Liraglutide and its Neuroprotective Properties—Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events


 , , , ,
, , , ,
Abstract
1. Introduction
2. Ischemia
3. CNS Inflammation and Atherosclerosis
4. Alzheimer’s Disease
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Health Estimates 2015: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2015 [Internet]; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Buse, J.B.; Rosenstock, J.; Sesti, G.; Schmidt, W.E.; Montanya, E.; Brett, J.H.; Zychma, M.; Blonde, L.; LEAD-6 Study Group. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: A 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet 2009, 374, 39–47. [Google Scholar] [CrossRef]
- Goldstein, B.J.; Müller-Wieland, D. (Eds.) Type 2 Diabetes: Principles and Practice; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Beglinger, C.; Degen, L. Gastrointestinal satiety signals in humans—Physiologic roles for GLP-1 and PYY? Physiol. Behav. 2006, 89, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.; Frid, A.; Hermansen, K.; Shah, N.S.; Tankova, T.; Mitha, I.H.; Zdravkovic, M.; Düring, M.; Matthews, D.R. Efficacy and safety comparison of liraglutide, glimepiride, and placebo, all in combination with metformin, in type 2 diabetes: The LEAD (liraglutide effect and action in diabetes)-2 study. Diabetes Care 2009, 32, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Shyangdan, D.; Cummins, E.; Royle, P.; Waugh, N. Liraglutide for the treatment of type 2 diabetes: A single technology appraisal. Health Technol. Assess. 2011, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.; Hölscher, C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. Bmc Neurosci. 2012, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Watson, E.; Jonker, D.M.; Jacobsen, L.V.; Ingwersen, S.H. Population pharmacokinetics of liraglutide, a once-daily human glucagon-like peptide-1 analog, in healthy volunteers and subjects with type 2 diabetes, and comparison to twice-daily exenatide. J. Clin. Pharmacol. 2010, 50, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.A.; Greig, N.H. A new Alzheimer’s disease interventive strategy: GLP-1. Curr. Drug Targets 2004, 5, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Fernagut, P.O.; Bezard, E.; Meissner, W.G. Insulin, IGF-1 and GLP-1 signaling in neurodegenerative disorders: Targets for disease modification? Prog. Neurobiol. 2014, 118, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Navarro, I.; Leibush, B.; Moon, T.W.; Plisetskaya, E.M.; Banos, N.; Mendez, E.; Planas, J.V.; Gutierrez, J. Insulin, insulin-like growth factor-I (IGF-I) and glucagon: The evolution of their receptors. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 1999, 122, 137–153. [Google Scholar] [CrossRef]
- Werner, H.; Weinstein, D.; Bentov, I. Similarities and differences between insulin and IGF-I: Structures, receptors, and signalling pathways. Arch. Physiol. Biochem. 2008, 114, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Torres-Aleman, I. Toward a comprehensive neurobiology of IGF-I. Dev. Neurobiol. 2010, 70, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [PubMed]
- Duarte, A.I.; Moreira, P.I.; Oliveira, C.R. Insulin in central nervous system: More than just a peripheral hormone. J. Aging Res. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Salehi, M.; Aulinger, B.A.; D’Alessio, D.A. Targeting β-Cell mass in type 2 diabetes: Promise and limitations of new drugs based on incretins. Endocr. Rev. 2008, 29, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhou, J.; Doyle, M.E.; Egan, J.M. Glucagon-like peptide-1 causes pancreatic duodenal homeobox-1 protein translocation from the cytoplasm to the nucleus of pancreatic β-cells by a cyclic adenosine monophosphate/protein kinase A-dependent mechanism. Endocrinology 2001, 142, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Copps, K.D.; White, M.F. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012, 55, 2565–2582. [Google Scholar] [CrossRef] [PubMed]
