An Integrated Transcriptome and Proteome Analysis Reveals Putative Regulators of Adventitious Root Formation in Taxodium ‘Zhongshanshan’

 ,
,
Abstract
1. Introduction
2. Results
2.1. Anatomical Changes During AR Formation
2.2. General Characterization of Transcriptome Data
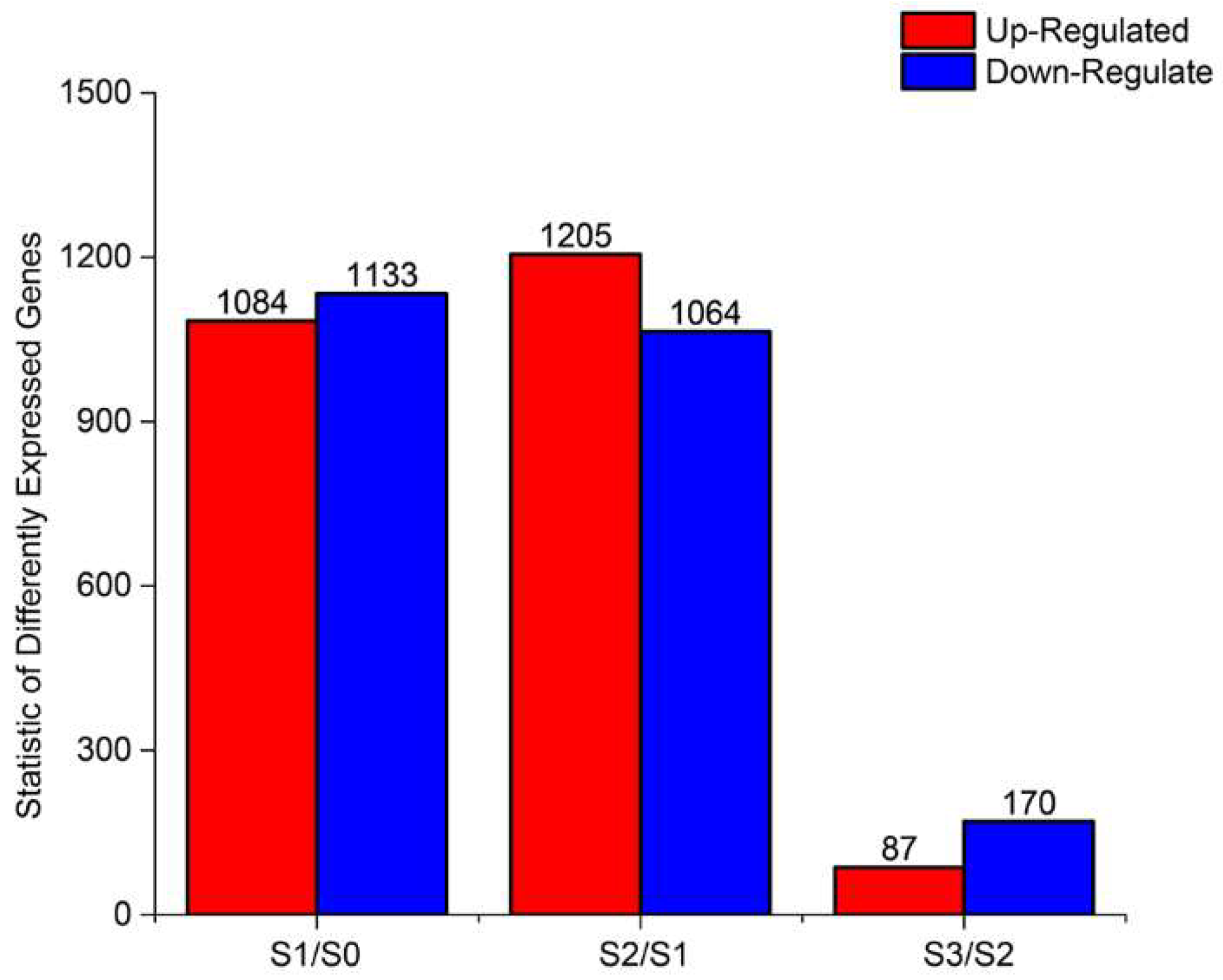
2.3. DEGs During AR Formation
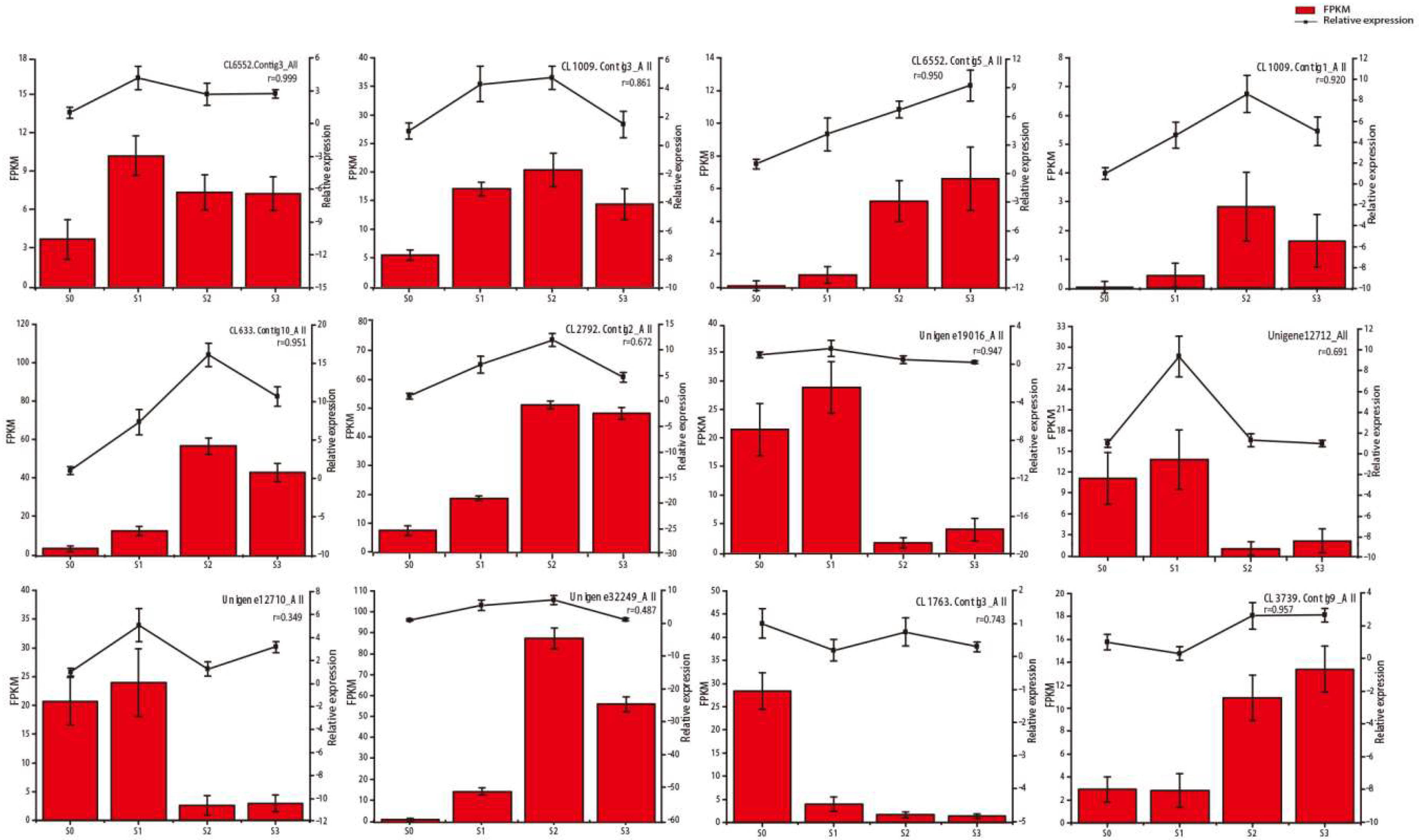
2.4. qRT‑PCR Confirmation of Selected Gene Expression Levels
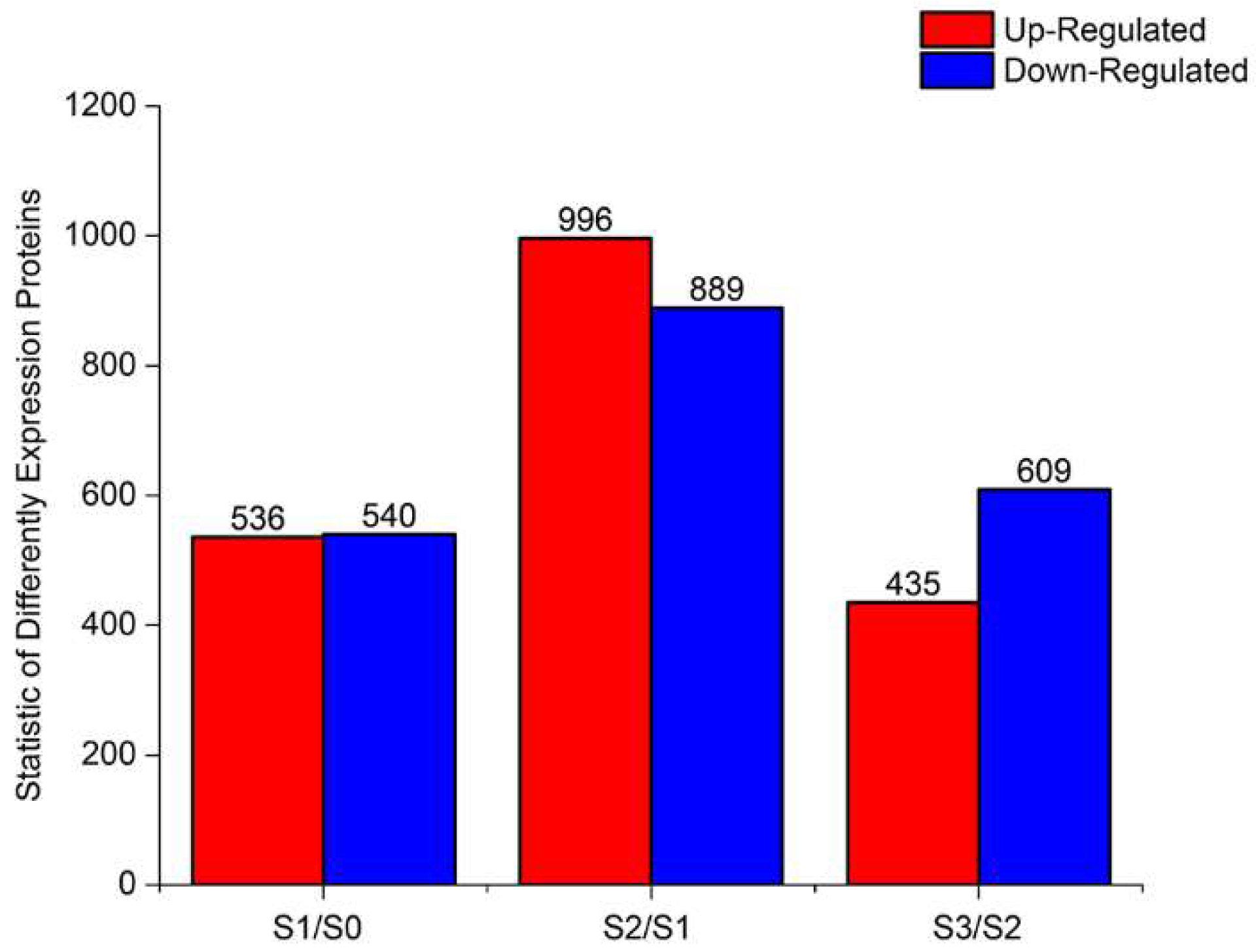
2.5. Proteomic Analysis of DEPs During AR Formation
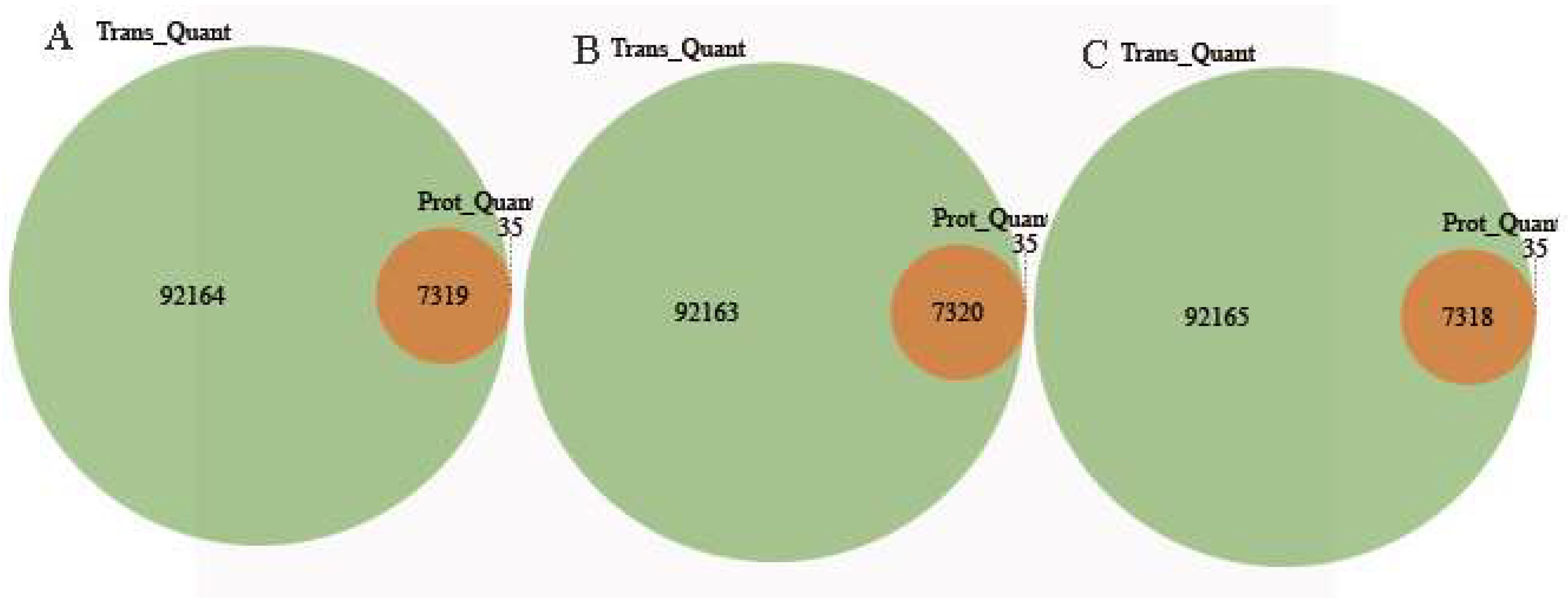
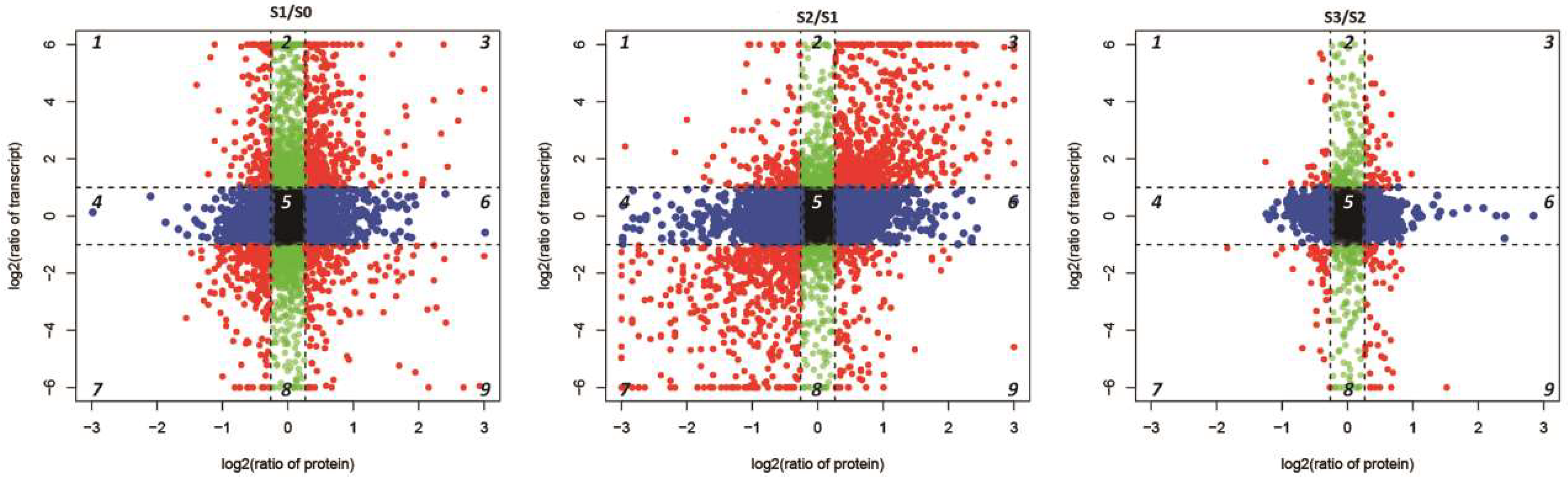
2.6. Conjoint Analysis of DEPs and DEGs During AR Formation
2.7. Identification of TFs During AR Formation
3. Discussion
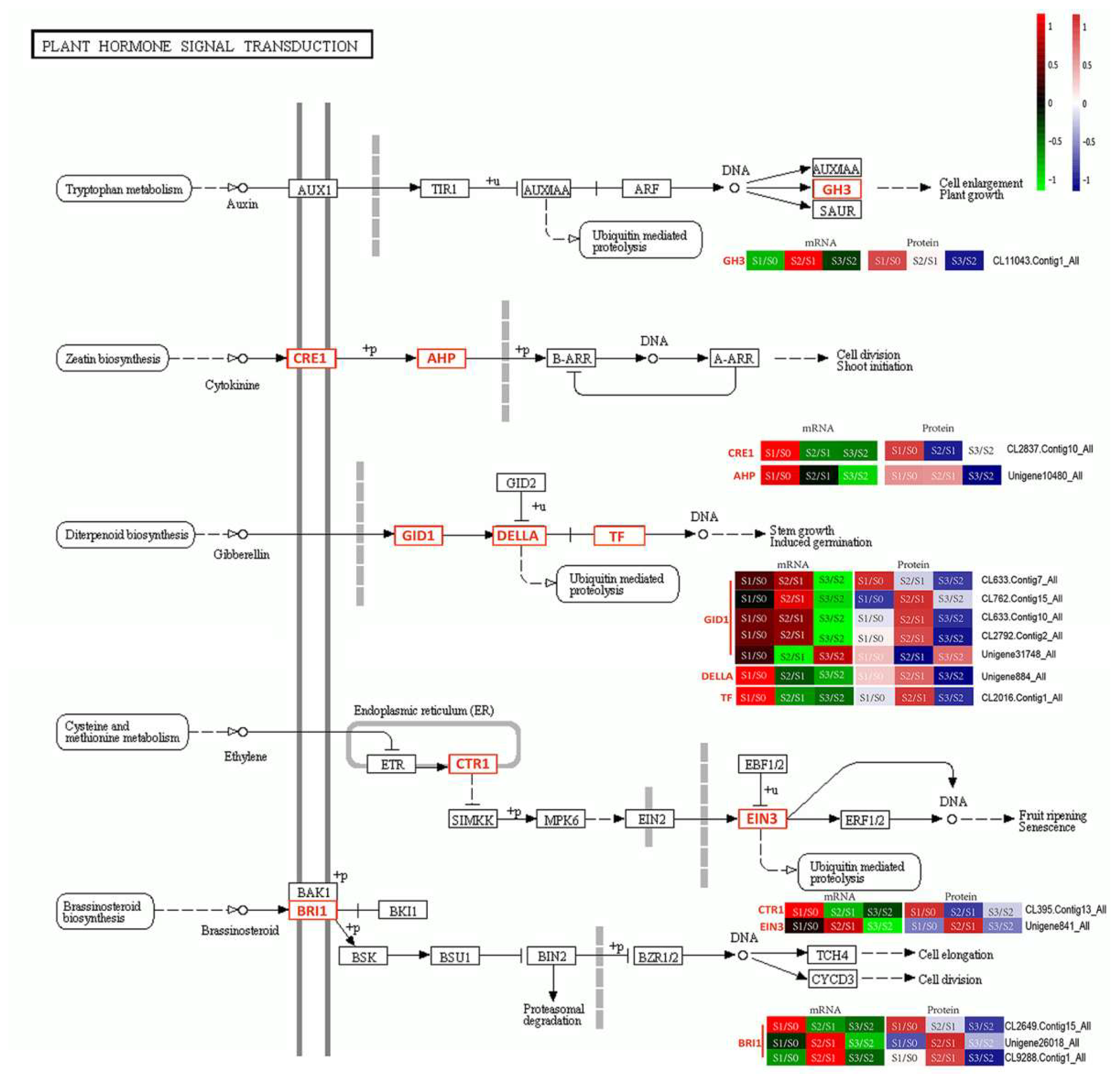
3.1. Plant Hormones in AR Formation
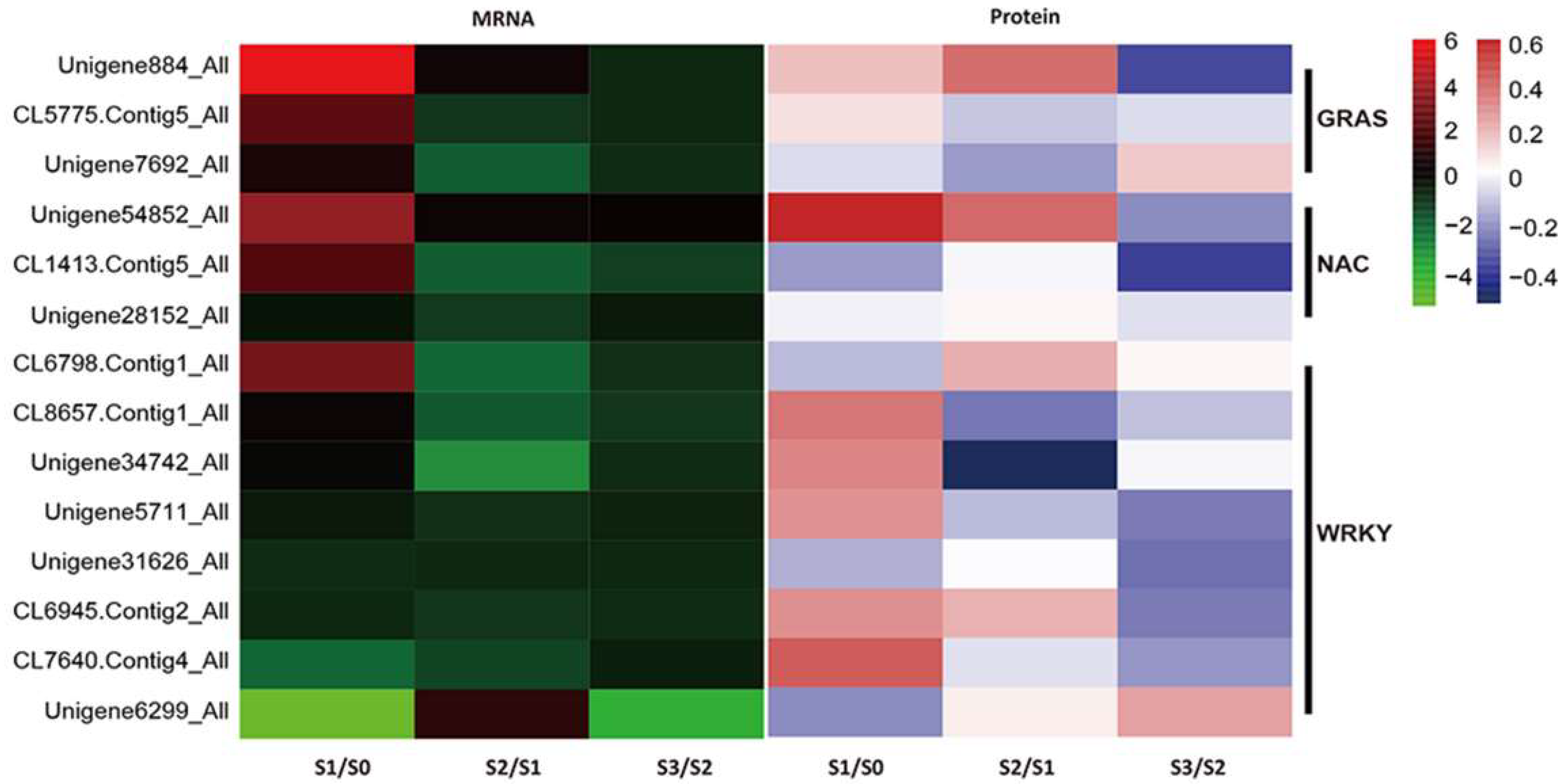
3.2. Identification of Other Potential Regulators Involved in AR Formation
3.3. TFs in AR Formation
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Isolation, Illumina Sequencing and Raw Data Processing
4.3. Functional Annotation of the Transcriptome
4.4. Differential Expression Analysis
4.5. Quantitative Real-Time PCR (qRT-PCR) Validation of DEGs
4.6. Protein Extraction and Isobaric Tags for Relative and Absolute Quantitation (iTRAQ) Reagent Labeling
4.7. Liquid Chromatography Linked to Tandem Mass Spectrometry (LC-MS/MS) Analysis, Protein Identification and Quantification
4.8. Conjoint Analysis of Transcriptomic and Proteomic Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | Adventitious root |
| T. ‘Zhongshanshan’ | Taxodium ‘Zhongshanshan’ |
| NT | NCBI non-redundant nucleotide sequences |
| NR | NCBI non-redundant protein sequences |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KOs | KEGG orthologs |
| COG | Cluster of Orthologous Groups of proteins |
| GO | gene ontology |
| TF | transcription factor |
| PCA | principal component analysis |
| DEGs | differentially expressed genes |
| qRT-PCR | Quantitative real-time PCR |
| iTRAQ | isobaric tags for relative and absolute quantitation |
| RNA-Seq | RNA sequencing |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| HPLC | high-performance liquid chromatography |
| LC-MS/MS | Liquid chromatography linked to tandem mass spectrometry |
| DEPs | differentially expressed proteins |
| TCA | tricarboxylic acid cycle |
References
- Haissig, B.E.; Davis, T.D. A historical evaluation of adventitious rooting research to 1993. In Biology of Adventitious Root Formation; Springer: Boston, MA, USA, 1994; Volume 62, pp. 275–331. [Google Scholar]
