Novel 8-Substituted Coumarins That Selectively Inhibit Human Carbonic Anhydrase IX and XII

 ,
, 
 and
and 
Abstract

1. Introduction
2. Results and Discussion
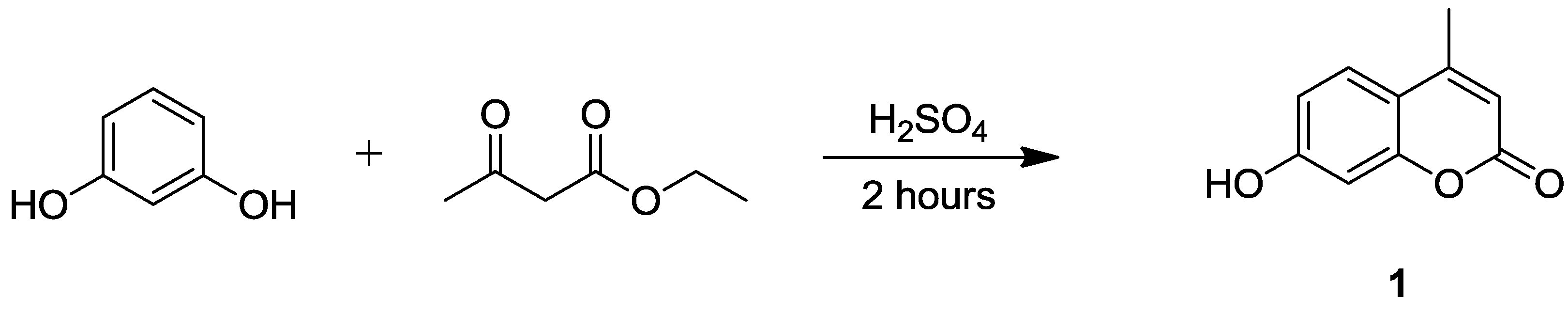
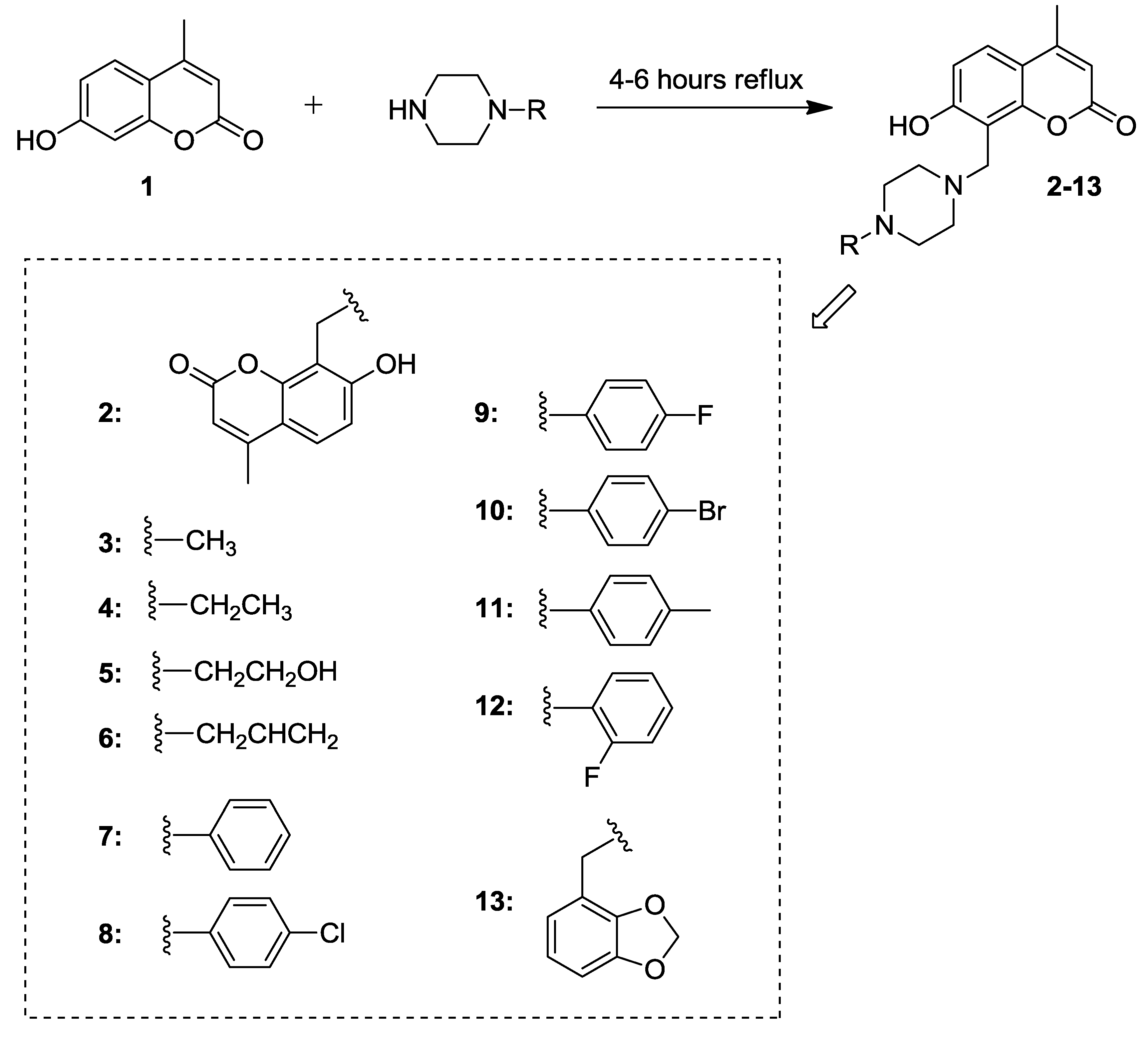
2.1. Synthesis of 7-Hydroxycoumarin Derivatives
2.2. Inhibitory Activity against hCA Isoforms
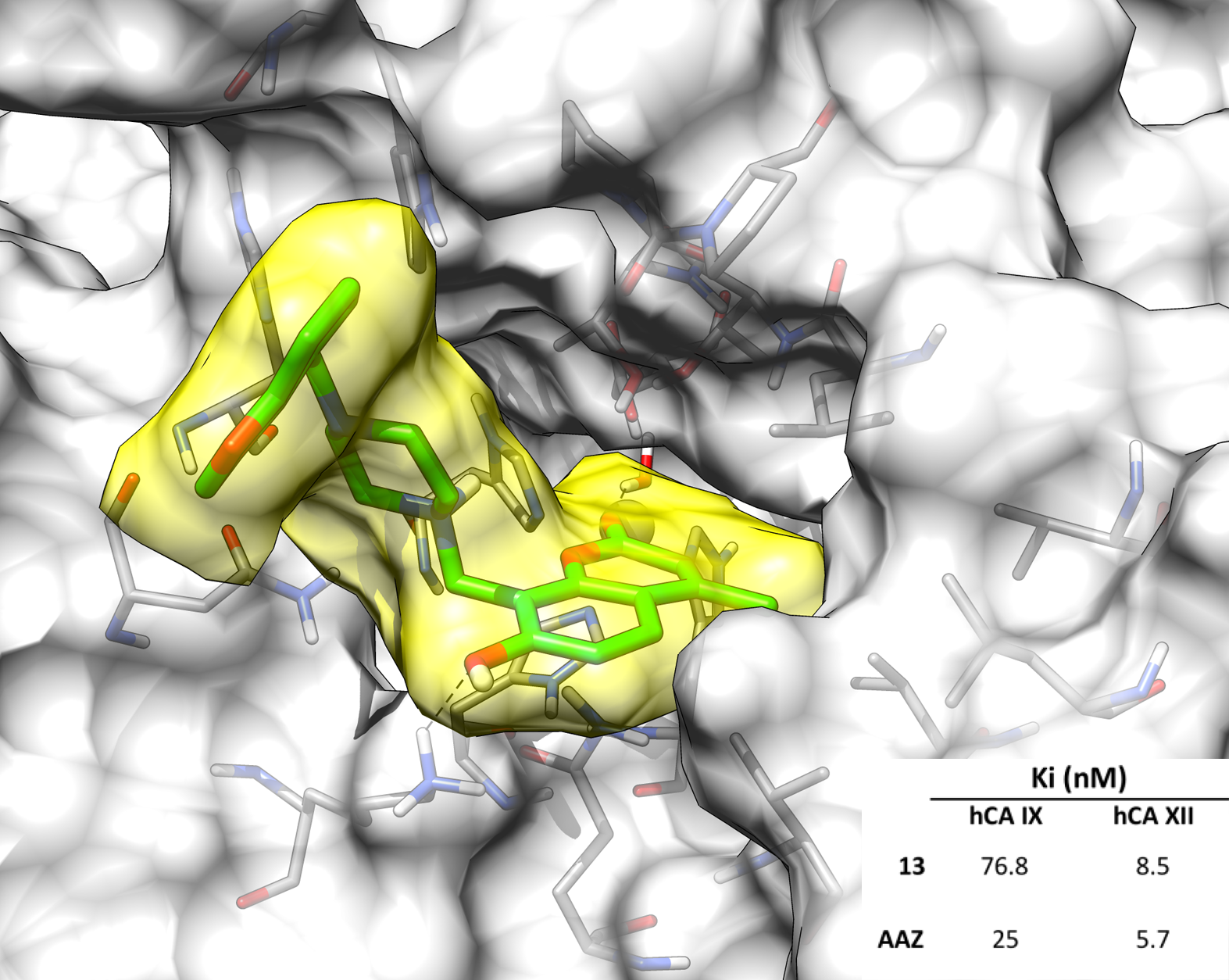
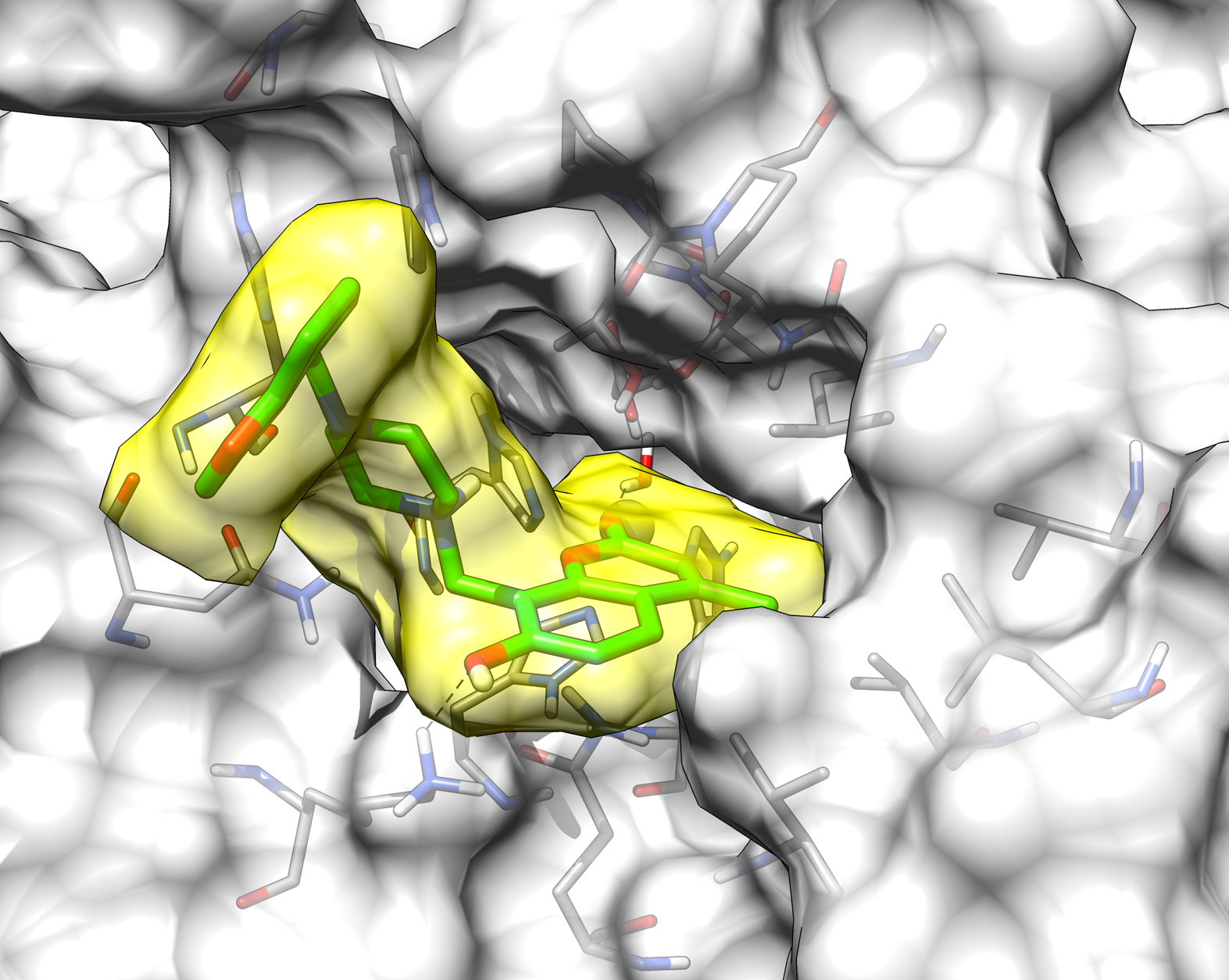
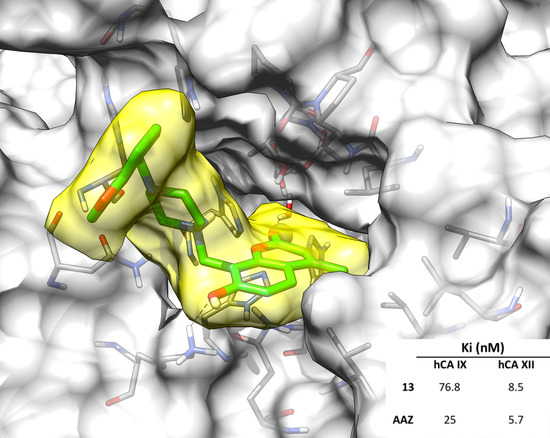
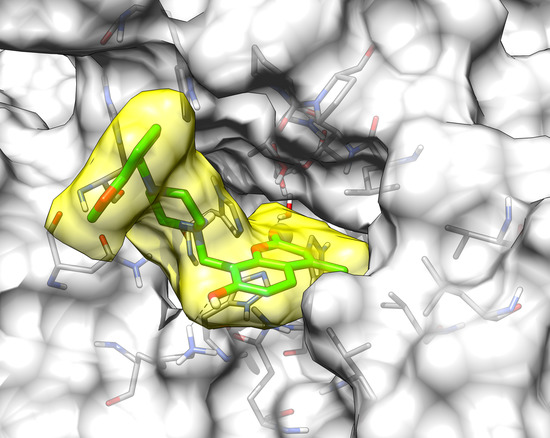
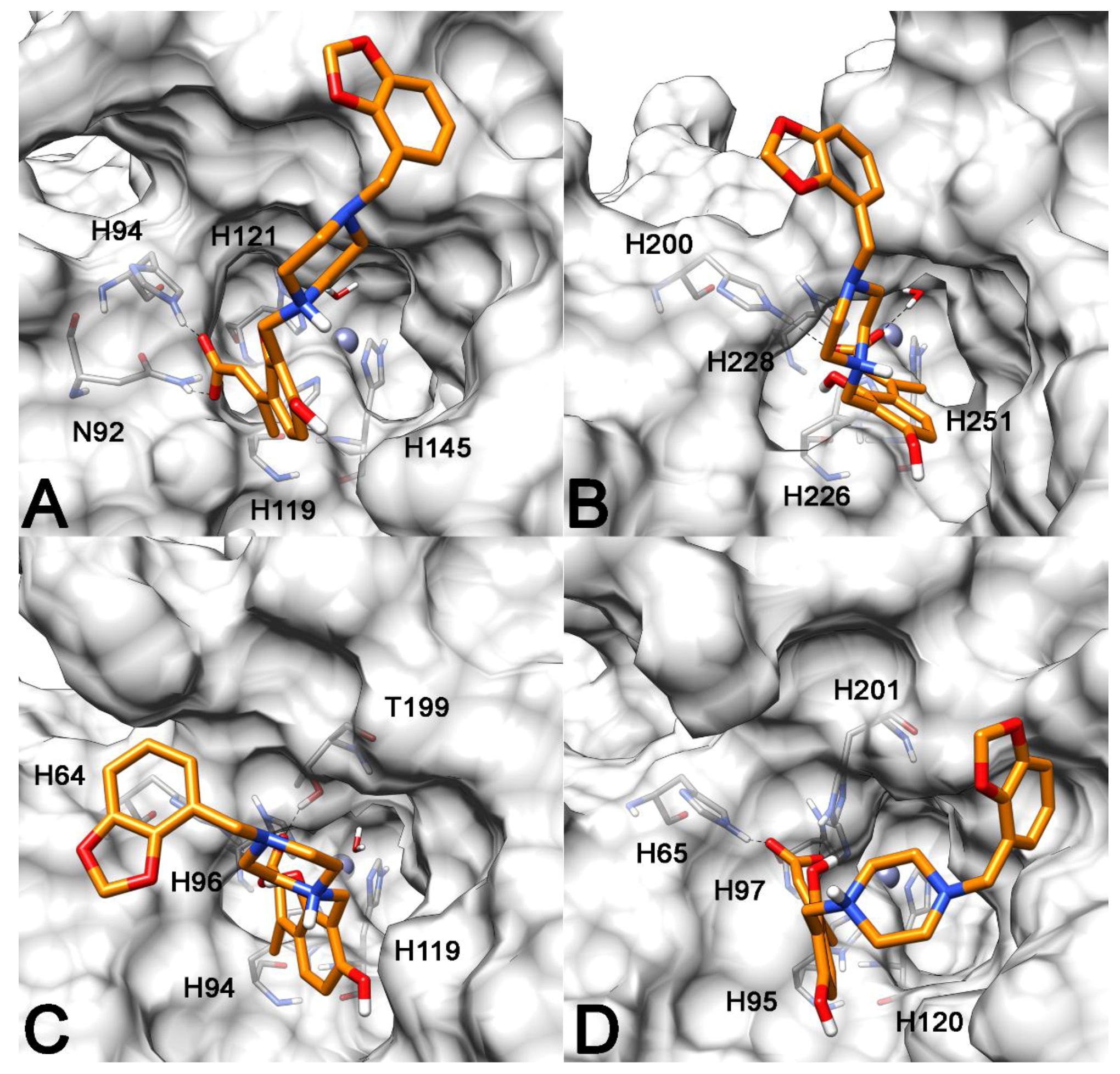
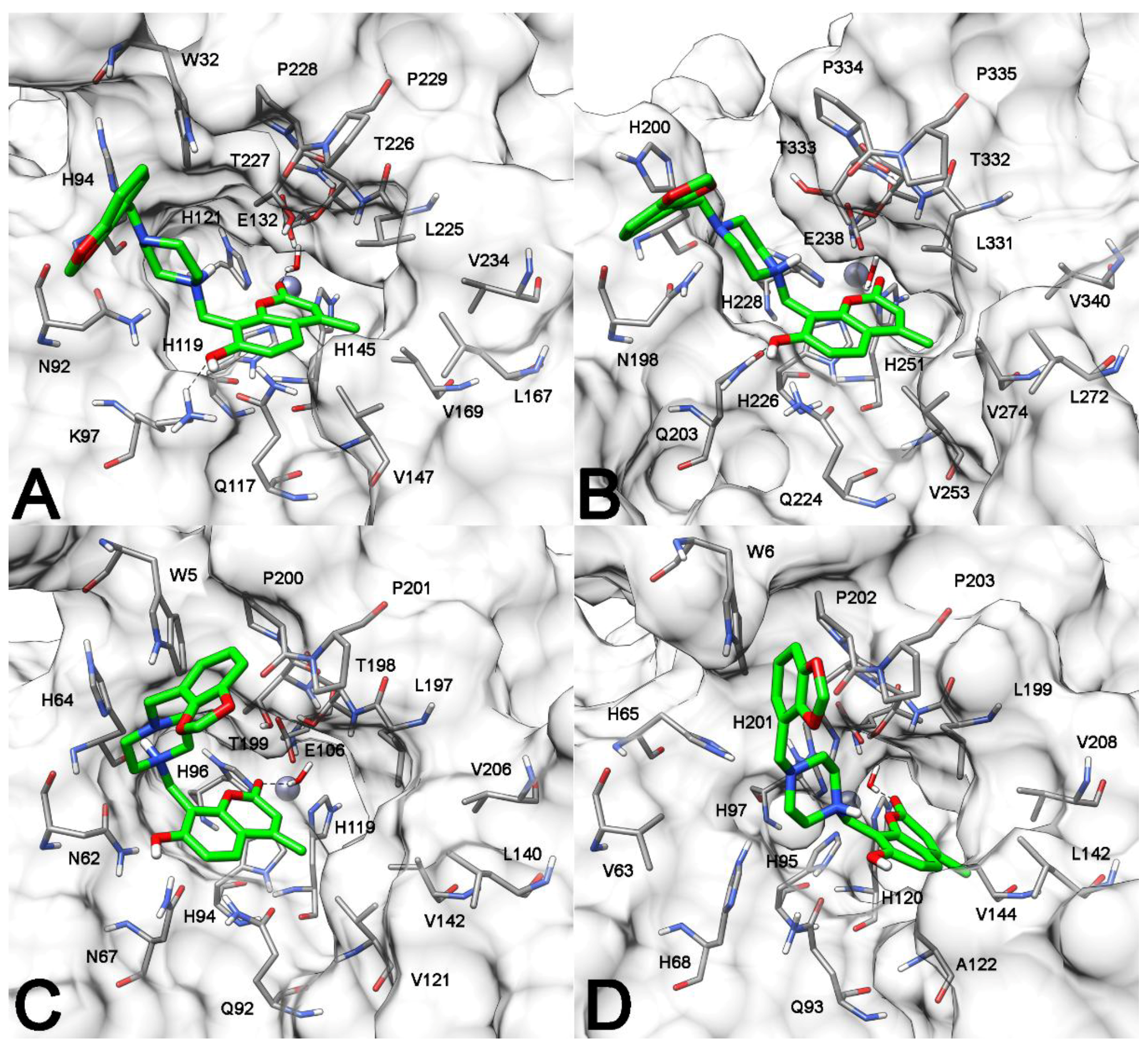
2.3. Molecular Modeling Studies
3. Materials and Methods
3.1. Chemistry
3.2. Carbonic Anhydrase Inhibition Assay
3.3. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Alterio, V.; di Fiore, A.; D’Ambrosio, K.; Supuran, C.T.; De Simone, G. Multiple Binding Modes of Inhibitors to Carbonic Anhydrases: How to Design Specific Drugs Targeting 15 Different Isoforms? Chem. Rev. 2012, 112, 4421–4468. