Characteristics of Gut Microbiota in Sows and Their Relationship with Apparent Nutrient Digestibility


Abstract
1. Introduction
2. Results
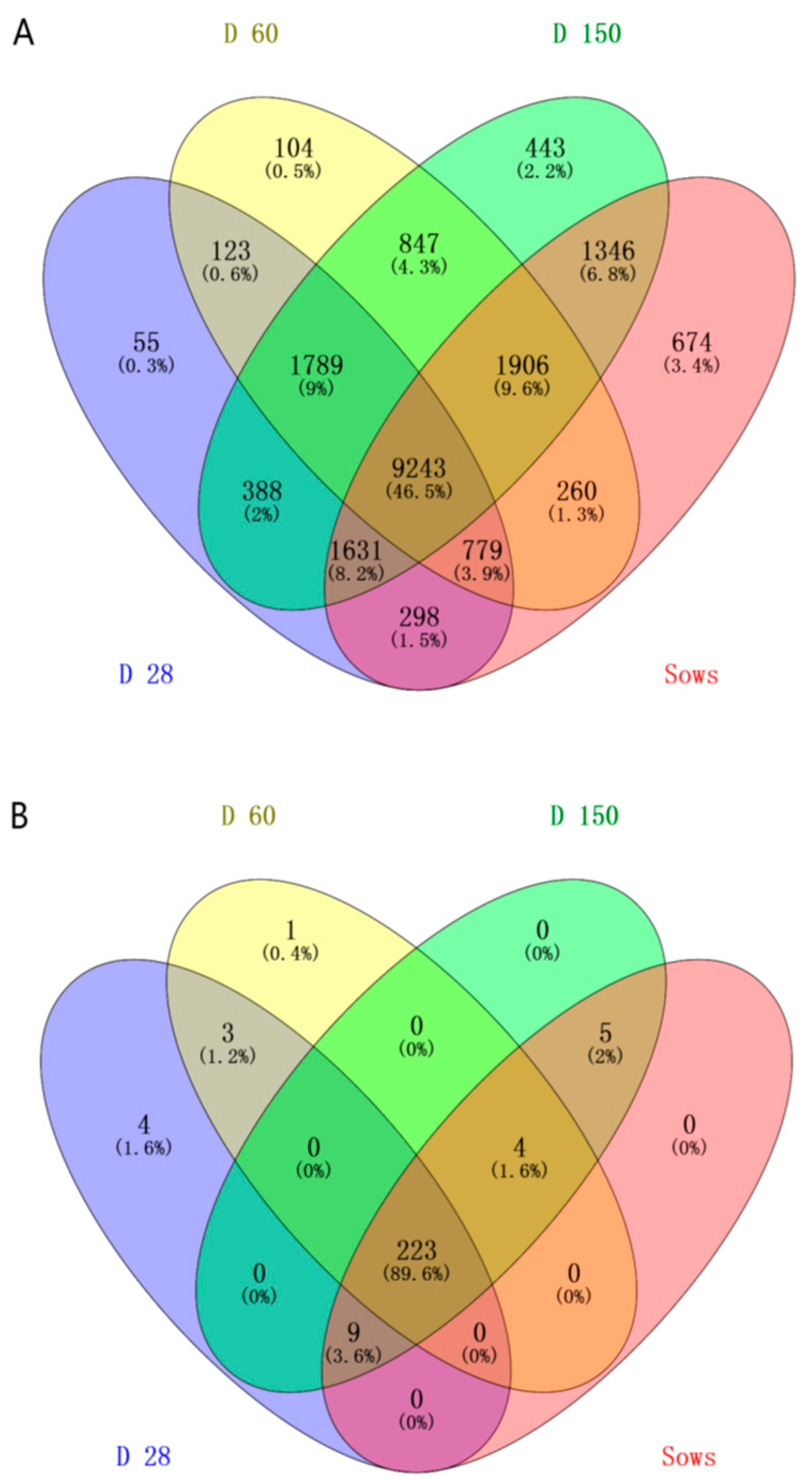
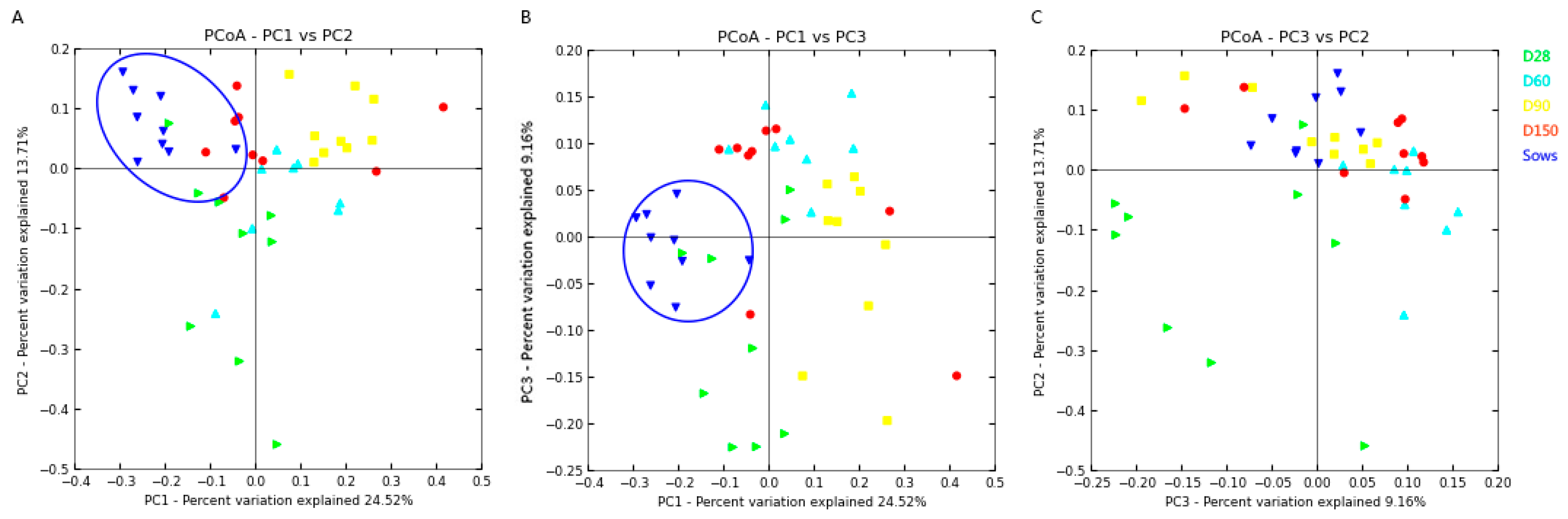
2.1. DNA Sequence Data and Bacterial Community Structure
2.2. Apparent Nutrient Digestibility in Sows and its Correlation with the Gut Microbiota
3. Discussion
4. Materials and Methods
4.1. Animals and Sample Collection
4.2. 16S rDNA Sequencing: DNA Extraction, PCR Amplification, Amplicon Sequence, Sequence Data Processing, Taxonomy Classification and Statistical Analysis
4.3. Experimental Feeds and Chemical Analysis
5. Data Analysis
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EE | ether extract |
| CP | crude protein |
| NDF | neutral detergent fiber |
| CF | crude fiber |
| ADF | acid detergent fiber |
| AIA | acid insoluble ash |
| OTUs | operational taxonomic units |
| RDP | ribosomal database project |
| PCoA | principal co-ordinates analysis |
References
- Isaacson, R.; Kim, H.B. The intestinal microbiome of the pig. Anim. Health Res. Rev. 2012, 13, 100–109. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Looft, T.; Allen, H.K.; Cantarel, B.L.; Levine, U.Y.; Bayles, D.O.; Alt, D.P.; Henrissat, B.; Stanton, T.B. Bacteria, phages and pigs: The effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014, 8, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Microbial shifts in the swine distal gut in response to the treatment with antimicrobial growth promoter, tylosin. Proc. Natl. Acad. Sci. USA 2012, 109, 15485–15490. [Google Scholar] [CrossRef]
- Yano, J.M.; Yu, K.; Donaldson, G.P.; Shastri, G.G.; Ann, P.; Ma, L.; Nagler, C.R.; Ismagilov, R.F.; Mazmanian, S.K.; Hsiao, E.Y. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell 2015, 161, 264–276. [Google Scholar] [CrossRef]
- Kelly, J.; Daly, K.; Moran, A.W.; Ryan, S.; Bravo, D.; Shirazi-Beechey, S.P. Composition and diversity of mucosa-associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences. Environ. Microbiol. 2017, 19, 1425–1438. [Google Scholar] [CrossRef]
- Green, G.L.; Brostoff, J.; Hudspith, B.; Michael, M.; Mylonaki, M.; Rayment, N.; Staines, N.; Sanderson, J.; Rampton, D.S.; Bruce, K.D. Molecular characterization of the bacteria adherent to human colorectal mucosa. J. Appl. Microbiol. 2006, 100, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Ashida, H.; Ogawa, M.; Kim, M.; Mimuro, H.; Sasakawa, C. Bacteria and host interactions in the gut epithelial barrier. Nat. Chem. Biol. 2011, 8, 36–45. [Google Scholar] [CrossRef]