- Barber, A.J.; Nakamura, M.; Wolpert, E.B.; Reiter, C.E.; Seigel, G.M.; Antonetti, D.A.; Gardner, T.W. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J. Biol. Chem. 2001, 276, 32814–32821. [Google Scholar] [CrossRef] [PubMed]
- Chin, P.C.; Majdzadeh, N.; D’Mello, S.R. Inhibition of GSK3β is a common event in neuroprotection by different survival factors. Mol. Brain Res. 2005, 137, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Plum, L.; Schubert, M.; Brüning, J.C. The role of insulin receptor signaling in the brain. Trends Endocrinol. Metab. 2005, 16, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Bonni, A.; Zigmond, M.J.; Lin, M.Z.; Juo, P.; Hu, L.S.; Anderson, M.J.; Arden, K.C.; Blenis, J.; Greenberg, M.E. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 1999, 96, 857–868. [Google Scholar] [CrossRef]
- Dupraz, S.; Grassi, D.; Karnas, D.; Guil, A.F.N.; Hicks, D.; Quiroga, S. The insulin-like growth factor 1 receptor is essential for axonal regeneration in adult central nervous system neurons. PLoS ONE 2013, 8, e54462. [Google Scholar] [CrossRef] [PubMed]
- Dagon, Y.; Avraham, Y.; Magen, I.; Gertler, A.; Ben-Hur, T.; Berry, E.M. Nutritional status, cognition and survival-a new role for leptin and AMPK. J. Biol. Chem. 2005, 280, 42142–42148. [Google Scholar] [CrossRef] [PubMed]
- Briyal, S.; Shah, S.; Gulati, A. Neuroprotective and anti-apoptotic effects of liraglutide in the rat brain following focal cerebral ischemia. Neuroscience 2014, 281, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Hölscher, C.; Xue, G.F.; Li, G.; Li, D. A novel dual-glucagon-like peptide-1 and glucose-dependent insulinotropic polypeptide receptor agonist is neuroprotective in transient focal cerebral ischemia in the rat. Neuroreport 2016, 27, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kameda, M.; Yasuhara, T.; Agari, T.; Baba, T.; Wang, F.; Shinko, A.; Wakamori, T.; Toyoshima, A.; Takeuchi, H.; et al. Neuroprotective effects of liraglutide for stroke model of rats. Int. J. Mol. Sci. 2013, 14, 21513–21524. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhang, Y.; Shi, Z.; Lu, D.; Li, T.; Ding, Y.; Ruan, Y.; Xu, A. The neuroprotection of liraglutide against ischaemia-induced apoptosis through the activation of the PI3K/AKT and MAPK pathways. Sci. Rep. 2016, 6, 26859. [Google Scholar] [CrossRef] [PubMed]
- Lozano, G.M.; Bejarano, I.; Espino, J.; Gonzalez, D.; Ortiz, A.; Garcia, J.F.; Rodríguez, A.B.; Pariente, J.A. Relationship between caspase activity and apoptotic markers in human sperm in response to hydrogen peroxide and progesterone. J. Reprod. Dev. 2009, 55, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Miao, Y.; Chen, A.; Cheng, M.; Ye, X.; Song, F.; Zheng, G. Delayed administration of the GLP-1 receptor agonist liraglutide improves metabolic and functional recovery after cerebral ischemia in rats. Neurosci. Lett. 2017, 641, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Schaar, K.L.; Brenneman, M.M.; Savitz, S.I. Functional assessments in the rodent stroke model. Exp. Transl. Stroke Med. 2010, 2, 13. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Hayakawa, K.; Lok, J.; Arai, K.; Lo, E.H. Injury and repair in the neurovascular unit. Neurol. Res. 2012, 34, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Zhang, K.; Li, P.; Zhu, L.; Xu, J.; Yang, B.; Hu, X.; Lu, Z.; Chen, J. Dysfunction of the neurovascular unit in ischemic stroke and neurodegenerative diseases: An aging effect. Ageing Res. Rev. 2017, 34, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Li, P.C.; Liu, L.F.; Jou, M.J.; Wang, H.K. The GLP-1 receptor agonists exendin-4 and liraglutide alleviate oxidative stress and cognitive and micturition deficits induced by middle cerebral artery occlusion in diabetic mice. BMC Neurosci. 2016, 17, 37. [Google Scholar] [CrossRef] [PubMed]