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Supuran, C.T. Diuretics with carbonic anhydrase inhibitory action: A patent and literature review (2005–2013). Expert Opin. Ther. Pat. 2013, 23, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Masini, E.; Carta, F.; Scozzafava, A.; Supuran, C.T. Antiglaucoma carbonic anhydrase inhibitors: A patent review. Expert Opin. Ther. Pat. 2013, 23, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.M.; Supuran, C.T.; De Simone, G. Anticancer carbonic anhydrase inhibitors: A patent review (2008–2013). Expert Opin. Ther. Pat. 2013, 23, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic Anhydrase Inhibition and the Management of Hypoxic Tumors. Metabolites 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Scozzafava, A.; Supuran, C.T.; Carta, F. Antiobesity carbonic anhydrase inhibitors: A literature and patent review. Expert Opin. Ther. Pat. 2013, 23, 725–735. [Google Scholar] [CrossRef] [PubMed]
- Carta, F.; Supuran, C.T.; Scozzafava, A. Sulfonamides and their isosters as carbonic anhydrase inhibitors. Future Med. Chem. 2014, 6, 1149–1165. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.; McKenna, R. Carbonic anhydrase inhibitors: A review on the progress of patent literature (2011–2016). Expert Opin. Ther. Pat. 2016, 26, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Shetnev, A.; Sharonova, T.; Baykov, S.; Kalinin, S.; Nocentini, A.; Sharoyko, V.; Poli, G.; Tuccinardi, T.; Presnukhina, S.; et al. Continued exploration of 1,2,4-oxadiazole periphery for carbonic anhydrase-targeting primary arene sulfonamides: Discovery of subnanomolar inhibitors of membrane-bound hCA IX isoform that selectively kill cancer cells in hypoxic environment. Eur. J. Med. Chem. 2019, 164, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Andreucci, E.; Ruzzolini, J.; Peppicelli, S.; Bianchini, F.; Laurenzana, A.; Carta, F.; Supuran, C.T.; Calorini, L. The carbonic anhydrase IX inhibitor SLC-0111 sensitises cancer cells to conventional chemotherapy. J. Enzyme Inhib. Med. Chem. 2019, 34, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Lomelino, C.; Supuran, C.; McKenna, R. Non-Classical Inhibition of Carbonic Anhydrase. Int. J. Mol. Sci. 2016, 17, 1150. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Vu, H.; Pham, N.B.; Poulsen, S.-A.; Scozzafava, A.; Quinn, R.J.; Supuran, C.T. Non-Zinc Mediated Inhibition of Carbonic Anhydrases: Coumarins Are a New Class of Suicide Inhibitors. J. Am. Chem. Soc. 2009, 131, 3057–3062. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Temperini, C.; Pochet, L.; Masereel, B.; Scozzafava, A.; Supuran, C.T. Deciphering the Mechanism of Carbonic Anhydrase Inhibition with Coumarins and Thiocoumarins. J. Med. Chem. 2010, 53, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Maresca, A.; Scozzafava, A.; Supuran, C.T. 7,8-Disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg. Med. Chem. Lett. 2010, 20, 7255–7258. [Google Scholar] [CrossRef] [PubMed]
- Touisni, N.; Maresca, A.; McDonald, P.C.; Lou, Y.; Scozzafava, A.; Dedhar, S.; Winum, J.-Y.; Supuran, C.T. Glycosyl Coumarin Carbonic Anhydrase IX and XII Inhibitors Strongly Attenuate the Growth of Primary Breast Tumors. J. Med. Chem. 2011, 54, 8271–8277. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; McDonald, P.C.; Oloumi, A.; Chia, S.; Ostlund, C.; Ahmadi, A.; Kyle, A.; auf dem Keller, U.; Leung, S.; Huntsman, D.; et al. Targeting Tumor Hypoxia: Suppression of Breast Tumor Growth and Metastasis by Novel Carbonic Anhydrase IX Inhibitors. Cancer Res. 2011, 71, 3364–3376. [Google Scholar] [CrossRef] [PubMed]
- Ferraroni, M.; Carta, F.; Scozzafava, A.; Supuran, C.T. Thioxocoumarins Show an Alternative Carbonic Anhydrase Inhibition Mechanism Compared to Coumarins. J. Med. Chem. 2016, 59, 462–473. [Google Scholar] [CrossRef] [PubMed]
- De Luca, L.; Mancuso, F.; Ferro, S.; Buemi, M.R.; Angeli, A.; del Prete, S.; Capasso, C.; Supuran, C.T.; Gitto, R. Inhibitory effects and structural insights for a novel series of coumarin-based compounds that selectively target human CA IX and CA XII carbonic anhydrases. Eur. J. Med. Chem. 2018, 143, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Yuan, M.; Zhang, S.; Wu, J.; Qi, S.; Li, Q. Complete assignments of 1H and 13C-NMR data for ten phenylpiperazine derivatives. Magn. Reson. Chem. 2005. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, S.; Xu, X.; Liu, X.; Yu, M.; Zhao, S.; Liu, S.; Qiu, Y.; Zhang, T.; Liu, B.-F.; et al. Synthesis and Biological Investigation of Coumarin Piperazine (Piperidine) Derivatives as Potential Multireceptor Atypical Antipsychotics. J. Med. Chem. 2013, 56, 4671–4690. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, M.; Nieddu, E.; Miele, M.; Balbi, A.; Ferrone, M.; Fermeglia, M.; Mazzei, M.T.; Pricl, S.; la Colla, P.; Marongiu, F.; et al. Activity of Mannich bases of 7-hydroxycoumarin against Flaviviridae. Bioorg. Med. Chem. 2008, 16, 2591–2605. [Google Scholar] [CrossRef] [PubMed]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martins, D.; Forli, S.; Ramos, M.J.; Olson, A.J. AutoDock4Zn: An improved AutoDock force field for small-molecule docking to zinc metalloproteins. J. Chem. Inf. Model. 2014, 54, 2371–2379. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Jha, V.; Martinelli, A.; Supuran, C.; Tuccinardi, T. Development of a Fingerprint-Based Scoring Function for the Prediction of the Binding Mode of Carbonic Anhydrase II Inhibitors. Int. J. Mol. Sci. 2018, 19, 1851. [Google Scholar] [CrossRef] [PubMed]
- Aghazadeh Tabrizi, M.; Baraldi, P.G.; Ruggiero, E.; Saponaro, G.; Baraldi, S.; Poli, G.; Tuccinardi, T.; Ravani, A.; Vincenzi, F.; Borea, P.A.; et al. Synthesis and structure activity relationship investigation of triazolo[1,5-a]pyrimidines as CB2 cannabinoid receptor inverse agonists. Eur. J. Med. Chem. 2016, 113, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Tuccinardi, T.; Ferrarini, P.L.; Manera, C.; Ortore, G.; Saccomanni, G.; Martinelli, A. Cannabinoid CB2/CB1 selectivity. Receptor modeling and automated docking analysis. J. Med. Chem. 2006, 49. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. The AMBER molecular modeling package. Available online: casegroup.rutgers.edu/casegr-sh-2.html (accessed on 9 March 2019).