- Wang, Z.; Yin, Y.; Hua, J.; Fan, W.; Yu, C.; Xuan, L.; Yu, F. Cloning and Characterization of ThSHRs and ThSCR Transcription Factors in Taxodium Hybrid ‘Zhongshanshan 406’. Genes 2017, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Rout, G.; Mohapatra, A.; Jain, S.M. Tissue culture of ornamental pot plant: A critical review on present scenario and future prospects. Biotechnol. Adv. 2006, 24, 531–560. [Google Scholar] [CrossRef] [PubMed]
- Villacorta-Martín, C.; Sánchez-García, A.B.; Villanova, J.; Cano, A.; van de Rhee, M.; de Haan, J.; Acosta, M.; Passarinho, P.; Pérez-Pérez, J.M. Gene expression profiling during adventitious root formation in carnation stem cuttings. BMC Genom. 2015, 16, 789. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Xie, W.; Huang, M. Two WUSCHEL-related HOMEOBOX genes, PeWOX11a and PeWOX11b, are involved in adventitious root formation of poplar. Physiol. Plant. 2015, 155, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Guan, L.; Murphy, A.S.; Peer, W.A.; Gan, L.; Li, Y.; Cheng, Z.-M. Physiological and molecular regulation of adventitious root formation. Crit. Rev. Plant Sci. 2015, 34, 506–521. [Google Scholar] [CrossRef]
- Zhang, B.; Tong, C.; Yin, T.; Zhang, X.; Zhuge, Q.; Huang, M.; Wang, M.; Wu, R. Detection of quantitative trait loci influencing growth trajectories of adventitious roots in Populus using functional mapping. Tree Genet. Genomes 2009, 5, 539–552. [Google Scholar] [CrossRef]
- Da Costa, C.T.; de Almeida, M.R.; Ruedell, C.M.; Schwambach, J.; Maraschin, F.S.; Fett-Neto, A.G. When stress and development go hand in hand: Main hormonal controls of adventitious rooting in cuttings. Front. Plant Sci. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Porfírio, S.; da Silva, M.D.G.; Cabrita, M.J.; Azadi, P.; Peixe, A. Reviewing current knowledge on olive (Olea europaea L.) adventitious root formation. Sci. Hortic. 2016, 198, 207–226. [Google Scholar] [CrossRef]
- Yu, C.; Xu, S.; Yin, Y. Transcriptome analysis of the Taxodium ‘Zhongshanshan 405’ roots in response to salinity stress. Plant Physiol. Biochem. 2016, 100, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gu, C.; Xuan, L.; Hua, J.; Shi, Q.; Fan, W.; Yin, Y.; Yu, F. Identification of suitable reference genes in Taxodium ‘Zhongshanshan’ under abiotic stresses. Trees 2017, 31, 1519–1530. [Google Scholar] [CrossRef]
- Qi, B.; Yang, Y.; Yin, Y.; Xu, M.; Li, H. De novo sequencing, assembly, and analysis of the Taxodium ‘Zhongshansa’roots and shoots transcriptome in response to short-term waterlogging. BMC Plant Biol. 2014, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yu, C. Hybridization between Taxodium Species; China Forestry Publishing House: Beijing, China, 2005. [Google Scholar]