- Shiraki, A.; Oyama, J.I.; Komoda, H.; Asaka, M.; Komatsu, A.; Sakuma, M.; Kodama, K.; Sakamoto, Y.; Kotooka, N.; Hirase, T.; et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis 2012, 221, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Cao, J.; Han, J.; Li, J.; Li, Z.; Shi, N.; He, J. Liraglutide Activates the Nrf2/HO-1 Antioxidant Pathway and Protects Brain Nerve Cells against Cerebral Ischemia in Diabetic Rats. Comput. Intell. Neurosci. 2018, 2018, 3094504. [Google Scholar] [CrossRef] [PubMed]
- Zagoura, D.; Canovas-Jorda, D.; Pistollato, F.; Bremer-Hoffmann, S.; Bal-Price, A. Evaluation of the rotenone-induced activation of the Nrf2 pathway in a neuronal model derived from human induced pluripotent stem cells. Neurochem. Int. 2017, 106, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Xue, F.; Huang, J.W.; Ding, P.Y.; Zang, H.G.; Kou, Z.J.; Li, T.; Fan, J.; Peng, Z.; Yan, W.J. Nrf2/antioxidant defense pathway is involved in the neuroprotective effects of Sirt1 against focal cerebral ischemia in rats after hyperbaric oxygen preconditioning. Behav. Brain Res. 2016, 309, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Liu, X.; Wang, M.; Chen, H.; Chen, Z.; Qiu, T. Oxymatrine ameliorates renal ischemia-reperfusion injury from oxidative stress through Nrf2/HO-1 pathway. Acta Cir. Bras. 2015, 30, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Radak, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Niture, S.K.; Jaiswal, A.K. Nrf2 protein up-regulates antiapoptotic protein Bcl-2 and prevents cellular apoptosis. J. Biol. Chem. 2012, 287, 9873–9886. [Google Scholar] [CrossRef] [PubMed]
- Szadujkis-Szadurska, K.; Malinowski, B.; Piotrowska, M.; Grześk, G.; Wiciński, M.; Gajdus, M. The Modulatory Effect of Ischemia and Reperfusion on Arginine Vasopressin-Induced Arterial Reactions. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, A.K.; Loucks, F.A.; Schroeder, E.K.; Bouchard, R.J.; Tyler, K.L.; Linseman, D.A. Glutathione binding to the Bcl-2 homology-3 domain groove: A molecular basis for Bcl-2 antioxidant function at mitochondria. J. Biol. Chem. 2007, 282, 29296–29304. [Google Scholar] [CrossRef] [PubMed]
- Bratton, M.R.; Duong, B.N.; Elliott, S.; Weldon, C.B.; Beckman, B.S.; McLachlan, J.A.; Burow, M.E. Regulation of ERalpha-mediated transcription of Bcl-2 by PI3K-AKT crosstalk: Implications for breast cancer cell survival. Int. J. Oncol. 2010, 37, 541–550. [Google Scholar] [PubMed]
- Creson, T.K.; Yuan, P.; Manji, H.K.; Chen, G. Evidence for involvement of ERK, PI3K, and RSK in induction of Bcl-2 by valproate. J. Mol. Neurosci. 2009, 37, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Xu, D.; Wang, D.; Wu, R.; Zhang, L.; Zhu, H.; He, Q.; Yang, B. ROS-driven Akt dephosphorylation at Ser-473 is involved in 4-HPR-mediated apoptosis in NB4 cells. Free Radic. Biol. Med. 2009, 47, 536–547. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Devadas, S.; Williams, M.S. T cell receptor-stimulated generation of hydrogen peroxide inhibits MEK-ERK activation and lck serine phosphorylation. Free Radic. Biol. Med. 2003, 35, 406–417. [Google Scholar] [CrossRef]
- Shao, S.; Nie, M.; Chen, C.; Chen, X.; Zhang, M.; Yuan, G.; Yu, X.; Yang, Y. Protective action of liraglutide in beta cells under lipotoxic stress via PI3K/Akt/FoxO1 pathway. J. Cell. Biochem. 2014, 115, 1166–1175. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, S.Y.; Yin, T.; Tian, F.; Wang, S.; Chen, Y. Liraglutide promotes proliferation and migration of cardiac microvascular endothelial cells through PI3K/Akt and MAPK/ERK signaling pathways. J. South. Med. Univ. 2015, 35, 1221–1226. [Google Scholar]