- Dal Piaz, F.; Vera Saltos, M.B.; Franceschelli, S.; Forte, G.; Marzocco, S.; Tuccinardi, T.; Poli, G.; Nejad Ebrahimi, S.; Hamburger, M.; De Tommasi, N.; et al. Drug Affinity Responsive Target Stability (DARTS) Identifies Laurifolioside as a New Clathrin Heavy Chain Modulator. J. Nat. Prod. 2016, 79, 2681–2692. [Google Scholar] [CrossRef] [PubMed]
- De Leo, M.; Huallpa, C.G.; Alvarado, B.; Granchi, C.; Poli, G.; De Tommasi, N.; Braca, A. New diterpenes from Salvia pseudorosmarinus and their activity as inhibitors of monoacylglycerol lipase (MAGL). Fitoterapia 2018, 130, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Abraham, K.; Wöhrlin, F.; Lindtner, O.; Heinemeyer, G.; Lampen, A. Toxicology and risk assessment of coumarin: Focus on human data. Mol. Nutr. Food Res. 2010, 54, 228–239. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Ki (nM) | |||
|---|---|---|---|---|
| hCA I | hCA II | hCA IX | hCA XII | |
| 1 | >10,000 | >10,000 | 32.1 | 5.8 |
| 2 | >10,000 | >10,000 | 67.5 | 8.5 |
| 3 | >10,000 | >10,000 | 88.7 | 9.6 |
| 4 | >10,000 | >10,000 | 32.0 | 6.4 |
| 5 | >10,000 | >10,000 | 294.9 | 7.1 |
| 6 | >10,000 | >10,000 | 27.1 | 5.6 |
| 7 | >10,000 | >10,000 | 114.7 | 7.7 |
| 8 | >10,000 | >10,000 | 190.3 | 8.9 |
| 9 | >10,000 | >10,000 | 92.8 | 8.1 |
| 10 | >10,000 | >10,000 | 162.3 | 9.4 |
| 11 | >10,000 | >10,000 | 307.7 | 9.6 |
| 12 | >10,000 | >10,000 | 120.7 | 26.4 |
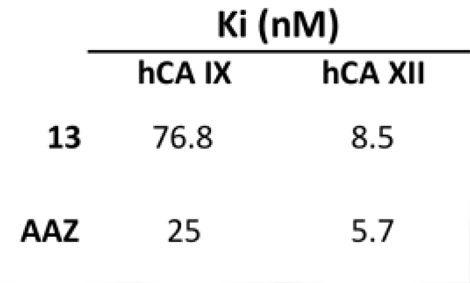
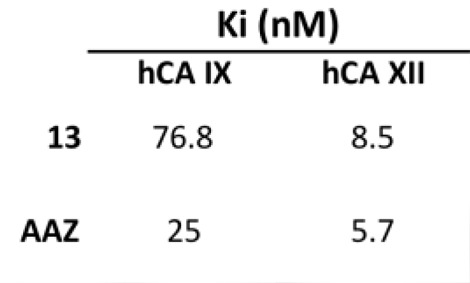
| 13 | >10,000 | >10,000 | 76.8 | 8.5 |
| AAZ | 250 | 12 | 25 | 5.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buran, K.; Bua, S.; Poli, G.; Önen Bayram, F.E.; Tuccinardi, T.; Supuran, C.T. Novel 8-Substituted Coumarins That Selectively Inhibit Human Carbonic Anhydrase IX and XII. Int. J. Mol. Sci. 2019, 20, 1208. https://doi.org/10.3390/ijms20051208
Buran K, Bua S, Poli G, Önen Bayram FE, Tuccinardi T, Supuran CT. Novel 8-Substituted Coumarins That Selectively Inhibit Human Carbonic Anhydrase IX and XII. International Journal of Molecular Sciences. 2019; 20(5):1208. https://doi.org/10.3390/ijms20051208
Chicago/Turabian StyleBuran, Kerem, Silvia Bua, Giulio Poli, F. Esra Önen Bayram, Tiziano Tuccinardi, and Claudiu T. Supuran. 2019. "Novel 8-Substituted Coumarins That Selectively Inhibit Human Carbonic Anhydrase IX and XII" International Journal of Molecular Sciences 20, no. 5: 1208. https://doi.org/10.3390/ijms20051208
APA StyleBuran, K., Bua, S., Poli, G., Önen Bayram, F. E., Tuccinardi, T., & Supuran, C. T. (2019). Novel 8-Substituted Coumarins That Selectively Inhibit Human Carbonic Anhydrase IX and XII. International Journal of Molecular Sciences, 20(5), 1208. https://doi.org/10.3390/ijms20051208