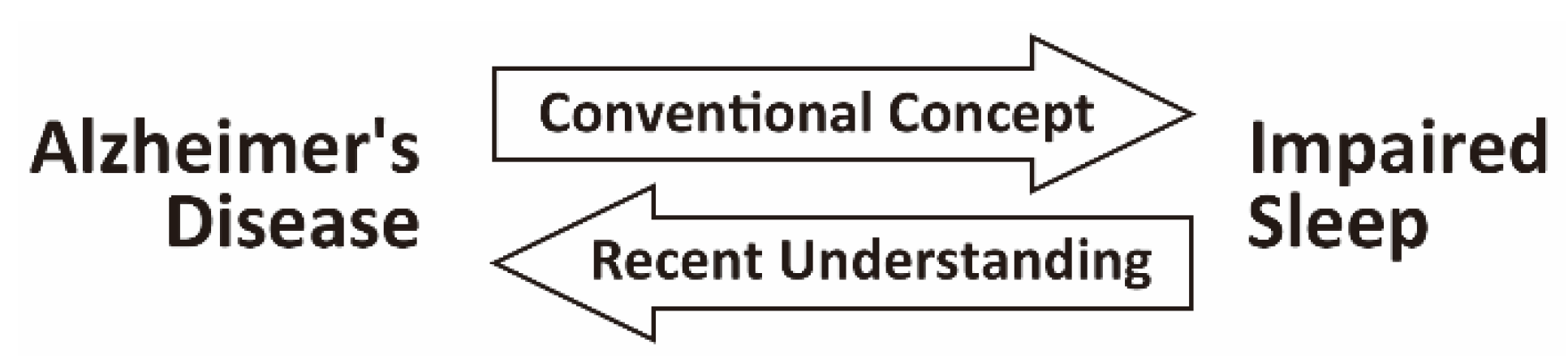
Sleep Disturbance as a Potential Modifiable Risk Factor for Alzheimer’s Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Age-Related Sleep Alterations
3. Sleep Disturbance in Alzheimer’s Disease (AD)
3.1. Sleep Abnormalities in Patients with Alzheimer’s Disease (AD)
3.2. Sleep Disturbance as a Consequence of AD Pathology
4. Sleep Disturbance as a Risk Factor of AD
4.1. Epidemiological Studies
4.2. The Causal Relationship between Sleep Disturbance and AD Pathology
5. Molecular/Cellular Mechanisms that Link AD and Sleep
5.1. Impaired Sleep Alters the Dynamics of Aβ and Tau in the Brain
5.2. Prolonged Wakefulness Induces Impaired Proteostasis, a Common Pathomechanism Underlying Neurodegenerative Diseases
5.3. Impaired Sleep May Aggravate the Propagation of AD-Related Pathology via Impaired Functional Connectivity in the Brain
5.4. Other Mechanisms that May Link Impaired Sleep and AD-Related Pathology
6. Conclusions and Future Directions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| REM | Rapid Eye Movement |
| NREM | Non-Rapid Eye Movement |
| EEG | Electroencephalogram |
| SWS | Slow-wave Sleep |
| SRBD | Sleep-related breathing disorders |
| SCN | Suprachiasmatic Nucleus |
| RR | Risk Ratio |
| ICD-9 | International Classification of Diseases, Ninth Edition |
| DSM-IV | Diagnostic Statistical Manual, Fourth Edition |
| Aβ | Amyloid Aβ |
| BACE1 | β-site amyloid precursor protein cleaving enzyme 1 |
| NFT | Neurofibrillary Tangles |
| CSF | Cerebrospinal Fluid |
| ISF | Interstitial Fluid |
| PD | Parkinson’s Disease |
| DLB | Dementia with Lewy Bodies |
| CNS | Central Nervous System |
| UPR | Unfolded Protein Response |
| ER | Endoplasmic Reticulum |
| RSN | Resting State Network |
| DMN | Default Mode Network |
| TNF-α | Tumor Necrosis Factor-α |
| OSA | Obstructive Sleep Apnea |
| CBT-I | Cognitive Behavioral Therapy for Insomnia |
| GABA | γ-aminobutyric Acid |
References
- Brzecka, A.; Leszek, J.; Ashraf, G.M.; Ejma, M.; Ávila-Rodriguez, M.F.; Yarla, N.S.; Tarasov, V.V.; Chubarev, V.N.; Samsonova, A.N.; Barreto, G.E.; et al. Sleep disorders associated with Alzheimer’s disease: A perspective. Front. Neurosci. 2018, 12, 330. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, B.; Adorni, F.; Musicco, M.; Appollonio, I.; Bonanni, E.; Caffarra, P.; Caltagirone, C.; Cerroni, G.; Concari, L.; Cosentino, F.I.I.; et al. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: A multicenter italian clinical cross-sectional study on 431 patients. Dement. Geriatr. Cogn. Disord. 2012, 33, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.-E.S.; Lucey, B.P.; Holtzman, D.M. Sleep and Alzheimer disease pathology—A bidirectional relationship. Nat. Rev. Neurol. 2014, 10, 115–119. [Google Scholar] [CrossRef]
- Dufort-Gervais, J.; Mongrain, V.; Brouillette, J. Bidirectional relationships between sleep and amyloid-beta in the hippocampus. Neurobiol. Learn. Mem. 2018, (in press). [CrossRef] [PubMed]
- Moser, D.; Anderer, P.; Gruber, G.; Parapatics, S.; Loretz, E.; Boeck, M.; Kloesch, G.; Heller, E.; Schmidt, A.; Danker-Hopfe, H.; et al. Sleep classification according to AASM and Rechtschaffen & Kales: Effects on sleep scoring parameters. Sleep 2009, 32, 139–149. [Google Scholar] [PubMed]
- Léger, D.; Debellemaniere, E.; Rabat, A.; Bayon, V.; Benchenane, K.; Chennaoui, M. Slow-wave sleep: From the cell to the clinic. Sleep Med. Rev. 2018, 41, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Ohayon, M.M.; Carskadon, M.A.; Guilleminault, C.; Vitiello, M.V. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: Developing normative sleep values across the human lifespan. Sleep 2004, 27, 1255–1273. [Google Scholar] [CrossRef]
