-sherbiny, Y.M.; Vital, E.M.; Tobin, D.J.; Turner, N.A. IFN λ Stimulates MxA Production in Human Dermal Fibroblasts via a MAPK-Dependent STAT1-Independent Mechanism. J. Investig. Dermatol. 2015, 135, 2935–2943. [Google Scholar] [CrossRef]
- Chatterjee-Kishore, M.; Wright, K.L.; Ting, J.P.; Stark, G.R. How Stat1 mediates constitutive gene expression: A complex of unphosphorylated Stat1 and IRF1 supports transcription of the LMP2 gene. EMBO J. 2000, 19, 4111–4122. [Google Scholar] [CrossRef]
- Nakaya, T.; Sato, M.; Hata, N.; Asagiri, M.; Suemori, H.; Noguchi, S.; Tanaka, N.; Taniguchi, T. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem. Biophys. Res. Commun. 2001, 283, 1150–1156. [Google Scholar] [CrossRef]
- Tamura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF Family Transcription Factors in Immunity and Oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhou, X.; Wang, W.; Wang, Y.; Yin, Y.; Van Der Laan, L.J.W.; Sprengers, D.; Metselaar, H.J. IFN regulatory factor 1 restricts hepatitis E virus replication by activating STAT1 to induce antiviral IFN-stimulated genes. FASEB J. 2017, 30, 3352–3367. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Takahashi, E.; Itoh, S.; Harada, K.; Hori, T.A.; Taniguchi, T. Structure and regulation of the human interferon regulatory factor 1 (IRF-1) and IRF-2 genes: Implications for a gene network in the interferon system. Mol. Cell. Biol. 1994, 14, 1500–1509. [Google Scholar] [CrossRef] [PubMed]
- Hida, S.; Ogasawara, K.; Sato, K.; Abe, M.; Takayanagi, H.; Yokochi, T.; Sato, T.; Hirose, S.; Shirai, T.; Taki, S.; et al. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity 2000, 13, 643–655. [Google Scholar] [CrossRef]
- Taki, S. Type I interferons and autoimmunity: Lessons from the clinic and from IRF-2-deficient mice. Cytokine Growth Factor Rev. 2002, 13, 379–391. [Google Scholar] [CrossRef]
- Porritt, R.A.; Hertzog, P.J. Dynamic control of type I IFN signalling by an integrated network of negative regulators. Trends Immunol. 2015, 36, 150–160. [Google Scholar] [CrossRef] [PubMed]
- van Boxel-Dezaire, A.H.H.; Rani, M.R.S.; Stark, G.R. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006, 25, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Kanno, Y.; Levi, B.-Z.; Tamura, T.; Ozato, K. Immune cell-specific amplification of interferon signaling by the IRF-4/8-PU.1 complex. J. Interferon Cytokine Res. 2005, 25, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Marie, I.; Smith, E.; Prakash, A. Enhancement and diversification of IFN induction by IRF-7-mediated positive feedback. J. Interferon Cytokine Res. 2002, 22, 87–93. [Google Scholar] [CrossRef]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717–726. [Google Scholar] [CrossRef]
- Novatt, H.; Renn, L.; Theisen, T.; Massie, T.; Massie, T.; Rabin, R.L. Interferon regulatory factor 1 (IRF1) expression patterns by respiratory epithelial cells reveal non-redundancy of type I versus type III interferon. J. Immunol. 2016, 196, 68.12. [Google Scholar]
- Novatt, H.; Theisen, T.C.; Massie, T.; Massie, T.; Simonyan, V.; Voskanian-Kordi, A.; Renn, L.A.; Rabin, R.L. Distinct Patterns of Expression of Transcription Factors in Response to Interferonbeta and Interferonlambda1. J. Interferon Cytokine Res. 2016, 36, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.C.; Schaefer, U.; Mecklenbrauker, I.; Stienen, A.; Dewell, S.; Chen, M.S.; Rioja, I.; Parravicini, V.; Prinjha, R.K.; Chandwani, R.; et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 2012, 209, 661–669. [Google Scholar] [CrossRef]
- Marcos-Villar, L.; Diaz-Colunga, J.; Sandoval, J.; Zamarreno, N.; Landeras-Bueno, S.; Esteller, M.; Falcon, A.; Nieto, A. Epigenetic control of influenza virus: Role of H3K79 methylation in interferon-induced antiviral response. Sci. Rep. 2018, 8, 1230. [Google Scholar] [CrossRef]
