The Contribution of the Immune System in Bone Metastasis Pathogenesis



Abstract
1. Introduction
2. Crosstalk among Cancer Cell, Immune Cells and the Bone Microenvironment
2.1. Bone Microenvironment
2.2. Cancer Cells Metastasize to Bone
2.3. Interaction between Immune Cells and Bone Microenvironment
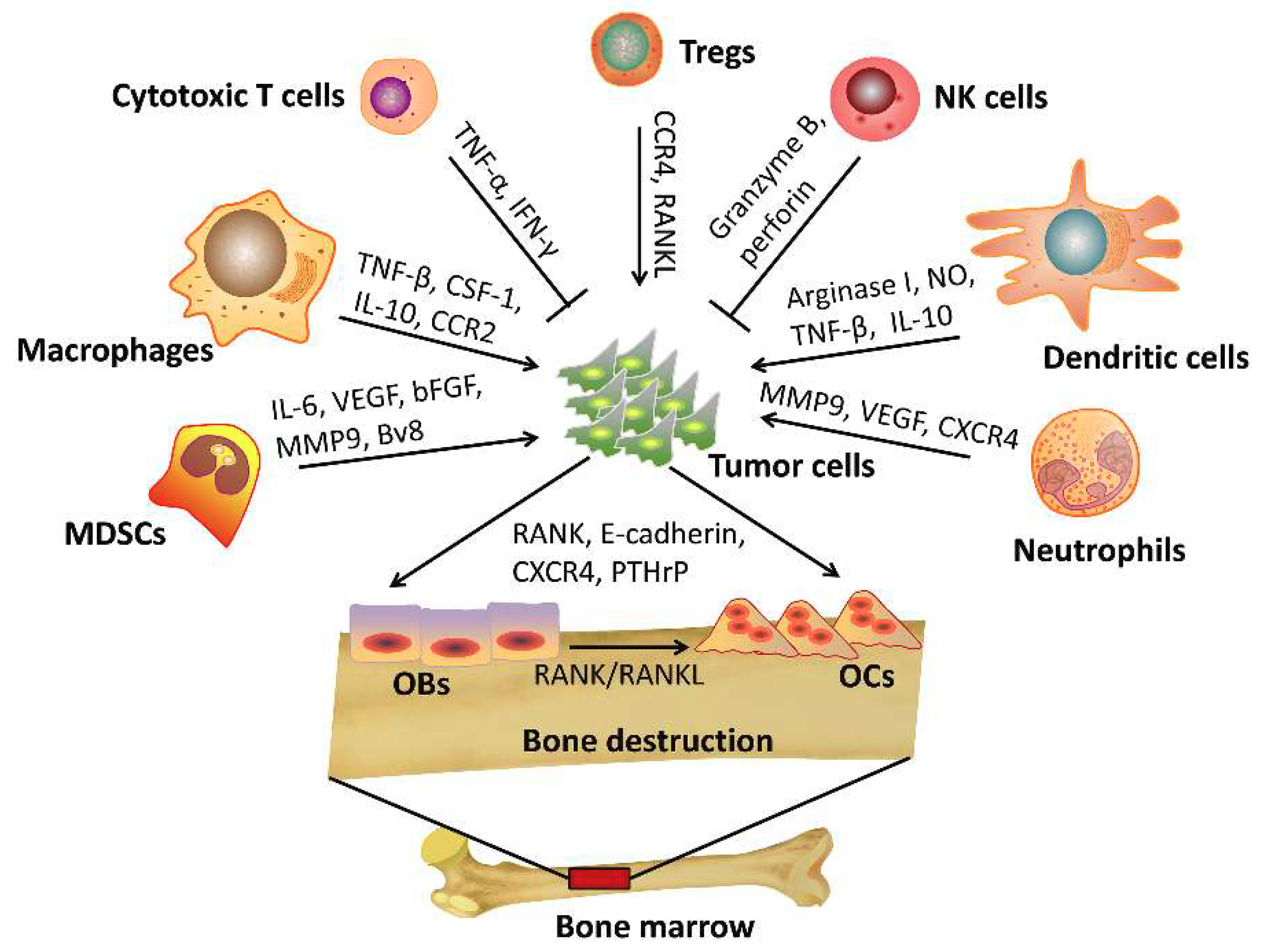
3. The Role of Immune Cells in Bone Metastasis
3.1. T Cells
3.2. NK Cells
3.3. Macrophages
3.4. Dendritic Cells
3.5. Myeloid-Derived Suppressor Cells
3.6. Neutrophils
4. Therapeutic Potential for Bone Metastasis by Modulating the Immune System
4.1. Targeting T Cells
4.2. Targeting NK Cells
4.3. Targeting Macrophages
4.4. Targeting Myeloid-Derived Suppressor Cells
4.5. Targeting Dendritic Cells and Tumor-Associated Neutrophils
5. Other Drugs Inhibiting Bone Metastasis
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Soeharno, H.; Povegliano, L.; Choong, P.F. Multimodal Treatment of Bone Metastasis—A Surgical Perspective. Front. Endocrinol. 2018, 9, 578. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, L.A.; Fournier, P.G.J.; Chirgwin, J.M.; Guise, T.A. Molecular biology of bone metastasis. Mol. Cancer Ther. 2007, 6, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Janssen, L.M.E.; Ramsay, E.E.; Logsdon, C.D.; Overwijk, W.W. The immune system in cancer metastasis: Friend or foe? J. Immunother. Cancer 2017, 5, 79. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.A.; Kang, Y.B. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Capietto, A.H.; Faccio, R. Immune regulation of bone metastasis. Bonekey Rep. 2014, 3, 600. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Metastasis to bone: Causes, consequences and therapeutic opportunities. Nat. Rev. Cancer 2002, 2, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Sleeman, J.P. The metastatic niche and stromal progression. Cancer Metastasis Rev. 2012, 31, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Fidler, I.J. The pathogenesis of cancer metastasis: The ‘seed and soil’ hypothesis revisited. Nat. Rev. Cancer 2003, 3, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Marks, S.C., Jr.; Popoff, S.N. Bone cell biology: The regulation of development, structure, and function in the skeleton. Am. J. Anat. 1988, 183, 1–44. [Google Scholar] [PubMed]
- Bussard, K.M.; Gay, C.V.; Mastro, A.M. The bone microenvironment in metastasis; what is special about bone? Cancer Metastasis Rev. 2008, 27, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Suva, L.J.; Washam, C.; Nicholas, R.W.; Griffin, R.J. Bone metastasis: Mechanisms and therapeutic opportunities. Nat. Rev. Endocrinol. 2011, 7, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, C.K.; Andrews, L.R.; Thomas, K.G.; Tindall, D.J.; Fitzpatrick, L.A. The effects of growth factors associated with osteoblasts on prostate carcinoma proliferation and chemotaxis: Implications for the development of metastatic disease. Endocrinology 1997, 138, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.L.; Bafico, A.; Dai, J.; Aaronson, S.A.; Keller, E.T. Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 2005, 65, 7554–7560. [Google Scholar] [CrossRef] [PubMed]
- Sottnik, J.L.; Hall, C.L.; Zhang, J.; Keller, E.T. Wnt and Wnt inhibitors in bone metastasis. BoneKEy Rep. 2012, 1, 101. [Google Scholar] [CrossRef] [PubMed]
- Reddington, J.A.; Mendez, G.A.; Ching, A.; Kubicky, C.D.; Klimo, P., Jr.; Ragel, B.T. Imaging characteristic analysis of metastatic spine lesions from breast, prostate, lung, and renal cell carcinomas for surgical planning: Osteolytic versus osteoblastic. Surg. Neurol. Int. 2016, 7 (Suppl. 13), S361–S365. [Google Scholar]
- Wu, M.Y.; Li, C.J.; Yiang, G.T.; Cheng, Y.L.; Tsai, A.P.; Hou, Y.T.; Ho, Y.C.; Hou, M.F.; Chu, P.Y. Molecular Regulation of Bone Metastasis Pathogenesis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 46, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- Lipton, A. Future treatment of bone metastases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6305s–6308s. [Google Scholar] [CrossRef] [PubMed]
- Loberg, R.D.; Logothetis, C.J.; Keller, E.T.; Pienta, K.J. Pathogenesis and treatment of prostate cancer bone metastases: Targeting the lethal phenotype. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 8232–8241. [Google Scholar] [CrossRef] [PubMed]
- Michigami, T.; Shimizu, N.; Williams, P.J.; Niewolna, M.; Dallas, S.L.; Mundy, G.R.; Yoneda, T. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and alpha(4)beta(1)-integrin enhances production of osteoclast-stimulating activity. Blood 2000, 96, 1953–1960. [Google Scholar] [PubMed]
- Chavez-Macgregor, M.; Aviles-Salas, A.; Green, D.; Fuentes-Alburo, A.; Gomez-Ruiz, C.; Aguayo, A. Angiogenesis in the bone marrow of patients with breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 5396–5400. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Philippe, B.; Fouqueray, B.; Perez, J.; Lebret, M.; Baud, L. Protection from tumor necrosis factor-mediated cytolysis by platelets. Am. J. Pathol. 1993, 143, 1713–1723. [Google Scholar] [PubMed]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Akfirat, C.; Zhang, X.; Ventura, A.; Berel, D.; Colangelo, M.E.; Miranti, C.K.; Krajewska, M.; Reed, J.C.; Higano, C.S.; True, L.D.; et al. Tumour cell survival mechanisms in lethal metastatic prostate cancer differ between bone and soft tissue metastases. J. Pathol. 2013, 230, 291–297. [Google Scholar] [CrossRef] [PubMed]