- Gao, H.; Zeng, Z.; Zhang, H.; Zhou, X.; Guan, L.; Deng, W.; Xu, L. The glucagon-like peptide-1 analogue liraglutide inhibits oxidative stress and inflammatory response in the liver of rats with diet-induced non-alcoholic fatty liver disease. Biol. Pharm. Bull. 2015, 38, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.K.; Liu, Y.C.; Shi, L.L.; Lu, K.D. Glucagon-like peptide-1 receptor agonists inhibit hepatic stellate cell activation by blocking the p38 MAPK signaling pathway. Genet. Mol. Res. 2015, 14, 19087–19093. [Google Scholar] [CrossRef] [PubMed]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Després, J.; Fullerton, H.J.; et al. Executive summary: Heart disease and stroke statistics-2016 update: A report from the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Park, S.H.; Kim, N.; Kim, W.J.; Park, J.H.; Ko, Y.; Yang, M.H.; Jang, M.S.; Han, M.K.; Jung, C.; et al. Trial of ORG 10172 in Acute Stroke Treatment (TOAST) classification and vascular territory of ischemic stroke lesions diagnosed by diffusion-weighted imaging. J. Am. Heart Assoc. 2014, 3, e001119. [Google Scholar] [CrossRef] [PubMed]
- Gaspari, T.; Welungoda, I.; Widdop, R.E.; Simpson, R.W.; Dear, A.E. The GLP-1 receptor agonist liraglutide inhibits progression of vascular disease via effects on atherogenesis, plaque stability and endothelial function in an ApoE−/− mouse model. Diabetes Vasc. Dis. Res. 2013, 10, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Mehta, J.L.; Chen, M. Glucagon-like peptide-1 receptor agonist liraglutide inhibits endothelin-1 in endothelial cell by repressing nuclear factor-kappa B activation. Cardiovasc. Drugs Ther. 2013, 27, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Szadujkis-Szadurska, K.; Węclewicz, M.M.; Malinowski, B.; Matusiak, G.; Walczak, M.; Wódkiewicz, E.; Grześk, G.; Pawlak-Osińska, K. The role of Rho-kinase and calcium ions in constriction triggered by ET-1. Microvasc. Res. 2018, 119, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Socha, M.; Walczak, M.; Wódkiewicz, E.; Malinowski, B.; Rewerski, S.; Górski, K.; Pawlak-Osińska, K. Beneficial Effects of Resveratrol Administration—Focus on Potential Biochemical Mechanisms in Cardiovascular Conditions. Nutrients 2018, 10, 1813. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; García-Cardeña, G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Hernandez, W.; Ansari, R.A.; Ferder, L. Inflammation, oxidative stress and renin angiotensin system in atherosclerosis. World J. Biol. Chem. 2015, 6, 209. [Google Scholar] [CrossRef] [PubMed]
- Madden, J.A. Role of the vascular endothelium and plaque in acute ischemic stroke. Neurology 2012, 79 (Suppl. 1), S58–S62. [Google Scholar] [CrossRef]
- Tashiro, Y.; Sato, K.; Watanabe, T.; Nohtomi, K.; Terasaki, M.; Nagashima, M.; Hirano, T. A glucagon-like peptide-1 analog liraglutide suppresses macrophage foam cell formation and atherosclerosis. Peptides 2014, 54, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Xin, C.; Yan-Fu, W.; Ping, H.; Jing, G.; Jing-Jing, W.; Chun-Li, M.; Wei, L.; Bei, C. Study of the insulin signaling pathways in the regulation of ACAT1 expression in cultured macrophages. Cell Biol. Int. 2009, 33, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Hyperinsulinemia and risk of Alzheimer disease. Neurology 2004, 63, 1187–1192. [Google Scholar] [CrossRef] [PubMed]
- Ohara, T.; Doi, Y.; Ninomiya, T.; Hirakawa, Y.; Hata, J.; Iwaki, T.; Kanba, S.; Kiyohara, Y. Glucose tolerance status and risk of dementia in the community: The Hisayama study. Neurology 2011, 77, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; Cardoso, S.; Correia, S.C.; Santos, R.X.; Santos, M.S.; Baldeiras, I.; Oliveira, C.R.; Moreira, P.I. Metabolic alterations induced by sucrose intake and Alzheimer’s disease promote similar brain mitochondrial abnormalities. Diabetes 2012, 61, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-M.; Kim, J.-J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.