- Moran, C.P.; Shanahan, F. Gut microbiota and obesity: Role in aetiology and potential therapeutic target. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 585–597. [Google Scholar] [CrossRef]
- Hasan, S.; Junnikkala, S.; Peltoniemi, O.; Paulin, L.; Lyyski, A.; Vuorenmaa, J.; Oliviero, C. Dietary supplementation with yeast hydrolysate in pregnancy influences colostrum yield and gut microbiota of sows and piglets after birth. PLoS ONE 2018, 13, e0197586. [Google Scholar] [CrossRef]
- Janssen, A.W.; Kersten, S. The role of the gut microbiota in metabolic health. FASEB 2015, 29, 3111–3123. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Li, P.; Hao, S.; Zhang, Y.; Kim, S.W.; Li, H.; Ma, X.; Gao, S.; He, L.; Wu, W.; et al. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 2015, 5, 9938. [Google Scholar] [CrossRef] [PubMed]
- Dierick, N.M.; Vervaeke, I.J.; Decuypere, J.A. Approach to the energentic importace of fiber digestion in pigs. Importance of fermentation in the overall energy supply. Anim. Feed. Sci. Tech. 1989, 23, 141–167. [Google Scholar] [CrossRef]
- Veum, T.L.; Crenshaw, J.D.; Crenshaw, T.D.; Cromwell, G.L.; Easter, R.A.; Ewan, R.C.; Nelssen, J.L.; Miller, E.R.; Pettigrew, J.E.; Ellersieck, M.R.; et al. The addition of ground wheat straw as a fiber source in the gestation diet of sows and the effect on sow and litter performance for three successive parities. J. Anim. Sci. 2009, 87, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Everts, H. The effect of feeding different sources of crude fiber during pregnancy on the reproductive performance of sows. Anim. Sci. 1991, 52, 175–184. [Google Scholar]
- Kim, H.B.; Borewicz, K.; White, B.A.; Singer, R.S.; Sreevatsan, S.; Tu, Z.J.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.M.; Lu, P.Z.; Zhang, H. Bacterial communities in manures of piglets and adult pigs bred with different feeds revealed by 16S rDNA 454 pyrosequencing. Appl. Microbiol. Biot. 2014, 98, 2657–2665. [Google Scholar] [CrossRef]
- Lamendella, R.; Domingo, J.W.; Ghosh, S.; Martinson, J.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef]
- Ye, L.; Zhang, T. Bacterial communities in different sections of a municipal wastewater treatment plant revealed by 16S rDNA 454 pyrosequencing. Appl. Microbiol. Biot. 2013, 97, 2681–2690. [Google Scholar] [CrossRef]