- Bubenik, J. Tumour MHC class I downregulation and immunotherapy (Review). Oncol. Rep. 2003, 10, 2005–2008. [Google Scholar] [CrossRef] [PubMed]
- Baschuk, N.; Rautela, J.; Parker, B.S. Bone specific immunity and its impact on metastasis. BoneKEy Rep. 2015, 4, 665. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.; Homey, B.; Soto, H.; Ge, N.; Catron, D.; Buchanan, M.E.; McClanahan, T.; Murphy, E.; Yuan, W.; Wagner, S.N.; et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 2001, 410, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Golan, K.; Kollet, O.; Lapidot, T. Dynamic Cross Talk between S1P and CXCL12 Regulates Hematopoietic Stem Cells Migration, Development and Bone Remodeling. Pharmaceuticals 2013, 6, 1145–1169. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Havens, A.M.; Jung, Y.; Ziegler, A.M.; Pedersen, E.A.; Wang, J.; Wang, J.; Lu, G.; Roodman, G.D.; Loberg, R.D.; et al. Annexin II/annexin II receptor axis regulates adhesion, migration, homing, and growth of prostate cancer. J. Cell. Biochem. 2008, 105, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Gunasinghe, N.P.; Wells, A.; Thompson, E.W.; Hugo, H.J. Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev. 2012, 31, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.H.; Nakashima, T.; Sanchez, O.H.; Kozieradzki, I.; Komarova, S.V.; Sarosi, I.; Morony, S.; Rubin, E.; Sarao, R.; Hojilla, C.V.; et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006, 440, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, C.; De Luca, P.; Gilardi, M.; Previdi, S.; Broggini, M.; Moretti, M. Direct but not indirect co-culture with osteogenically differentiated human bone marrow stromal cells increases RANKL/OPG ratio in human breast cancer cells generating bone metastases. Mol. Cancer 2014, 13, 238. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Bachelier, R.; Treilleux, I.; Pujuguet, P.; Peyruchaud, O.; Baron, R.; Clement-Lacroix, P.; Clezardin, P. Tumor alphavbeta3 integrin is a therapeutic target for breast cancer bone metastases. Cancer Res. 2007, 67, 5821–5830. [Google Scholar] [CrossRef] [PubMed]
- Clezardin, P. Integrins in bone metastasis formation and potential therapeutic implications. Curr. Cancer Drug Targets 2009, 9, 801–806. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Friedrichs, J.; Bonin, M.V.; Bejestani, E.P.; Werner, C.; Wobus, M.; Chavakis, T.; Bornhauser, M. Breast cancer cells compete with hematopoietic stem and progenitor cells for intercellular adhesion molecule 1-mediated binding to the bone marrow microenvironment. Carcinogenesis 2016, 37, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-beta2 dictates disseminated tumour cell fate in target organs through TGF-beta-RIII and p38alpha/beta signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Xing, F.; Liu, Y.; Wu, K.; Said, N.; Pochampally, R.; Shiozawa, Y.; Lin, H.K.; Balaji, K.C.; Watabe, K. Secreted Protein Acidic and Rich in Cysteine (SPARC) Mediates Metastatic Dormancy of Prostate Cancer in Bone. J. Biol. Chem. 2016, 291, 19351–19363. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Mu, E.; Wei, Y.; Riethdorf, S.; Yang, Q.; Yuan, M.; Yan, J.; Hua, Y.; Tiede, B.J.; Lu, X.; et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging alpha4beta1-positive osteoclast progenitors. Cancer Cell 2011, 20, 701–714. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.B.; Chabner, K.T.; Alley, I.R.; Olson, D.P.; Szczepiorkowski, Z.M.; Poznansky, M.C.; Kos, C.H.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Stem cell engraftment at the endosteal niche is specified by the calcium-sensing receptor. Nature 2006, 439, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Buijs, J.T.; Stayrook, K.R.; Guise, T.A. The role of TGF-beta in bone metastasis: Novel therapeutic perspectives. BoneKEy Rep. 2012, 1, 96. [Google Scholar] [CrossRef] [PubMed]
- Le Pape, F.; Vargas, G.; Clezardin, P. The role of osteoclasts in breast cancer bone metastasis. J. Bone Oncol. 2016, 5, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Weilbaecher, K.N.; Guise, T.A.; McCauley, L.K. Cancer to bone: A fatal attraction. Nat. Rev. Cancer 2011, 11, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Hensel, J.A.; Chanda, D.; Harris, B.A.; Siegal, G.P.; Maheshwari, A.; Ponnazhagan, S. Depletion of plasmacytoid dendritic cells inhibits tumor growth and prevents bone metastasis of breast cancer cells. J. Immunol. 2012, 189, 4258–4265. [Google Scholar] [CrossRef] [PubMed]