-K. Upregulated NLRP3 inflammasome activation in patients with type2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Leietal, Y. Fattyacid-inducedNLRP3-ASC inflammasome activation interferes with insulin signalling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Wódkiewicz, E.; Słupski, M.; Walczak, M.; Socha, M.; Malinowski, B.; Pawlak-Osińska, K. Neuroprotective activity of sitagliptin via reduction of neuroinflammation beyond the incretin effect: Focus on Alzheimer’s disease. Biomed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G. Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications. J. Clin. Investig. 2013, 123, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Perry, T.; Lahiri, D.K.; Chen, D.; Zhou, J.; Shaw, K.T.; Egan, J.M.; Greig, N.H. A novel neurotrophic property of glucagon-like peptide 1: A promoter of nerve growth factor-mediated differentiation in PC12 cells. J. Pharmacol. Exp. Ther. 2002, 300, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Bertilsson, G.; Patrone, C.; Zachrisson, O.; Andersson, A.; Dannaeus, K.; Heidrich, J.; Kortesmaa, J.; Mercer, A.; Nielsen, E.; Rönnholm, H.; et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Cork, S.C.; Richards, J.E.; Holt, M.K.; Gribble, F.M.; Reimann, F.; Trapp, S. Distribution and characterisation of Glucagon-like peptide-1 receptor expressing cells in the mouse brain. Mol. Metab. 2015, 4, 718–731. [Google Scholar] [CrossRef] [PubMed]
- Holt, M.K.; Trapp, S. The physiological role of the brain GLP-1 system in stress. Cogent Biol. 2016, 2, 1229086. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Martínez, M.D.; Roncero, I.; Chowen, J.A.; García-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vázquez, P.; Maldonado, A.; de Cáceres, J.; et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.; Hölscher, C. Receptors for the incretin glucagon-like peptide-1 are expressed on neurons in the central nervous system. Neuroreport 2009, 20, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.; Tracy, L.M.; Patrone, C.; Iverfeldt, K.; Sjöholm, Å. GLP-1 secretion by microglial cells and decreased CNS expression in obesity. J. Neuroinflammation 2012, 9, 276. [Google Scholar] [CrossRef] [PubMed]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. The impact of dietary energy intake on cognitive aging. Front. Aging Neurosci. 2010, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Vilsbøll, T.; Christensen, M.; Junker, A.E.; Knop, F.K.; Gluud, L.L. Effects of glucagon-like peptide-1 receptor agonists on weight loss: Systematic review and meta-analyses of randomised controlled trials. BMJ 2012, 344, d7771. [Google Scholar] [CrossRef] [PubMed]
- Lovshin, J.A.; Drucker, D.J. Incretin-based therapies for type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 262. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, I.; Tweedie, D.; Li, Y.; Greig, N.H. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: An emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br. J. Pharmacol. 2012, 166, 1586–1599. [Google Scholar] [CrossRef] [PubMed]
- Wiciński, M.; Malinowski, B.; Węclewicz, M.M.; Grześk, E.; Grześk, G. Resveratrol increases serum BDNF concentrations and reduces vascular smooth muscle cells contractility via a NOS-3-independent mechanism. Biomed Res. Int. 2017, 2017, 9202954. [Google Scholar] [CrossRef] [PubMed]
- Cunha, C.; Brambilla, R.; Thomas, K.L. A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 2010, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Pezet, S.; Malcangio, M. Brain-derived neurotrophic factor as a drug target for CNS disorders. Expert Opin. Ther. Targets 2004, 8, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Torres, A.I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur. J. Pharmacol. 2004, 490, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Alldred, M.J.; Ginsberg, S.D.; Ohno, M. Mechanisms underlying insulin deficiency-induced acceleration of β-amyloidosis in a mouse model of Alzheimer’s disease. PLoS ONE 2012, 7, e32792. [Google Scholar] [CrossRef] [PubMed]