- Wang, Z.; Xu, J.; Li, H.; Yu, C.; Yin, Y. Rooting capabilities for Taxodium ‘Zhongshanshan’ 302, 118, and 405. J. Zhejiang A F Univ. 2015, 32, 648–654. [Google Scholar] [CrossRef]
- Ribeiro, C.L.; Silva, C.M.; Drost, D.R.; Novaes, E.; Novaes, C.R.; Dervinis, C.; Kirst, M. Integration of genetic, genomic and transcriptomic information identifies putative regulators of adventitious root formation in Populus. BMC Plant Biol. 2016, 16, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Ma, L.; Li, Y.; Wang, S.a.; Li, L.; Yang, R.; Ma, Y.; Wang, Q. Transcriptome profiling of indole-3-butyric acid-induced adventitious root formation in softwood cuttings of the Catalpa bungei variety ‘YU-1’ at different developmental stages. Genes Genom. 2016, 38, 145–162. [Google Scholar] [CrossRef]
- Álvarez, C.; Valledor, L.; Sáez, P.; Sánchez-Olate, M.; Ríos, D. Proteomic Analysis through Adventitious Rooting of Pinus radiata Stem Cuttings with Different Rooting Capabilities. Am. J. Plant Sci. 2016, 7, 1888–1904. [Google Scholar] [CrossRef]
- Muers, M. Gene expression: Transcriptome to proteome and back to genome. Nat. Rev. Genet. 2011, 12, 518. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Wang, H.-Y.; Zhang, T. Comprehensive and Quantitative Proteomic Analysis of Metamorphosis-Related Proteins in the Veined Rapa Whelk, Rapana venosa. Int. J. Mol. Sci. 2016, 17, 924. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Saha, K.; Choudhuri, M. Role of auxin and polyamines in adventitious root formation in relation to changes in compounds involved in rooting. J. Plant Growth Regul. 2001, 20, 182–194. [Google Scholar] [CrossRef]
- Ramanathan, A.; Kiran, P.; Arnold, G.; Wang, W.; Abboud, R. Repeatability of the Pedar-X® in-shoe pressure measuring system. Foot Ankle Surg. 2010, 16, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Bellini, C.; Pacurar, D.I.; Perrone, I. Adventitious roots and lateral roots: Similarities and differences. Annu. Rev. Plant Biol. 2014, 65, 639–666. [Google Scholar] [CrossRef] [PubMed]
- Tiberia, I.P.; Pamfil, D.; Bellini, C. Auxin control in the formation of adventitious roots. Not. Bot. Horti Agrobot. 2011, 39, 307. [Google Scholar] [CrossRef]
- Preece, J.E. A Century of Progress with Vegetative Plant Propagation. Hortscience 2003, 38, 1015–1025. [Google Scholar] [CrossRef]
- Rademacher, W. Plant Growth Regulators: Backgrounds and Uses in Plant Production. J. Plant Growth Regul. 2015, 34, 845–872. [Google Scholar] [CrossRef]