- Roato, I.; Grano, M.; Brunetti, G.; Colucci, S.; Mussa, A.; Bertetto, O.; Ferracini, R. Mechanisms of spontaneous osteoclastogenesis in cancer with bone involvement. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.T.; Nguyen, H.; Gao, X.; Kong, Y.Y.; Gorczynski, R.M.; Singh, B.; Ellen, R.P.; Penninger, J.M. Functional human T-cell immunity and osteoprotegerin ligand control alveolar bone destruction in periodontal infection. J. Clin. Investig. 2000, 106, R59–R67. [Google Scholar] [CrossRef] [PubMed]
- Takayanagi, H.; Ogasawara, K.; Hida, S.; Chiba, T.; Murata, S.; Sato, K.; Takaoka, A.; Yokochi, T.; Oda, H.; Tanaka, K.; et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 2000, 408, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Grassi, F.; Ryan, M.R.; Terauchi, M.; Page, K.; Yang, X.; Weitzmann, M.N.; Pacifici, R. IFN-gamma stimulates osteoclast formation and bone loss in vivo via antigen-driven T cell activation. J. Clin. Investig. 2007, 117, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hurchla, M.A.; Deng, H.; Uluckan, O.; Bu, F.; Berdy, A.; Eagleton, M.C.; Heller, E.A.; Floyd, D.H.; Dirksen, W.P.; et al. Interferon-gamma targets cancer cells and osteoclasts to prevent tumor-associated bone loss and bone metastases. J. Biol. Chem. 2009, 284, 4658–4666. [Google Scholar] [CrossRef] [PubMed]
- Kiesel, J.R.; Buchwald, Z.S.; Aurora, R. Cross-presentation by osteoclasts induces FoxP3 in CD8+ T cells. J. Immunol. 2009, 182, 5477–5487. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, L.; Roato, I. The Impact of Immune System in Regulating Bone Metastasis Formation by Osteotropic Tumors. J. Immunol. Res. 2015, 2015, 143526. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.K.; Raggatt, L.J.; Alexander, K.A.; Kuliwaba, J.S.; Fazzalari, N.L.; Schroder, K.; Maylin, E.R.; Ripoll, V.M.; Hume, D.A.; Pettit, A.R. Osteal tissue macrophages are intercalated throughout human and mouse bone lining tissues and regulate osteoblast function in vitro and in vivo. J. Immunol. 2008, 181, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.C.; He, Y.; Broomfield, A.; Paatan, N.J.; Harrington, B.S.; Tseng, H.W.; Beaven, E.A.; Kiernan, D.M.; Swindle, P.; Clubb, A.B.; et al. CD169(+) macrophages mediate pathological formation of woven bone in skeletal lesions of prostate cancer. J. Pathol. 2016, 239, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Dejbakhsh-Jones, S.; Jerabek, L.; Weissman, I.L.; Strober, S. Extrathymic maturation of alpha beta T cells from hemopoietic stem cells. J. Immunol. 1995, 155, 3338–3344. [Google Scholar] [PubMed]
- Schirrmacher, V.; Feuerer, M.; Fournier, P.; Ahlert, T.; Umansky, V.; Beckhove, P. T-cell priming in bone marrow: The potential for long-lasting protective anti-tumor immunity. Trends Mol. Med. 2003, 9, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewski, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar] [PubMed]
- Mazo, I.B.; Honczarenko, M.; Leung, H.; Cavanagh, L.L.; Bonasio, R.; Weninger, W.; Engelke, K.; Xia, L.; McEver, R.P.; Koni, P.A.; et al. Bone marrow is a major reservoir and site of recruitment for central memory CD8+ T cells. Immunity 2005, 22, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Hoffmann, P.; Lan, F.; Huie, P.; Higgins, J.; Strober, S. Unique patterns of surface receptors, cytokine secretion, and immune functions distinguish T cells in the bone marrow from those in the periphery: Impact on allogeneic bone marrow transplantation. Blood 2002, 99, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Price, P.W.; Cerny, J. Characterization of CD4+ T cells in mouse bone marrow. I. Increased activated/memory phenotype and altered TCR Vbeta repertoire. Eur. J. Immunol. 1999, 29, 1051–1056. [Google Scholar] [CrossRef]
- Fournier, P.G.; Chirgwin, J.M.; Guise, T.A. New insights into the role of T cells in the vicious cycle of bone metastases. Curr. Opin. Rheumatol. 2006, 18, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Contribution of regulatory T cells to cancer: A review. J. Cell. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.J.; Fox, S.B.; Han, C.; Leek, R.D.; Garcia, J.F.; Harris, A.L.; Banham, A.H. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006, 24, 5373–5380. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.; Wolf, D.; Steurer, M.; Gastl, G.; Gunsilius, E.; Grubeck-Loebenstein, B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 606–612. [Google Scholar]
- Zhao, E.; Wang, L.; Dai, J.; Kryczek, I.; Wei, S.; Vatan, L.; Altuwaijri, S.; Sparwasser, T.; Wang, G.; Keller, E.T.; et al. Regulatory T cells in the bone marrow microenvironment in patients with prostate cancer. Oncoimmunology 2012, 1, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Barnett, B.; Safah, H.; Larussa, V.F.; Evdemon-Hogan, M.; Mottram, P.; Wei, S.; David, O.; Curiel, T.J.; Zou, W. Bone marrow is a reservoir for CD4+CD25+ regulatory T cells that traffic through CXCL12/CXCR4 signals. Cancer Res. 2004, 64, 8451–8455. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kryczek, I.; Zou, W. Regulatory T-cell compartmentalization and trafficking. Blood 2006, 108, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kryczek, I.; Edwards, R.P.; Zou, L.; Szeliga, W.; Banerjee, M.; Cost, M.; Cheng, P.; Chang, A.; Redman, B.; et al. Interleukin-2 administration alters the CD4+FOXP3+ T-cell pool and tumor trafficking in patients with ovarian carcinoma. Cancer Res. 2007, 67, 7487–7494. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Zhang, W.; Strasner, A.; Grivennikov, S.; Cheng, J.Q.; Hoffman, R.M.; Karin, M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature 2011, 470, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, T. Bone metastasis and RANKL. Clin. Calcium 2014, 24, 1201–1208. [Google Scholar] [PubMed]