- Parthsarathy, V.; Hölscher, C. Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS ONE 2013, 8, e58784. [Google Scholar] [CrossRef] [PubMed]
- Belsham, D.D.; Fick, L.J.; Dalvi, P.S.; Centeno, M.L.; Chalmers, J.A.; Lee, P.K.; Wang, Y.; Drucker, D.J.; Koletar, M.M. Ciliary neurotrophic factor recruitment of glucagon-like peptide-1 mediates neurogenesis, allowing immortalization of adult murine hypothalamic neurons. FASEB J. 2009, 23, 4256–4265. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Halabisky, B.; Zhou, Y.; Palop, J.J.; Yu, G.; Mucke, L.; Gan, L. Imbalance between GABAergic and glutamatergic transmission impairs adult neurogenesis in an animal model of Alzheimer’s disease. Cell Stem Cell 2009, 5, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Sakamoto, M.; Ohtsuka, T.; Takao, K.; Miyakawa, T.; Yamaguchi, M.; Mori, K.; Ikeda, T.; Itohara, S.; Kageyama, R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008, 11, 1153–1161. [Google Scholar] [CrossRef] [PubMed]
- Saxe, M.D.; Battaglia, F.; Wang, J.W.; Malleret, G.; David, D.J.; Monckton, J.E.; Garcia, A.D.; Sofroniew, M.V.; Kandel, E.R.; Santarelli, L.; et al. Ablation of hippocampal neurogenesis impairs contextual fear conditioning and synaptic plasticity in the dentate gyrus. Proc. Natl. Acad. Sci. USA 2006, 103, 17501–17506. [Google Scholar] [CrossRef] [PubMed]
- Babateen, O.; Korol, S.V.; Jin, Z.; Bhandage, A.K.; Ahemaiti, A.; Birnir, B. Liraglutide modulates GABAergic signaling in rat hippocampal CA3 pyramidal neurons predominantly by presynaptic mechanism. Bmc Pharmacol. Toxicol. 2017, 18, 83. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.; Dahiya, R.; Dua, K.; Chellappan, D.K.; Tiwari, J.; Sharma, G.N.; Singh, S.K.; Mishra, A.; Sharma, R.K.; Agrawal, M. Anticonvulsant effect of liraglutide, GLP-1 agonist by averting a change in GABA and brain glutathione level on picrotoxin-induced seizures. EXCLI J. 2017, 16, 752–754. [Google Scholar] [PubMed]
- Gilman, C.P.; Perry, T.; Furukawa, K.; Grieg, N.H.; Egan, J.M.; Mattson, M.P. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J. Neurochem. 2003, 87, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Hölscher, C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology 2013, 76, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra e Silva, N.M.; Brito-Moreira, J.; Boehnke, S.E.; Marques, S.A. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Niehoff, M.L.; Morley, J.E.; Jelsing, J.; Pyke, C.; Knudsen, L.B.; Farr, S.A.; Vrang, N. The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 46, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Barkholt, P.; Fabricius, K.; Jelsing, J.; Terwel, D.; Pyke, C.; Knudsen, L.B.; Vrang, N. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 2016, 1634, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Holubová, M.; Hrubá, L.; Popelová, A.; Bencze, M.; Pražienková, V.; Gengler, S.; Kratochvílová, H.; Haluzík, M.; Železná, B.; Kuneš, J.; et al. Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of β-amyloid pathology. Neuropharmacology 2019, 144, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Bohm, C.; Chen, F.; Sevalle, J.; Qamar, S.; Dodd, R.; Li, Y.; Schmitt-Ulms, G.; Fraser, P.E.; St George-Hyslop, P.H. Current and future implications of basic and translational research on amyloid-β peptide production and removal pathways. Mol. Cell. Neurosci. 2015, 66, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Clearance of amyloid-beta in Alzheimer’s disease: Progress, problems and perspectives. Drug Discov. Today 2006, 11, 931–938. [Google Scholar] [CrossRef] [PubMed]