- Sorin, C.; Negroni, L.; Balliau, T.; Corti, H.; Jacquemot, M.-P.; Davanture, M.; Sandberg, G.; Zivy, M.; Bellini, C. Proteomic analysis of different mutant genotypes of Arabidopsis led to the identification of 11 proteins correlating with adventitious root development. Plant Physiol. 2006, 140, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, L.; Mongelard, G.; Floková, K.; Păcurar, D.I.; Novák, O.; Staswick, P.; Kowalczyk, M.; Păcurar, M.; Demailly, H.; Geiss, G. Auxin controls Arabidopsis adventitious root initiation by regulating jasmonic acid homeostasis. Plant Cell 2012, 24, 2515–2527. [Google Scholar] [CrossRef] [PubMed]
- Laplaze, L.; Benkova, E.; Casimiro, I.; Maes, L.; Vanneste, S.; Swarup, R.; Weijers, D.; Calvo, V.; Parizot, B.; Herrera-Rodriguez, M.B. Cytokinins act directly on lateral root founder cells to inhibit root initiation. Plant Cell 2007, 19, 3889–3900. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Carvajal, G.A.; Morse, A.M.; Dervinis, C.; Davis, J.M. The cytokinin type-B response regulator PtRR13 is a negative regulator of adventitious root development in Populus. Plant Physiol. 2009, 150, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Ricci, A.; Carra, A.; Torelli, A.; Maggiali, C.A.; Morini, G.; Branca, C. Cytokinin-like activity of N,N′-diphenylureas. N,N′-bis-(2,3-methylenedioxyphenyl) urea and N,N′-bis-(3,4-methylenedioxyphenyl) urea enhance adventitious root formation in apple rootstock M26 (Malus pumila Mill.). Plant Sci. 2001, 160, 1055–1065. [Google Scholar] [CrossRef]
- Ricci, A.; Rolli, E.; Dramis, L.; Diaz-Sala, C. N,N′-bis-(2,3-methylenedioxyphenyl) urea and N,N′-bis-(3,4-methylenedioxyphenyl) urea enhance adventitious rooting in Pinus radiata and affect expression of genes induced during adventitious rooting in the presence of exogenous auxin. Plant Sci. 2008, 175, 356–363. [Google Scholar] [CrossRef]
- Duan, D.; Jia, Y.; Yang, J.; Li, Z.-H. Comparative Transcriptome Analysis of Male and Female Conelets and Development of Microsatellite Markers in Pinus bungeana, an Endemic Conifer in China. Genes 2017, 8, 393. [Google Scholar] [CrossRef] [PubMed]
- Imin, N.; Goffard, N.; Nizamidin, M.; Rolfe, B.G. Genome-wide transcriptional analysis of super-embryogenic Medicago truncatula explant cultures. BMC Plant Biol. 2008, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Zhang, X.; Nie, H.; Liu, M.; Cheng, J.L. Transcript profiling analysis reveals crucial genes regulating main metabolism during adventitious root formation in cuttings of Morus alba L. Plant Growth Regul. 2016, 79, 251–262. [Google Scholar] [CrossRef]