- Roy, L.D.; Ghosh, S.; Pathangey, L.B.; Tinder, T.L.; Gruber, H.E.; Mukherjee, P. Collagen induced arthritis increases secondary metastasis in MMTV-PyV MT mouse model of mammary cancer. BMC Cancer 2011, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Takayanagi, H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res. Ther. 2011, 13, 219. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.C.; Leal, A.C.; Goncalves-Silva, T.; Mercadante, A.C.; Kestelman, F.; Chaves, S.B.; Azevedo, R.B.; Monteiro, J.P.; Bonomo, A. T cells induce pre-metastatic osteolytic disease and help bone metastases establishment in a mouse model of metastatic breast cancer. PLoS ONE 2013, 8, e68171. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zeng, D.; Gazit, G.; Dejbakhsh-Jones, S.; Balk, S.P.; Snapper, S.; Taniguchi, M.; Strober, S. Heterogeneity of NK1.1+ T cells in the bone marrow: Divergence from the thymus. J. Immunol. 1999, 163, 5338–5345. [Google Scholar] [PubMed]
- Higuchi, M.; Zeng, D.; Shizuru, J.; Gworek, J.; Dejbakhsh-Jones, S.; Taniguchi, M.; Strober, S. Immune tolerance to combined organ and bone marrow transplants after fractionated lymphoid irradiation involves regulatory NK T cells and clonal deletion. J. Immunol. 2002, 169, 5564–5570. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lanier, L.L. Natural killer cells and cancer. Adv. Cancer Res. 2003, 90, 127–156. [Google Scholar] [PubMed]
- Bidwell, B.N.; Slaney, C.Y.; Withana, N.P.; Forster, S.; Cao, Y.; Loi, S.; Andrews, D.; Mikeska, T.; Mangan, N.E.; Samarajiwa, S.A.; et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P.; et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, T.; Yoneyama, M.S.; Hatakeyama, S.; Mori, K.; Yamamoto, H.; Koie, T.; Saitoh, H.; Yamaya, K.; Funyu, T.; Fukuda, M.; et al. Core2 O-glycan-expressing prostate cancer cells are resistant to NK cell immunity. Mol. Med. Rep. 2013, 7, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Kono, K.; Kawaguchi, Y.; Mizukami, Y.; Mimura, K.; Maruyama, T.; Izawa, S.; Fujii, H. NK cell dysfunction with down-regulated CD16 and up-regulated CD56 molecules in patients with esophageal squamous cell carcinoma. Dis. Esophagus Off. J. Int. Soc. Dis. Esophagus 2010, 23, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Izawa, S.; Kono, K.; Mimura, K.; Kawaguchi, Y.; Watanabe, M.; Maruyama, T.; Fujii, H. H(2)O(2) production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: Possible mechanisms of NK cell dysfunction. Cancer Immunol. Immunother. CII 2011, 60, 1801–1810. [Google Scholar] [CrossRef] [PubMed]
- Sinder, B.P.; Pettit, A.R.; McCauley, L.K. Macrophages: Their Emerging Roles in Bone. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2015, 30, 2140–2149. [Google Scholar] [CrossRef] [PubMed]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A. Macrophages, innate immunity and cancer: Balance, tolerance, and diversity. Curr. Opin. Immunol. 2010, 22, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Reviews. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Karnevi, E.; Andersson, R.; Rosendahl, A.H. Tumour-educated macrophages display a mixed polarisation and enhance pancreatic cancer cell invasion. Immunol. Cell Biol. 2014, 92, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Choi, H.J.; Ha, S.J.; Lee, K.T.; Kwon, Y.G. Recruitment of monocytes/macrophages in different tumor microenvironments. Biochim. Biophys. Acta 2013, 1835, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Zabuawala, T.; Taffany, D.A.; Sharma, S.M.; Merchant, A.; Adair, B.; Srinivasan, R.; Rosol, T.J.; Fernandez, S.; Huang, K.; Leone, G.; et al. An ets2-driven transcriptional program in tumor-associated macrophages promotes tumor metastasis. Cancer Res. 2010, 70, 1323–1333. [Google Scholar] [CrossRef] [PubMed]
- Sawa-Wejksza, K.; Kandefer-Szerszen, M. Tumor-Associated Macrophages as Target for Antitumor Therapy. Arch. Immunol. Ther. Exp. 2018, 66, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Grossman, J.G.; Nywening, T.M.; Belt, B.A.; Panni, R.Z.; Krasnick, B.A.; DeNardo, D.G.; Hawkins, W.G.; Goedegebuure, S.P.; Linehan, D.C.; Fields, R.C. Recruitment of CCR2(+) tumor associated macrophage to sites of liver metastasis confers a poor prognosis in human colorectal cancer. Oncoimmunology 2018, 7, e1470729. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Cui, W.Q.; Wei, Y.; Cui, J.; Qiu, J.; Hu, L.L.; Gong, W.Y.; Dong, J.C.; Liu, B.J. Astragaloside IV inhibits lung cancer progression and metastasis by modulating macrophage polarization through AMPK signaling. J. Exp. Clin. Cancer Res. 2018, 37, 207. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, L.; Zhang, J.; Zheng, C.; Ding, K.; Xiao, H.; Wang, L.; Zhang, Z. C-C Chemokine Ligand 2 (CCL2) Recruits Macrophage-Membrane-Camouflaged Hollow Bismuth Selenide Nanoparticles to Facilitate Photothermal Sensitivity and Inhibit Lung Metastasis of Breast Cancer. ACS Appl. Mater. Interfaces 2018, 10, 31124–31135. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Lynch, C.C. Multifaceted Roles for Macrophages in Prostate Cancer Skeletal Metastasis. Front. Endocrinol. 2018, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Soki, F.N.; Cho, S.W.; Kim, Y.W.; Jones, J.D.; Park, S.I.; Koh, A.J.; Entezami, P.; Daignault-Newton, S.; Pienta, K.J.; Roca, H.; et al. Bone marrow macrophages support prostate cancer growth in bone. Oncotarget 2015, 6, 35782–35796. [Google Scholar] [CrossRef] [PubMed]
- Lindholm, P.F.; Lu, Y.; Adley, B.P.; Vladislav, T.; Jovanovic, B.; Sivapurapu, N.; Yang, X.J.; Kajdacsy-Balla, A. Role of monocyte-lineage cells in prostate cancer cell invasion and tissue factor expression. Prostate 2010, 70, 1672–1682. [Google Scholar] [CrossRef] [PubMed]
- Tang, X. Tumor-associated macrophages as potential diagnostic and prognostic biomarkers in breast cancer. Cancer Lett. 2013, 332, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Chemokine (C-C motif) ligand 2 engages CCR2+ stromal cells of monocytic origin to promote breast cancer metastasis to lung and bone. J. Biol. Chem. 2009, 284, 29087–29096. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, A.R.; Pixley, F.J. CSF-1R signaling in health and disease: A focus on the mammary gland. J. Mammary Gland Biol. Neoplasia 2014, 19, 149–159. [Google Scholar] [CrossRef] [PubMed]
- DeNardo, D.G.; Brennan, D.J.; Rexhepaj, E.; Ruffell, B.; Shiao, S.L.; Madden, S.F.; Gallagher, W.M.; Wadhwani, N.; Keil, S.D.; Junaid, S.A.; et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011, 1, 54–67. [Google Scholar] [CrossRef] [PubMed]
- Fend, L.; Accart, N.; Kintz, J.; Cochin, S.; Reymann, C.; Le Pogam, F.; Marchand, J.B.; Menguy, T.; Slos, P.; Rooke, R.; et al. Therapeutic effects of anti-CD115 monoclonal antibody in mouse cancer models through dual inhibition of tumor-associated macrophages and osteoclasts. PLoS ONE 2013, 8, e73310. [Google Scholar] [CrossRef] [PubMed]
- Schuler, G.; Steinman, R.M. Dendritic cells as adjuvants for immune-mediated resistance to tumors. J. Exp. Med. 1997, 186, 1183–1187. [Google Scholar] [CrossRef] [PubMed]
- Porgador, A.; Snyder, D.; Gilboa, E. Induction of antitumor immunity using bone marrow-generated dendritic cells. J. Immunol. 1996, 156, 2918–2926. [Google Scholar] [PubMed]
- Abdul Hafid, S.R.; Chakravarthi, S.; Nesaretnam, K.; Radhakrishnan, A.K. Tocotrienol-adjuvanted dendritic cells inhibit tumor growth and metastasis: A murine model of breast cancer. PLoS ONE 2013, 8, e74753. [Google Scholar] [CrossRef] [PubMed]
- Cavanagh, L.L.; Bonasio, R.; Mazo, I.B.; Halin, C.; Cheng, G.; van der Velden, A.W.; Cariappa, A.; Chase, C.; Russell, P.; Starnbach, M.N.; et al. Activation of bone marrow-resident memory T cells by circulating, antigen-bearing dendritic cells. Nat. Immunol. 2005, 6, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Corak, J.; Ciernik, I.F.; Kavanaugh, D.; Carbone, D.P. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1997, 3, 483–490. [Google Scholar]
- Shurin, M.R.; Naiditch, H.; Zhong, H.; Shurin, G.V. Regulatory dendritic cells: New targets for cancer immunotherapy. Cancer Biol. Ther. 2011, 11, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Reviews. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Ponnazhagan, S. Myeloid-derived suppressor cells as osteoclast progenitors: A novel target for controlling osteolytic bone metastasis. Cancer Res. 2013, 73, 4606–4610. [Google Scholar] [CrossRef] [PubMed]
- Danilin, S.; Merkel, A.R.; Johnson, J.R.; Johnson, R.W.; Edwards, J.R.; Sterling, J.A. Myeloid-derived suppressor cells expand during breast cancer progression and promote tumor-induced bone destruction. Oncoimmunology 2012, 1, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, J.; Zhang, J.; Lwin, S.T.; Edwards, J.R.; Edwards, C.M.; Mundy, G.R.; Yang, X. Osteoclasts in multiple myeloma are derived from Gr-1+CD11b+myeloid-derived suppressor cells. PLoS ONE 2012, 7, e48871. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Deshane, J.; Jules, J.; Lee, C.M.; Harris, B.A.; Feng, X.; Ponnazhagan, S. Myeloid-derived suppressor cells function as novel osteoclast progenitors enhancing bone loss in breast cancer. Cancer Res. 2013, 73, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Jenne, C.N.; Liao, S.; Singh, B. Neutrophils: Multitasking first responders of immunity and tissue homeostasis. Cell Tissue Res. 2018, 371, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Burdon, P.C.; Bridger, G.; Gutierrez-Ramos, J.C.; Williams, T.J.; Rankin, S.M. Chemokines acting via CXCR2 and CXCR4 control the release of neutrophils from the bone marrow and their return following senescence. Immunity 2003, 19, 583–593. [Google Scholar] [CrossRef]