- Pflanzner, T.; Kuhlmann, C.R.; Pietrzik, C.U. Blood-brain-barrier models for the investigation of transporter-and receptor-mediated amyloid-β clearance in Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [PubMed]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cao, L.; Ren, Y.; Jiang, Y.; Xie, W.; Li, D. GLP-1 receptor regulates cell growth through regulating IDE expression level in Aβ1–42-treated PC12 cells. Biosci. Rep. 2018, 38, BSR20171284. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Ferreira-da-Silva, F.; Saraiva, M.J.; Cardoso, I. Transthyretin protects against A-beta peptide toxicity by proteolytic cleavage of the peptide: A mechanism sensitive to the Kunitz protease inhibitor. PLoS ONE 2008, 3, e2899. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.; Goncalves, A.; Saraiva, M.J.; Cardoso, I. Transthyretin binding to A-Beta peptide–Impact on A-Beta fibrillogenesis and toxicity. FEBS Lett. 2008, 582, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Trejo, J.L.; Gerber, A.; Loetscher, H.; Torrado, J.; Metzger, F.; Torres-Aleman, I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol. Aging 2006, 27, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Torres-Aleman, I. Serum insulin-like growth factor I in brain function. KEIO J. Med. 2006, 55, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, S.; Wu, J.; Ji, L.; Zhu, L.; Cao, L.; Huang, J.; Jiang, Q.; Wei, J.; Liu, M.; et al. cAMP/PKA signaling pathway contributes to neuronal apoptosis via regulating IDE expression in a mixed model of type 2 diabetes and Alzheimer’s disease. J. Cell. Biochem. 2018, 119, 1616–1626. [Google Scholar] [CrossRef] [PubMed]
- Padurariu, M.; Ciobica, A.; Mavroudis, I.; Fotiou, D.; Baloyannis, S. Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr. Danub. 2012, 24, 152–158. [Google Scholar] [PubMed]
- Ayala-Grosso, C.; Ng, G.; Roy, S.; Robertson, G.S. Caspase-cleaved amyloid precursor protein in Alzheimer’s disease. Brain Pathol. 2002, 12, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Płóciennik, A.; Prendecki, M.; Zuba, E.; Siudzinski, M.; Dorszewska, J. Activated caspase-3 and neurodegeneration and synaptic plasticity in Alzheimer’s disease. Adv. Alzheimer’s Dis. 2015, 4, 63–77. [Google Scholar]
- Liang, Z.; Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J. Neurochem. 2007, 103, 2462–2470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.W.; Rowan, M.J.; Anwyl, R. Inhibition of LTP by beta-amyloid is prevented by activation of beta2 adrenoceptors and stimulation of the cAMP/PKA signalling pathway. Neurobiol. Aging 2009, 30, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- van der Harg, J.M.; Eggels, L.; Bangel, F.N.; Ruigrok, S.R.; Zwart, R.; Hoozemans, J.J.M.; la Fleur, S.E.; Scheper, W. Insulin deficiency results in reversible protein kinase A activation and tau phosphorylation. Neurobiol. Dis. 2017, 103, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Heras-Sandoval, D.; Pérez-Rojas, J.M.; Hernández-Damián, J.; Pedraza-Chaverri, J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell. Signal. 2014, 26, 2694–2701. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, J.; Ma, D.; Zhang, M.; Hu, S.; Shao, S.; Gong, C.X. Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J. Alzheimer’s Dis. 2013, 37, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chen, F.Q.; Xu, W.J.; Yue, W.Z.; Yuan, G.; Yang, Y. Early intervention with glucagon-like peptide 1 analog liraglutide prevents tau hyperphosphorylation in diabetic db/db mice. J. Neurochem. 2015, 135, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.; Ke, L.; Liu, X.; Liao, L.; Ke, S.; Liu, X.; Wang, Y.; Lin, X.; Zhou, Y.; Wu, L.; et al. Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced alzheimer disease mouse model. Eur. J. Pharmacol. 2016, 783, 23–32. [Google Scholar] [CrossRef] [PubMed]



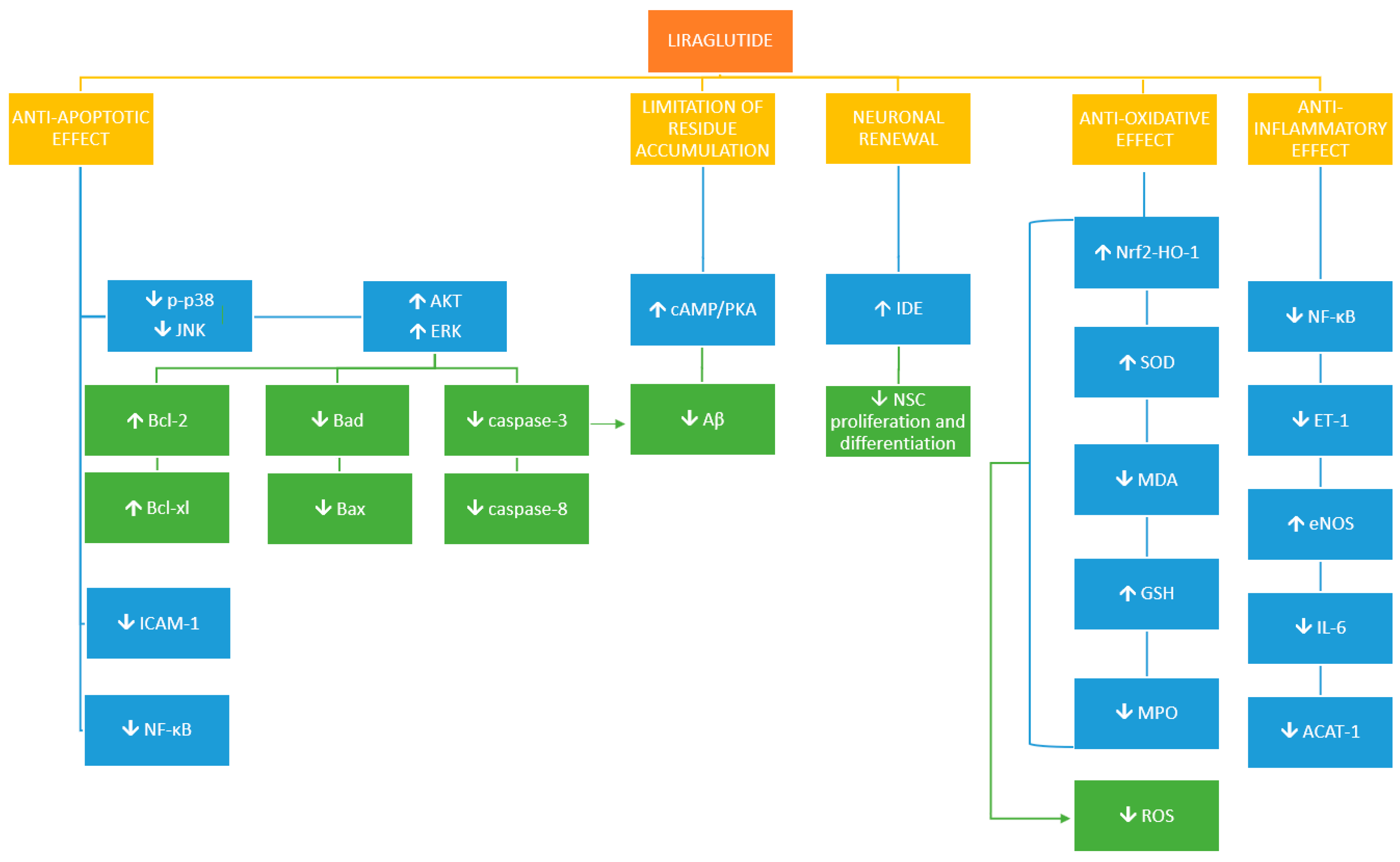{kind=link}