- Druege, U.; Franken, P.; Lischewski, S.; Ahkami, A.H.; Zerche, S.; Hause, B.; Hajirezaei, M.R. Transcriptomic analysis reveals ethylene as stimulator and auxin as regulator of adventitious root formation in petunia cuttings. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Xuan, L.; Xu, L.-A.; Huang, M.; Xu, M. Molecular cloning, expression analysis and subcellular localization of four DELLA genes from hybrid poplar. SpringerPlus 2016, 5, 1129. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Yao, T.; Li, M.; Guo, X.; Zhang, Y.; Zhu, S.; He, Y. PIF3 is involved in the primary root growth inhibition of Arabidopsis induced by nitric oxide in the light. Mol. Plant 2014, 7, 616–625. [Google Scholar] [CrossRef] [PubMed]
- Leivar, P.; Quail, P.H. PIFs: Pivotal components in a cellular signaling hub. Trends Plant Sci. 2011, 16, 19. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H. Brassinolide promotes adventitious shoot regeneration from cauliflower hypocotyl segments. PCTOC 2002, 71, 111–116. [Google Scholar] [CrossRef]
- Bao, F.; Shen, J.; Brady, S.R.; Muday, G.K.; Asami, T.; Yang, Z. Brassinosteroids interact with auxin to promote lateral root development in Arabidopsis. Plant Physiol. 2004, 134, 1624–1631. [Google Scholar] [CrossRef] [PubMed]
- Rönsch, H.; Adam, G.; Matschke, J.; Schachler, G. Influence of (22S, 23S)-homobrassinolide on rooting capacity and survival of adult Norway spruce cuttings. Tree Physiol. 1993, 12, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Sharma, I.; Kaur, N.; Pati, P.K. Auxin: A master regulator in plant root development. Plant Cell Rep. 2013, 32, 741–757. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.; Maharjan, P.M.; Lee, O.; Fujioka, S.; Jang, S.; Kim, B.; Takatsuto, S.; Tsujimoto, M.; Kim, H.; Cho, S. Auxin stimulates DWARF4 expression and brassinosteroid biosynthesis in Arabidopsis. Plant J. 2011, 66, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, C.; Hess, C.E. Isozymic changes in relation to root initiation in mung bean. Can. J. Bot. 1974, 52, 295–297. [Google Scholar] [CrossRef]
- Upadhyaya, A.; Davis, T.D.; Sankhla, N. Some biochemical changes associated with paclobutrazol-induced adventitious root formation on bean hypocotyl cuttings. Ann. Bot. 1986, 57, 309–315. [Google Scholar] [CrossRef]
- Nodzon, L.A.; Xu, W.H.; Wang, Y.; Pi, L.Y.; Chakrabarty, P.K.; Song, W.Y. The ubiquitin ligase XBAT32 regulates lateral root development in Arabidopsis. Plant J. 2004, 40, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A. The ubiquitin–proteasome pathway: On protein death and cell life. EMBO J. 1998, 17, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- Brinker, M.; van Zyl, L.; Liu, W.; Craig, D.; Sederoff, R.R.; Clapham, D.H.; von Arnold, S. Microarray analyses of gene expression during adventitious root development in Pinus contorta. Plant Physiol. 2004, 135, 1526–1539. [Google Scholar] [CrossRef] [PubMed]