- Di Carlo, E.; Forni, G.; Lollini, P.; Colombo, M.P.; Modesti, A.; Musiani, P. The intriguing role of polymorphonuclear neutrophils in antitumor reactions. Blood 2001, 97, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Shaul, M.E.; Levy, L.; Sun, J.; Mishalian, I.; Singhal, S.; Kapoor, V.; Horng, W.; Fridlender, G.; Albelda, S.M.; Fridlender, Z.G. Tumor-associated neutrophils display a distinct N1 profile following TGFbeta modulation: A transcriptomics analysis of pro- vs. antitumor TANs. Oncoimmunology 2016, 5, e1232221. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Schernberg, A.; Blanchard, P.; Chargari, C.; Deutsch, E. Neutrophils, a candidate biomarker and target for radiation therapy? Acta Oncol. 2017, 56, 1522–1530. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gu, Y.; Han, Y.; Zhang, Q.; Jiang, Z.; Zhang, X.; Huang, B.; Xu, X.; Zheng, J.; Cao, X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell 2016, 30, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Chua, W.; Charles, K.A.; Baracos, V.E.; Clarke, S.J. Neutrophil/lymphocyte ratio predicts chemotherapy outcomes in patients with advanced colorectal cancer. Br. J. Cancer 2011, 104, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Oh, S.Y.; Kim, S.H.; Lee, J.H.; Kim, M.C.; Kim, K.H.; Kim, H.J. Prognostic significance of neutrophil lymphocyte ratio and platelet lymphocyte ratio in advanced gastric cancer patients treated with FOLFOX chemotherapy. BMC Cancer 2013, 13, 350. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Z.; Fang, F.; Gao, X.; Sun, W.; Liu, H. The neutrophil/lymphocyte ratio is an independent prognostic indicator in patients with bone metastasis. Oncol. Lett. 2011, 2, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. CII 2007, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.; Villard, J.; Means, T.K.; Alexander, S.; Dombkowski, D.; Dey, B.R.; McAfee, S.; Ballen, K.K.; Saidman, S.; Preffer, F.I.; et al. Regulatory T-cell recovery in recipients of haploidentical nonmyeloablative hematopoietic cell transplantation with a humanized anti-CD2 mAb, MEDI-507, with or without fludarabine. Exp. Hematol. 2007, 35, 1140–1152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dermawan, K.; Jin, M.; Liu, R.; Zheng, H.; Xu, L.; Zhang, Y.; Cai, Y.; Chu, Y.; Xiong, S. Differential impairment of regulatory T cells rather than effector T cells by paclitaxel-based chemotherapy. Clin. Immunol. 2008, 129, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Vonderheide, R.H. Clinical use of anti-CD25 antibody daclizumab to enhance immune responses to tumor antigen vaccination by targeting regulatory T cells. Ann. N. Y. Acad. Sci. 2009, 1174, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.D.; Li, H.Y.; Li, B.H.; Xie, T.; Zhu, T.; Sun, L.L.; Ren, H.Y.; Ye, Z.M. The role of CTLA-4 and PD-1 in anti-tumor immune response and their potential efficacy against osteosarcoma. Int. Immunopharmacol. 2016, 38, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Busse, A.; Asemissen, A.M.; Nonnenmacher, A.; Braun, F.; Ochsenreither, S.; Stather, D.; Fusi, A.; Schmittel, A.; Miller, K.; Thiel, E.; et al. Immunomodulatory effects of sorafenib on peripheral immune effector cells in metastatic renal cell carcinoma. Eur. J. Cancer 2011, 47, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Oberg, H.H.; Juricke, M.; Kabelitz, D.; Wesch, D. Regulation of T cell activation by TLR ligands. Eur. J. Cell Biol. 2011, 90, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Bayry, J.; Tchilian, E.Z.; Davies, M.N.; Forbes, E.K.; Draper, S.J.; Kaveri, S.V.; Hill, A.V.; Kazatchkine, M.D.; Beverley, P.C.; Flower, D.R.; et al. In silico identified CCR4 antagonists target regulatory T cells and exert adjuvant activity in vaccination. Proc. Natl. Acad. Sci. USA 2008, 105, 10221–10226. [Google Scholar] [CrossRef] [PubMed]
- Klingemann, H. Improving natural killer cells. Cytotherapy 2008, 10, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.V.; Alici, E.; Aints, A.; Christensson, B.; Ljunggren, H.G.; Dilber, M.S. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp. Hematol. 2005, 33, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Leong, J.W.; Fehniger, T.A. Utilizing cytokines to function-enable human NK cells for the immunotherapy of cancer. Scientifica 2014, 2014, 205796. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Linde, N.; Casanova-Acebes, M.; Sosa, M.S.; Mortha, A.; Rahman, A.; Farias, E.; Harper, K.; Tardio, E.; Reyes Torres, I.; Jones, J.; et al. Macrophages orchestrate breast cancer early dissemination and metastasis. Nat. Commun. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Ruttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Gong, Y.; Meng, W.; Yuan, M.; Zhu, H.; Ying, M.; He, Q.; Cao, J.; Yang, B. The involvement of M2 macrophage polarization inhibition in fenretinide-mediated chemopreventive effects on colon cancer. Cancer Lett. 2017, 388, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.P.; Emens, L.A. The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE(2) in murine macrophages. Int. Immunopharmacol. 