- Whitehead, T.R.; Cotta, M.A. Characterisation and Comparison of Microbial Populations in Swine Faeces and Manure Storage Pits by 16S rDNA Gene Sequence Analyses. Anaerobe 2001, 7, 181–187. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, Y.; Liu, S.; Huang, J.; Zhai, Z.; He, C.; Ding, J.; Wang, J.; Wang, H.; Fan, W.; et al. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the human intestinal microbial flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Woting, A.; Pfeiffer, N.; Loh, G.; Klaus, S.; Blaut, M. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio 2014, 5, e01530-14. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Nyman, M.; Fak, F. Modulation of gut microbiota in rats fed high-fat diets by processing whole-grain barley to barley malt. Mol. Nutr. Food Res. 2015, 59, 2066–2076. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Kaser, A. Gut microbiome, obesity, and metabolic dysfunction. J. Clin. Investig. 2011, 121, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, T.H.; Hobson, P.N. Further studies on the isolation of proteolytic bacteria from the sheep rumen. J. Gen. Microbio. 1962, 29, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Conde, M.S.; Rozas, A.P.D.; Badiola, I.; Pérez-Alba, L.; Blas, C.D.; Carabaño, R.; García, J. Effect of neutral detergent soluble fibre on digestion, intestinal microbiota and performance in twenty five day old weaned rabbits. Livest. Sci. 2009, 125, 192–198. [Google Scholar] [CrossRef]
- Chen, H.; Mao, X.B.; Che, L.Q.; Yu, B.; He, J.; Yu, J.; Han, G.Q.; Huang, Z.Q.; Zheng, P.; Chen, D.W. Impact of fiber types on gut microbiota, gut environment and gut function in fattening pigs. Anim. Sci. Tech. 2014, 195, 101–111. [Google Scholar] [CrossRef]
- Varel, V.H.; Richardson, A.J.; Stewart, C.S. Degradation of Barley Straw, Ryegrass, and Alfalfa Cell Walls by Clostridium longisporum and Ruminococcus albus. Appl. Environ. Microb. 1989, 55, 3080–3084. [Google Scholar]
- Medellin-Pena, M.J.; Griffiths, M.W. Effect of molecules secreted by Lactobacillus acidophilus strain La-5 on Escherichia coli O157:H7 colonization. Appl. Environ. Microb. 2009, 75, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Ransom-Jones, E.; Jones, D.L.; McCarthy, A.J.; McDonald, J.E. The Fibrobacteres: An important phylum of cellulose-degrading bacteria. Microb. Ecol. 2012, 63, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Jha, R.; Berrocoso, J.F.D. Dietary fiber and protein fermentation in the intestine of swine and their interactive effects on gut health and on the environment: A review. Anim. Feed. Sci. Tech. 2016, 212, 18–26. [Google Scholar] [CrossRef]