- Fogaça, C.M.; Fett-Neto, A.G. Role of auxin and its modulators in the adventitious rooting of Eucalyptus species differing in recalcitrance. Plant Growth Regul. 2005, 45, 1–10. [Google Scholar] [CrossRef]
- Rapaka, V.K.; Bessler, B.; Schreiner, M.; Druege, U. Interplay between initial carbohydrate availability, current photosynthesis, and adventitious root formation in Pelargonium cuttings. Plant Sci. 2005, 168, 1547–1560. [Google Scholar] [CrossRef]
- Liu, R.; Chen, S.; Jiang, J.; Zhu, L.; Zheng, C.; Han, S.; Gu, J.; Sun, J.; Li, H.; Wang, H. Proteomic changes in the base of chrysanthemum cuttings during adventitious root formation. BMC Genom. 2013, 14, 919. [Google Scholar] [CrossRef] [PubMed]
- Haissig, B. Metabolism during adventitious root primordium initiation and development. N. Z. J. For. Sci. 1974, 4, 324–337. [Google Scholar]
- Rigal, A.; Yordanov, Y.S.; Perrone, I.; Karlberg, A.; Tisserant, E.; Bellini, C.; Busov, V.B.; Martin, F.; Kohler, A.; Bhalerao, R. The AINTEGUMENTA LIKE1 homeotic transcription factor PtAIL1 controls the formation of adventitious root primordia in poplar. Plant Physiol. 2012, 160, 1996–2006. [Google Scholar] [CrossRef] [PubMed]
- Vielba, J.M.; Díaz-Sala, C.; Ferro, E.; Rico, S.; Lamprecht, M.; Abarca, D.; Ballester, A.; Sánchez, C. CsSCL1 is differentially regulated upon maturation in chestnut microshoots and is specifically expressed in rooting-competent cells. Tree Physiol. 2011, 31, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Abarca, D.; Pizarro, A.; Hernández, I.; Sánchez, C.; Solana, S.P.; del Amo, A.; Carneros, E.; Díaz-Sala, C. The GRAS gene family in pine: Transcript expression patterns associated with the maturation-related decline of competence to form adventitious roots. BMC Plant Biol. 2014, 14, 354. [Google Scholar] [CrossRef] [PubMed]
- Legué, V.; Rigal, A.; Bhalerao, R.P. Adventitious root formation in tree species: Involvement of transcription factors. Physiol. Plant. 2014, 151, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Guo, H.-S.; Dallman, G.; Fang, S.; Weissman, A.M.; Chua, N.-H. SINAT5 promotes ubiquitin-related degradation of NAC1 to attenuate auxin signals. Nature 2002, 419, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.-S.; Xie, Q.; Fei, J.-F.; Chua, N.-H. MicroRNA directs mRNA cleavage of the transcription factor NAC1 to downregulate auxin signals for Arabidopsis lateral root development. Plant Cell 2005, 17, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Pijut, P.M.; Woeste, K.E.; Michler, C.H. 6 Promotion of Adventitious Root Formation of Difficult-to-Root Hardwood Tree Species. Hortic. Rev. 2011, 38, 213. [Google Scholar] [CrossRef]