2010, 10, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Lerman, B.; Sakamaki, I.; Wei, G.; Cha, S.C.; Rao, S.S.; Qian, J.; Hailemichael, Y.; Nurieva, R.; Dwyer, K.C.; et al. Generation of a new therapeutic peptide that depletes myeloid-derived suppressor cells in tumor-bearing mice. Nat. Med. 2014, 20, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, H.; Mabuchi, S.; Kozasa, K.; Yokoi, E.; Matsumoto, Y.; Komura, N.; Kawano, M.; Hashimoto, K.; Sawada, K.; Kimura, T. PM01183 inhibits myeloid-derived suppressor cells in vitro and in vivo. Immunotherapy 2017, 9, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Yokoi, E.; Komura, N.; Kimura, T. Myeloid-derived suppressor cells and their role in gynecological malignancies. Tumour Biol. 2018, 40. [Google Scholar] [CrossRef] [PubMed]
- Bu, L.L.; Li, Y.C.; Yu, G.T.; Liu, J.F.; Deng, W.W.; Zhang, W.F.; Zhang, L.; Sun, Z.J. Targeting phosphorylation of STAT3 delays tumor growth in HPV-negative anal squamous cell carcinoma mouse model. Sci. Rep. 2017, 7, 6629. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhao, Y.; Wang, H.; Li, Y.; Shao, L.; Wang, R.; Lu, J.; Yang, Z.; Wang, J.; Zhao, Y. mTOR masters monocytic myeloid-derived suppressor cells in mice with allografts or tumors. Sci. Rep. 2016, 6, 20250. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells-a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc. Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Heine, A.; Schilling, J.; Grunwald, B.; Kruger, A.; Gevensleben, H.; Held, S.A.; Garbi, N.; Kurts, C.; Brossart, P.; Knolle, P.; et al. The induction of human myeloid derived suppressor cells through hepatic stellate cells is dose-dependently inhibited by the tyrosine kinase inhibitors nilotinib, dasatinib and sorafenib, but not sunitinib. Cancer Immunol. Immunother. CII 2016, 65, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Huang, J.; Ren, X.; Gorska, A.E.; Chytil, A.; Aakre, M.; Carbone, D.P.; Matrisian, L.M.; Richmond, A.; Lin, P.C.; et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell 2008, 13, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Zollo, M.; Di Dato, V.; Spano, D.; De Martino, D.; Liguori, L.; Marino, N.; Vastolo, V.; Navas, L.; Garrone, B.; Mangano, G.; et al. Targeting monocyte chemotactic protein-1 synthesis with bindarit induces tumor regression in prostate and breast cancer animal models. Clin. Exp. Metastasis 2012, 29, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Muller, P.; Martin, K.; Theurich, S.; von Bergwelt-Baildon, M.; Zippelius, A. Cancer chemotherapy agents target intratumoral dendritic cells to potentiate antitumor immunity. Oncoimmunology 2014, 3, e954460. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; He, Y.; Zhao, Q.; Yu, X. Immune Cells Act as Promising Targets for the Treatment of Bone Metastasis. Recent Pat. Anti-Cancer Drug Discov. 2017, 12, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Rennard, S.I.; Dale, D.C.; Donohue, J.F.; Kanniess, F.; Magnussen, H.; Sutherland, E.R.; Watz, H.; Lu, S.; Stryszak, P.; Rosenberg, E.; et al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2015, 191, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Benevides, L.; da Fonseca, D.M.; Donate, P.B.; Tiezzi, D.G.; De Carvalho, D.D.; de Andrade, J.M.; Martins, G.A.; Silva, J.S. IL17 Promotes Mammary Tumor Progression by Changing the Behavior of Tumor Cells and Eliciting Tumorigenic Neutrophils Recruitment. Cancer Res. 2015, 75, 3788–3799. [Google Scholar] [CrossRef] [PubMed]
- Guislain, A.; Gadiot, J.; Kaiser, A.; Jordanova, E.S.; Broeks, A.; Sanders, J.; van Boven, H.; de Gruijl, T.D.; Haanen, J.B.; Bex, A.; et al. Sunitinib pretreatment improves tumor-infiltrating lymphocyte expansion by reduction in intratumoral content of myeloid-derived suppressor cells in human renal cell carcinoma. Cancer Immunol. Immunother. CII 2015, 64, 1241–1250. [Google Scholar] [CrossRef] [PubMed]
- Raje, N.; Terpos, E.; Willenbacher, W.; Shimizu, K.; Garcia-Sanz, R.; Durie, B.; Legiec, W.; Krejci, M.; Laribi, K.; Zhu, L.; et al. Denosumab versus zoledronic acid in bone disease treatment of newly diagnosed multiple myeloma: An international, double-blind, double-dummy, randomised, controlled, phase 3 study. Lancet Oncol. 2018, 19, 370–381. [Google Scholar] [CrossRef]
- Lipton, A.; Fizazi, K.; Stopeck, A.T.; Henry, D.H.; Brown, J.E.; Yardley, D.A.; Richardson, G.E.; Siena, S.; Maroto, P.; Clemens, M.; et al. Superiority of denosumab to zoledronic acid for prevention of skeletal-related events: A combined analysis of 3 pivotal, randomised, phase 3 trials. Eur. J. Cancer 2012, 48, 3082–3092. [Google Scholar] [CrossRef] [PubMed]
- Yee, A.J.; Raje, N.S. Denosumab for the treatment of bone disease in solid tumors and multiple myeloma. Future Oncol. 2018, 14, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Gul, G.; Sendur, M.A.; Aksoy, S.; Sever, A.R.; Altundag, K. A comprehensive review of denosumab for bone metastasis in patients with solid tumors. Curr. Med Res. Opin. 2016, 32, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Herbertz, S.; Sawyer, J.S.; Stauber, A.J.; Gueorguieva, I.; Driscoll, K.E.; Estrem, S.T.; Cleverly, A.L.; Desaiah, D.; Guba, S.C.; Benhadji, K.A.; et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des. Dev. Ther. 2015, 9, 4479–4499. [Google Scholar]