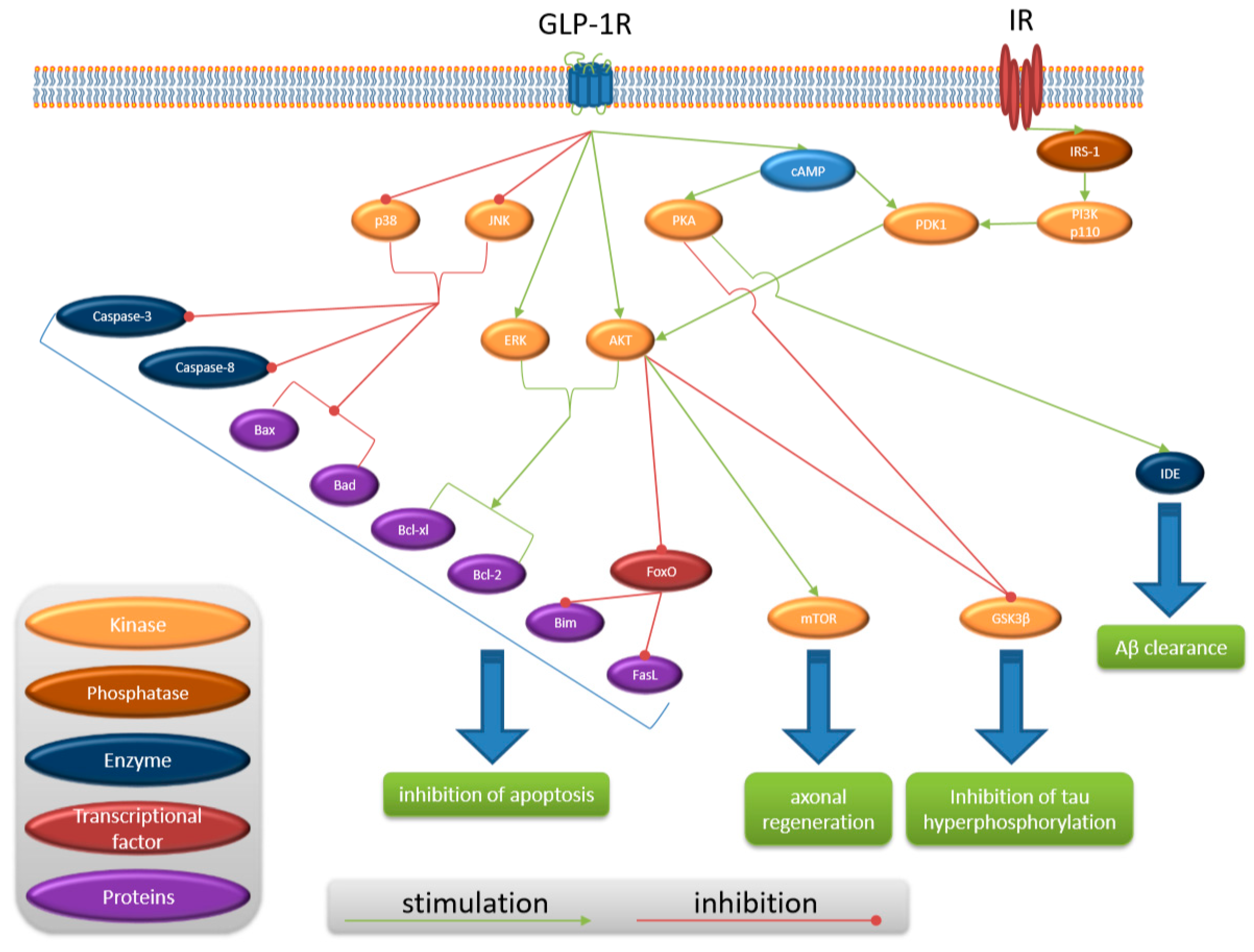{kind=link}
| Authors | Subject of Study | Dose of Liraglutide | Results |
|---|---|---|---|
| Batista et al. (2018) [100] | Non-human primate model, male Swiss mice | 0.006 mg/kg for the first week and 0.012mg/kg thereafter 25 nmol/kg/day for 7 days | ↑ cAMP/PKA ↓ Aβ plaques ↓ memory impairment ↓ synapse loss |
| Briyal et al. (2014) [25] | Sprague-Dawley rats | Pretreated 50 μg/kg per day for 14 days | ↓ infract size after MCAO, ↓ neurological deficit, ↑ Bcl-2, ↓ Bax, ↓MDA, ↑ GSH, ↓ TUNEL-positive cells, ↑ SOD |
| Dai et al. (2013) [53] | HUVECs | 10, 100, 1000 ng/mL for 6–24 h | ↓ NF-κB, ↓ ET-1, ↑ eNOS, ↓ IL-6 |
| Deng et al. (2018) [37] | Sprague-Dawley rats | 100 μg/kg twice daily for 7 days prior MCAO | ↓ infract size after MCAO, ↓ neurological deficit, ↑ SOD, ↓ MPO |
| Dong et al. (2017) [30] | Sprague-Dawley rats | 1 day after MCAO–50, 100, 200 μg/kg per day for 4 weeks | ↑ mNSS, ↑ 18F-FDG, ↑ NeuN, ↑ GFAP, ↑ vWF |
| Hansen et al. (2015) [101] | SAMP8 mice | 100 or 500 g/kg/day s.c. for 4 months | ↑ memory retention; ↑ CA1 neuron number |
| Hansen et al. (2016) [102] | hTauP301L transgenic mice | 500 mg/kg/day for 6 months | ↓ NFTs ↑motor function |
| Holubová et al. (2019) [103] | APP/PS1 mice | 0.2 mg/kg /day for 3 months | ↓ Aβ plaques ↓ caspase-3 |
| McClean et al. (2011) [98] | APP/PS1 mice | 25 nmol/kg for 2 months | ↓ synapse loss ↓ Aβ plaques ↓ memory impairment ↑ recognition test score |
| McClean et al. (2013) [99] | APP/PS1 mice | 25 nmol/kg for 2 months | ↓ Aβ plaques ↑ neuronal progenitor cell count ↓ inflammatory response in CNS |
| Li et al. (2016) [35] | db/db mouse | 0.1 mg/mL administered intraperitoneally during the 0, 3, 6, or 12 h reperfusion periods following MCAO | ↓ ROS, ↓ NF-κB, ↓ ICAM-1, ↓ caspase-3, ↓ TUNEL-positive cells ↑ p-AKT, ↑ p-eNOS |
| Parthsarathy et Hölscher (2013) [90] | APP/PS1 transgenic mice | 25 nmol/kg per day for 7 days 25 nmol/kg per day for 37 days | ↑NSC proliferation ↑NSC differentiation |
| Shiraki et al. (2012) [36] | HUVECs | Pre-incubated 30nM/mL for 1 h | ↓ ROS, ↑ SOD, ↑ catalase |
| Tashiro et al. (2014) [63] | Human macrophages and apoE−/− mice | 107 nmol/kg/day for 4 weeks | ↓ foam cells, ↓ macrophage-driven atherosclerotic lesions, |
| Zhu et al. (2016) [28] | Sprague-Dawley rats | 1 h after MCAO–100 μg/kg per day for 1, 3 and 7 days | ↓ infract size after MCAO, ↓ neurological deficit, ↑ Bcl-2, ↑ Bcl-xl, ↓ Bax, ↓ Bad, ↓ ROS |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wiciński, M.; Socha, M.; Malinowski, B.; Wódkiewicz, E.; Walczak, M.; Górski, K.; Słupski, M.; Pawlak-Osińska, K. Liraglutide and its Neuroprotective Properties—Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. Int. J. Mol. Sci. 2019, 20, 1050. https://doi.org/10.3390/ijms20051050
Wiciński M, Socha M, Malinowski B, Wódkiewicz E, Walczak M, Górski K, Słupski M, Pawlak-Osińska K. Liraglutide and its Neuroprotective Properties—Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. International Journal of Molecular Sciences. 2019; 20(5):1050. https://doi.org/10.3390/ijms20051050
Chicago/Turabian StyleWiciński, Michał, Maciej Socha, Bartosz Malinowski, Eryk Wódkiewicz, Maciej Walczak, Karol Górski, Maciej Słupski, and Katarzyna Pawlak-Osińska. 2019. "Liraglutide and its Neuroprotective Properties—Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events" International Journal of Molecular Sciences 20, no. 5: 1050. https://doi.org/10.3390/ijms20051050
APA StyleWiciński, M., Socha, M., Malinowski, B., Wódkiewicz, E., Walczak, M., Górski, K., Słupski, M., & Pawlak-Osińska, K. (2019). Liraglutide and its Neuroprotective Properties—Focus on Possible Biochemical Mechanisms in Alzheimer’s Disease and Cerebral Ischemic Events. International Journal of Molecular Sciences, 20(5), 1050. https://doi.org/10.3390/ijms20051050