- Ali, M.A.; Wieczorek, K.; Kreil, D.P.; Bohlmann, H. The beet cyst nematode Heterodera schachtii modulates the expression of WRKY transcription factors in syncytia to favour its development in Arabidopsis roots. PLoS ONE 2014, 9, e102360. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Somssich, I.E. Networks of WRKY transcription factors in defense signaling. Curr. Opin. Plant Biol. 2007, 10, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sun, Z.; Xu, M. Identification and characterization of long non-coding RNAs involved in the formation and development of poplar adventitious roots. Ind. Crops Prod. 2018, 118, 334–346. [Google Scholar] [CrossRef]
- Sun, Q.; Zhou, G.; Cai, Y.; Fan, Y.; Zhu, X.; Liu, Y.; He, X.; Shen, J.; Jiang, H.; Hu, D. Transcriptome analysis of stem development in the tumourous stem mustard Brassica juncea var. tumida Tsen et Lee by RNA sequencing. BMC Plant Biol. 2012, 12, 53. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Yu, G.; Chen, Y.; Jiang, S.; Wang, M.; Jin, Y.; Lan, X.; Liang, Y.; Sun, H. Genome-wide transcriptome and proteome analysis on different developmental stages of Cordyceps militaris. PLoS ONE 2012, 7, e51853. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR Gene Indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xiong, Y.; Xu, L.; Zhang, Q.; Luo, Z. An integrated analysis based on transcriptome and proteome reveals deastringency-related genes in CPCNA persimmon. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Xu, S.; Wang, Z.; Liu, L.; Zhang, Y.; Deng, Y.; Huang, S. De novo sequencing, assembly, and analysis of Iris lactea var. chinensisroots’ transcriptome in response to salt stress. Plant Physiol. Biochem. 2018, 125, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Iseli, C.; Jongeneel, C.V.; Bucher, P. ESTScan: A Program for Detecting, Evaluating, and Reconstructing Potential Coding Regions in EST Sequences; ISMB: Heidelberg, Germany, 1999; pp. 138–148. [Google Scholar]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-T.; Qi, Y.-P.; Lu, Y.-B.; Guo, P.; Sang, W.; Feng, H.; Zhang, H.-X.; Chen, L.-S. iTRAQ protein profile analysis of Citrus sinensis roots in response to long-term boron-deficiency. J. Proteom. 