- Song, B.; Park, S.H.; Zhao, J.C.; Fong, K.W.; Li, S.; Lee, Y.; Yang, Y.A.; Sridhar, S.; Lu, X.; Abdulkadir, S.A.; et al. Targeting FOXA1-mediated repression of TGF-beta signaling suppresses castration-resistant prostate cancer progression. J. Clin. Investig. 2019, 129, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Fields, S.Z.; Parshad, S.; Anne, M.; Raftopoulos, H.; Alexander, M.J.; Sherman, M.L.; Laadem, A.; Sung, V.; Terpos, E. Activin receptor antagonists for cancer-related anemia and bone disease. Expert Opin. Investig. Drugs 2013, 22, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Gore, J.; Imasuen-Williams, I.E.; Conteh, A.M.; Craven, K.E.; Cheng, M.; Korc, M. Combined targeting of TGF-beta, EGFR and HER2 suppresses lymphangiogenesis and metastasis in a pancreatic cancer model. Cancer Lett. 2016, 379, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Spender, L.C.; Ferguson, G.J.; Hughes, G.D.; Davies, B.R.; Goldberg, F.W.; Herrera, B.; Taylor, R.G.; Strathearn, L.S.; Sansom, O.J.; Barry, S.T.; et al. Preclinical Evaluation of AZ12601011 and AZ12799734, Inhibitors of Transforming Growth Factor beta Superfamily Type 1 Receptors. Mol. Pharmacol. 2019, 95, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M. Implications of Hypoxia in Breast Cancer Metastasis to Bone. Int. J. Mol. Sci. 2016, 17, 1669. [Google Scholar] [CrossRef] [PubMed]
- Futakuchi, M.; Fukamachi, K.; Suzui, M. Heterogeneity of tumor cells in the bone microenvironment: Mechanisms and therapeutic targets for bone metastasis of prostate or breast cancer. Adv. Drug Deliv. Rev. 2016, 99 Pt B, 206–211. [Google Scholar] [CrossRef]


{kind=link}
| Immune Cells | Functions | Therapeutic Strategies | |
|---|---|---|---|
| Anti-Tumoral | Pro-Tumoral | ||
| CD8+ T cells | Recognize and eliminate cancer cells | — | Enhance CD8+ T cells immune response by vaccination or engineering the T cell receptor or chimeric antigen receptor |
| Tregs | — | Suppress immune response of cytotoxic T cells and NK cells that promote cancer cells survival and metastasis | Suppression of Tregs by chemotherapy with/or anti-CD25 treatment, anti-CTLA-4 antagonist mAb, anti-PD-1 antibodies, tyrosine kinase inhibitors, PI3K-Akt inhibitors, toll-like receptors inhibitors, CCR4/CCL22 antagonists |
| NK cells | Recognize and eliminate cancer cells | — | Stimulation of NK cells by cytokines such as IL-2, IL-15 and IL-21, or immunomodulatory drugs (IMiDs), or genetic modification of NK cells |
| Macrophages | Killing of cancer cells directly; Antigen-presenting function | Immunosuppression functions: TAMs promote survival and metastasis of cancer cells and angiogenesis | Suppress the accumulation of TAMs by CSF-1 inhibitors, CCL2 inhibitors; Depletion of TAMs by trabectedin, clodronate, and zoledronic acid; Reprogramming of TAMs by CD40 antagonism, toll-like receptor inhibitors, VEGF inhibitors, PI3Kg and HDAC inhibition, tyrosine kinase inhibitors, and fenretinide |
| Dendritic cells | Antigen presentation to T cells to stimulate immune response | Suppress the cytotoxic capacity of T cells and recruit immunosuppressive immune cells to promote tumor progression and metastasis | Targeting of DCs by microtubule destabilizing agents (dolastatin 10 and ansamitocin P3), chemotherapy, and vaccine immunotherapy |
| MDSCs | — | Suppress T cell functions; Regulate the immunosuppression; Stimulate angiogenesis and lymphangiogenesis | Targeting of MDSCs by chemotherapy, PDE-5 inhibitors, mTOR inhibitors, STAT3 inhibitors, COX2 inhibitors, tyrosine kinase inhibitors, chemokine receptors antagonists, peptibodies, vemurafenib, zoledronic acid, differentiation agents (ATRA), and vitamin D |
| Neutrophils | Attack cancer cells through antigen 1-dependent recognition | Release protumoral factors including CXCR4, VEGF and MMP9 to promote metastasis | Targeting of TANs by CXCR2 or IL-17 inhibition, or combination of immunotherapy with sunitinib |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang, L.; Gilkes, D.M. The Contribution of the Immune System in Bone Metastasis Pathogenesis. Int. J. Mol. Sci. 2019, 20, 999. https://doi.org/10.3390/ijms20040999
Xiang L, Gilkes DM. The Contribution of the Immune System in Bone Metastasis Pathogenesis. International Journal of Molecular Sciences. 2019; 20(4):999. https://doi.org/10.3390/ijms20040999
Chicago/Turabian StyleXiang, Lisha, and Daniele M. Gilkes. 2019. "The Contribution of the Immune System in Bone Metastasis Pathogenesis" International Journal of Molecular Sciences 20, no. 4: 999. https://doi.org/10.3390/ijms20040999
APA StyleXiang, L., & Gilkes, D. M. (2019). The Contribution of the Immune System in Bone Metastasis Pathogenesis. International Journal of Molecular Sciences, 20(4), 999. https://doi.org/10.3390/ijms20040999