- Renteria-Flores, J.A.; Johnston, L.J.; Shurson, G.C.; Moser, R.L.; Webel, S.K. Effect of soluble and insoluble dietary fiber on embryo survival and sow performance. J. Anim Sci. 2008, 86, 2576–2584. [Google Scholar] [CrossRef] [PubMed]
- Fernández, J.A.; Jørgensen, H.; Just, A. Comparative digestibility experiments with growing pigs and adult sows. Anim. Prod. 2010, 43, 127–132. [Google Scholar] [CrossRef]
- Goff, G.L.; Milgen, J. v.; Noblet, J. Influence of dietary fibre on digestive utilization and rate of passage in growing pigs, finishing pigs and adult sows. Anim. Sci. 2016, 74, 503–515. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Interactions between gut microbiota and host metabolism predisposing to obesity and diabetes. Annu. Rev. Med. 2011, 62, 361–380. [Google Scholar] [CrossRef]
- Suzuki, T.; Yoshida, S.; Hara, H. Physiological concentrations of short-chain fatty acids immediately suppress colonic epithelial permeability. Brit. J. Nutr. 2008, 100, 297–305. [Google Scholar] [CrossRef]
- Bach Knudsen, K.E.; Hedemann, M.S.; Lærke, H.N. The role of carbohydrates in intestinal health of pigs. Anim Feed Sci Tech 2012, 173, 41–53. [Google Scholar] [CrossRef]
- Ma, J.W.; Yang, J.; Zhou, L.S.; Zhang, Z.Y.; Ma, H.B.; Xie, X.H.; Zhang, F.; Xiong, X.W.; Cui, L.L.; Yang, H.; et al. Genome-wide association study of meat quality traits in a white Duroc×Erhualian F2 intercross and Chinese Sutai pigs. PLoS ONE 2013, 8, e64047. [Google Scholar]
- Zhang, F.; Zhang, Z.Y.; Yan, X.M.; Chen, H.; Zhang, W.C.; Hong, Y.; Huang, L.S. Genome-wide association studies for hematological traits in Chinese Sutai pigs. BMC Genet. 2014, 15, 41. [Google Scholar] [CrossRef] [PubMed]
- Mao, S.Y.; Huo, W.J.; Zhu, W.Y. Microbiome-metabolome analysis reveals unhealthy alterations in the composition and metabolism of ruminal microbiota with increasing dietary grain in a goat model. Environ. Microbiol. 2016, 18, 525–541. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.H.; Bian, G.R.; Zhu, W.Y.; Mao, S.Y. High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front. Microbiol. 