2013, 93, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, B.; Wang, Q.; Tong, W.; Guo, J.; Bai, X.; Zhao, J.; Sun, Y.; Tang, Q.; Lin, Z. Quantitative proteomics reveals the temperature-dependent proteins encoded by a series of cluster genes in Thermoanaerobacter tengcongensis. Mol. Cell. Proteom. 2013, 12, 2266–2277. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Zhou, R.; Feng, Q.; Wang, Q.; Wang, J.; Liu, S. IQuant: An automated pipeline for quantitative proteomics based upon isobaric tags. Proteomics 2014, 14, 2280–2285. [Google Scholar] [CrossRef] [PubMed]








 , indirect effect;
, indirect effect;  , activation;
, activation;  , inhibition;
, inhibition;  , expression;
, expression;  , indirect effect;
, indirect effect;  , dissociation.
, indirect effect; , activation; , inhibition; , expression; , indirect effect; , dissociation.
, dissociation.
, indirect effect; , activation; , inhibition; , expression; , indirect effect; , dissociation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Values | Total | Nr Annotated | Nt Annotated | Swissprot Annotated | KEGG Annotated | COG Annotated | Interpro Annotated | GO Annotated | Overall |
|---|---|---|---|---|---|---|---|---|---|
| Number | 105,879 | 68,046 | 48,467 | 51,068 | 53,682 | 32,668 | 55,196 | 23,381 | 71,637 |
| Percentage | 100% | 64.27% | 45.78% | 48.23% | 50.70% | 30.85% | 52.13% | 22.08% | 67.66% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Z.; Hua, J.; Yin, Y.; Gu, C.; Yu, C.; Shi, Q.; Guo, J.; Xuan, L.; Yu, F. An Integrated Transcriptome and Proteome Analysis Reveals Putative Regulators of Adventitious Root Formation in Taxodium ‘Zhongshanshan’. Int. J. Mol. Sci. 2019, 20, 1225. https://doi.org/10.3390/ijms20051225
Wang Z, Hua J, Yin Y, Gu C, Yu C, Shi Q, Guo J, Xuan L, Yu F. An Integrated Transcriptome and Proteome Analysis Reveals Putative Regulators of Adventitious Root Formation in Taxodium ‘Zhongshanshan’. International Journal of Molecular Sciences. 2019; 20(5):1225. https://doi.org/10.3390/ijms20051225
Chicago/Turabian StyleWang, Zhiquan, Jianfeng Hua, Yunlong Yin, Chunsun Gu, Chaoguang Yu, Qin Shi, Jinbo Guo, Lei Xuan, and Fangyuan Yu. 2019. "An Integrated Transcriptome and Proteome Analysis Reveals Putative Regulators of Adventitious Root Formation in Taxodium ‘Zhongshanshan’" International Journal of Molecular Sciences 20, no. 5: 1225. https://doi.org/10.3390/ijms20051225
APA StyleWang, Z., Hua, J., Yin, Y., Gu, C., Yu, C., Shi, Q., Guo, J., Xuan, L., & Yu, F. (2019). An Integrated Transcriptome and Proteome Analysis Reveals Putative Regulators of Adventitious Root Formation in Taxodium ‘Zhongshanshan’. International Journal of Molecular Sciences, 20(5), 1225. https://doi.org/10.3390/ijms20051225