- Bhushal, S.; Wolfsmuller, M.; Selvakumar, T.A.; Kemper, L.; Wirth, D.; Hornef, M.W.; Hauser, H.; Koster, M. Cell Polarization and Epigenetic Status Shape the Heterogeneous Response to Type III Interferons in Intestinal Epithelial Cells. Front. Immunol. 2017, 8, 671. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Khoury-Hanold, W.; Iwasaki, A.; Robek, M.D. Epigenetic Reprogramming of the Type III Interferon Response Potentiates Antiviral Activity and Suppresses Tumor Growth. PLoS Biol. 2014, 12, e1001758. [Google Scholar] [CrossRef]
- Banaszynski, L.A.; Wen, D.; Dewell, S.; Whitcomb, S.J.; Lin, M.; Diaz, N.; Elsasser, S.J.; Chapgier, A.; Goldberg, A.D.; Canaani, E. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell 2013, 155, 107–120. [Google Scholar] [CrossRef]
- Tamura, T.; Smith, M.; Kanno, T.; Dasenbrock, H.; Nishiyama, A.; Ozato, K. Inducible deposition of the histone variant H3.3 in interferon-stimulated genes. J. Biol. Chem. 2009, 284, 12217–12225. [Google Scholar] [CrossRef]
- Kamada, R.; Yang, W.; Zhang, Y.; Patel, M.C.; Yang, Y.; Ouda, R.; Dey, A.; Wakabayashi, Y.; Sakaguchi, K.; Fujita, T. Interferon stimulation creates chromatin marks and establishes transcriptional memory. Proc. Natl. Acad. Sci. USA 2018, 115, E9162–E9171. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Kang, H.; Liu, R.; Chen, X.; Zhao, K. Maximal Induction of a Subset of Interferon Target Genes Requires the Chromatin-Remodeling Activity of the BAF Complex. Mol. Cell. Boil. 2002, 22, 6471–6479. [Google Scholar] [CrossRef]
- Cui, K.; Tailor, P.; Liu, H.; Chen, X.; Ozato, K.; Zhao, K. The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol. Cell. Biol. 2004, 24, 4476–4486. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Karaskov, E.; Yu, T.; Callaghan, S.M.; Der, S.; Park, D.S.; Xu, Z.; Pattenden, S.G.; Bremner, R. Apical role for BRG1 in cytokine-induced promoter assembly. Proc. Natl. Acad. Sci. USA 2005, 102, 14611–14616. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Cui, K.; Murray, D.M.; Ling, C.; Xue, Y.; Gerstein, A.; Parsons, R.; Zhao, K.; Wang, W. PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev. 2005, 19, 1662–1667. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Qian, F.; Hu, Y.; Ang, C.; Li, Z.; Wen, Z. Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes. Nat. Cell Biol. 2002, 4, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, S.; Eckner, R.; Grossman, S.; Oldread, E.; Arany, Z.; D’Andrea, A.; Livingston, D.M. Cooperation of Stat2 and p300/CBP in signalling induced by interferon-alpha. Nature 1996, 383, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Vinkemeier, U.; Gu, W.; Chakravarti, D.; Horvath, C.M.; Darnell, J.E.J. Two contact regions between Stat1 and CBP/p300 in interferon gamma signaling. Proc. Natl. Acad. Sci. USA 1996, 93, 15092–15096. [Google Scholar] [CrossRef]
- Paulson, M.; Press, C.; Smith, E.; Tanese, N.; Levy, D.E. IFN-Stimulated transcription through a TBP-free acetyltransferase complex escapes viral shutoff. Nat. Cell Boil. 2003, 4, 140. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. https://doi.org/10.3390/ijms20061445
Stanifer ML, Pervolaraki K, Boulant S. Differential Regulation of Type I and Type III Interferon Signaling. International Journal of Molecular Sciences. 2019; 20(6):1445. https://doi.org/10.3390/ijms20061445
Chicago/Turabian StyleStanifer, Megan L., Kalliopi Pervolaraki, and Steeve Boulant. 2019. "Differential Regulation of Type I and Type III Interferon Signaling" International Journal of Molecular Sciences 20, no. 6: 1445. https://doi.org/10.3390/ijms20061445
APA StyleStanifer, M. L., Pervolaraki, K., & Boulant, S. (2019). Differential Regulation of Type I and Type III Interferon Signaling. International Journal of Molecular Sciences, 20(6), 1445. https://doi.org/10.3390/ijms20061445