2015, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Ngoc, T.T.; Len, N.T.; Lindberg, J.E. Impact of fibre intake and fibre source on digestibility, gut development, retention time and growth performance of indigenous and exotic pigs. Animal 2013, 7, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Hess, M.; Sczyrba, A.; Egan, R.; Kim, T.W.; Chokhawala, H.; Schroth, G.; Luo, S.J.; Clark, D.S.; Chen, F.; Zhang, T.; et al. Metagenomic discovery of biomass-degrading genes and genomes from cow rumen. Science 2011, 331, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Sauer, W.C.; Fan, M.Z.; Mosenthin, R.; Drochner, W. Method for measuring ileal amino acid digestibility in pigs. In Farm Animal Metabolism and Nutrition; D’Mello, J.P.F., Ed.; Academic: Edinburgh, UK, 2000; pp. 279–306. [Google Scholar]
- Lan, Y.M.; Wang, Q.; James, R.C.; Rosen, G.L. Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Cardenas, E.; Fish, J.; Chai, B.; Farris, R.J.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Marsh, T.; Garrity, G.M.; et al. The Ribosomal Database Project: Improved alignments and new tools for rRNA analysis. Nucleic Acids. Res. 2009, 37, D141–D145. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Shen, Z.; Wang, D.; Ruan, Y.; Xue, C.; Zhang, J.; Li, R.; Shen, Q. Deep 16S rRNA pyrosequencing reveals a bacterial community associated with Banana Fusarium Wilt disease suppression induced by bio-organic fertilizer application. PLoS ONE 2014, 9, e98420. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| 28D | 60D | 90D | 150D | Sows | |
|---|---|---|---|---|---|
| Treponema | 241.0 ± 63.8 B | 202.5 ± 60.4 | 803.9 ± 194.0 | 1670.8 ± 923.7 | 4212.4 ± 601.6 A |
| Bifidobacterium | 188.5 ± 42.6 b | 65.9 ± 26.8 b | 135.5 ± 76.6 b | 97.2 ± 27.6 b | 1080.0 ±600.7 a |
| Fibrobacter | 19.0 ± 2.2 B | 22.3 ± 4.7 B | 103.4 ± 42.9 B | 53.7 ± 10.6 B | 590.0 ± 241.5 A |
| Anaeroplasma | 2.8 ± 0.4 B | 7.8 ± 4.9 B | 7.4 ± 3.4 B | 35.6 ± 6.9 B | 141.1 ± 38.9 A |
| Papillibacter | 0.4 ± 0.2 B | 0.5 ± 0.3 B | 0.3 ± 0.2 B | 0.3 ± 0.2 B | 20.2 ± 6.6 A |
| Sows | D60 | D90 | D150 | |
|---|---|---|---|---|
| EE | 55.34 ± 2.50 A | 26.83 ± 3.77 B | 20.83 ± 3.78 B | 26.97 ± 2.73 B |
| CP | 88.45 ± 1.05 A | 73.66 ± 1.14 B | 77.18 ± 0.90 B | 77.39 ± 0.68 B |
| CF | 61.61 ± 5.23 Aa | 37.05 ± 2.49 Bab | 49.11 ± 3.14 ABb | 63.10 ± 1.78 ABab |
| NDF | 65.83 ± 4.02 a | 56.74 ± 2.29 b | 69.35 ± 1.48 ab | 62.43 ± 3.53 ab |
| ADF | 54.47 ± 3.43 Aa | 28.46 ± 2.71 Bab | 30.53 ± 2.48Bab | 44.55 ± 2.70 ABb |
| Genera (Pearson’s Positive Correlation) | ||
|---|---|---|
| Sows | Grower Pigs | |
| Apparent EE digestibility | Clostridium (0.780 *) Turicibacter (0.723 *) | |
| Apparent CF digestibility | Anaerofustis (0.735 *) Robinsoniella (0.799 **) | Anaeroplasma (0.595 **) |
| Campylobacter (0.511 **) | ||
| Clostridium (0.453 **) | ||
| Enterococcus (0.415 *) | ||
| Janibacter (0.397 *) | ||
| Methanobrevibacter (0.568 **) | ||
| Nitrosospira (0.433 *) | ||
| Propionibacterium (0.500 *) | ||
| Pseudobutyrivibrio (0.505 *) | ||
| Robinsoniella (0.606 **) | ||
| Staphylococcus (0.402 *) | ||
| Treponema (0.542 **) | ||
| Turicibacter (0.418 *) | ||
| Apparent NDF digestibility | Collinsella (0.716 *) Sutterella (0.744 *) | Methanobrevibacter (0.449 *) |
| Parasporobacterium (0.469 *) | ||
| Sporobacter (0.435 *) | ||
| Treponema (0.553 **) | ||
| Apparent ADF digestibility | Clostridium (0.761 *) Collinsella (0.757 *) Robinsoniella (0.768 *) Turicibacter (0.781 *) | Anaeroplasma (0.481 *) |
| Campylobacter (0.437 *) | ||
| Caulobacter (0.443 *) | ||
| Cloacibacillus (0.546 **) | ||
| Enterococcus (0.434 *) | ||
| Lactobacillus (0.496 *) | ||
| Methanobrevibacter (0.561 **) | ||
| Nitrosospira (0.488 *) | ||
| Propionibacterium (0.461 *) | ||
| Pseudomonas (0.455 *) | ||
| Robinsoniella (0.651 **) | ||
| Staphylococcus (0.476 *) | ||
| Treponema (0.542 **) | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, Q.; Li, P.; Hao, S.; Kim, S.W.; Du, T.; Hua, J.; Huang, R. Characteristics of Gut Microbiota in Sows and Their Relationship with Apparent Nutrient Digestibility. Int. J. Mol. Sci. 2019, 20, 870. https://doi.org/10.3390/ijms20040870
Niu Q, Li P, Hao S, Kim SW, Du T, Hua J, Huang R. Characteristics of Gut Microbiota in Sows and Their Relationship with Apparent Nutrient Digestibility. International Journal of Molecular Sciences. 2019; 20(4):870. https://doi.org/10.3390/ijms20040870
Chicago/Turabian StyleNiu, Qing, Pinghua Li, Shuaishuai Hao, Sung Woo Kim, Taoran Du, Jindi Hua, and Ruihua Huang. 2019. "Characteristics of Gut Microbiota in Sows and Their Relationship with Apparent Nutrient Digestibility" International Journal of Molecular Sciences 20, no. 4: 870. https://doi.org/10.3390/ijms20040870
APA StyleNiu, Q., Li, P., Hao, S., Kim, S. W., Du, T., Hua, J., & Huang, R. (2019). Characteristics of Gut Microbiota in Sows and Their Relationship with Apparent Nutrient Digestibility. International Journal of Molecular Sciences, 20(4), 870. https://doi.org/10.3390/ijms20040870