- Mander, B.A.; Winer, J.R.; Walker, M.P. Sleep and human aging. Neuron 2017, 94, 19–36. [Google Scholar] [CrossRef]
- Peter-Derex, L.; Yammine, P.; Bastuji, H.; Croisile, B. Sleep and Alzheimer’s disease. Sleep Med. Rev. 2015, 19, 29–38. [Google Scholar] [CrossRef]
- Yaffe, K.; Falvey, C.M.; Hoang, T. Connections between sleep and cognition in older adults. Lancet. Neurol. 2014, 13, 1017–1028. [Google Scholar] [CrossRef]
- Petit, D.; Gagnon, J.-F.; Fantini, M.L.; Ferini-Strambi, L.; Montplaisir, J. Sleep and quantitative EEG in neurodegenerative disorders. J. Psychosom. Res. 2004, 56, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Prinz, P.N.; Peskind, E.R.; Vitaliano, P.P.; Raskind, M.A.; Eisdorfer, C.; Zemcuznikov, H.N.; Gerber, C.J. Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J. Am. Geriatr. Soc. 1982, 30, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Crowley, K.; Trinder, J.; Kim, Y.; Carrington, M.; Colrain, I.M. The effects of normal aging on sleep spindle and K-complex production. Clin. Neurophysiol. 2002, 113, 1615–1622. [Google Scholar] [CrossRef]
- Montplaisir, J.; Petit, D.; Lorrain, D.; Gauthier, S.; Nielsen, T. Sleep in Alzheimer’s disease: Further considerations on the role of brainstem and forebrain cholinergic populations in sleep–wake mechanisms. Sleep 1995, 18, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Bhimasani, M.; Zangrilli, M.A.; Morris, J.C.; Holtzman, D.M.; Ju, Y.-E.S. Circadian rest-activity pattern changes in aging and preclinical alzheimer disease. JAMA Neurol. 2018, 75, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Bedrosian, T.A.; Nelson, R.J. Sundowning syndrome in aging and dementia: Research in mouse models. Exp. Neurol. 2013, 243, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.S.P.; Ellison, B.A.; Wang, J.L.; Yu, L.; Schneider, J.A.; Buchman, A.S.; Bennett, D.A.; Saper, C.B. Sleep is related to neuron numbers in the ventrolateral preoptic/intermediate nucleus in older adults with and without Alzheimer’s disease. Brain 2014, 137, 2847–2861. [Google Scholar] [CrossRef]
- Sherin, J.E.; Shiromani, P.J.; McCarley, R.W.; Saper, C.B. Activation of ventrolateral preoptic neurons during sleep. Science 1996, 271, 216–219. [Google Scholar] [CrossRef]
- Sherin, J.E.; Elmquist, J.K.; Torrealba, F.; Saper, C.B. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J. Neurosci. 1998, 18, 4705–4721. [Google Scholar] [CrossRef]
- Swaab, D.F.; Fliers, E.; Partiman, T.S. The suprachiasmatic nucleus of the human brain in relation to sex, age and senile dementia. Brain Res. 1985, 342, 37–44. [Google Scholar] [CrossRef]
- Zhou, J.N.; Hofman, M.A.; Swaab, D.F. VIP neurons in the human SCN in relation to sex, age and Alzheimer’s disease. Neurobiol. Aging 1995, 16, 571–576. [Google Scholar] [CrossRef]
- Harper, D.G.; Stopa, E.G.; Kuo-Leblanc, V.; McKee, A.C.; Asayama, K.; Volicer, L.; Kowall, N.; Satlin, A. Dorsomedial SCN neuronal subpopulations subserve different functions in human dementia. Brain 2008, 131, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.H.; Huang, Y.; Bero, A.W.; Kasten, T.; Stewart, F.R.; Bateman, R.J.; Holtzman, D.M. Disruption of the sleep–wake cycle and diurnal fluctuation of β-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 2012, 4, 150ra122. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.J.; Smith, J.T.; Franklin, K.M.; Beckett, T.L.; Murphy, M.P.; St Clair, D.K.; Donohue, K.D.; Striz, M.; O’Hara, B.F. Effects of aging and genotype on circadian rhythms, sleep and clock gene expression in APPxPS1 knock-in mice, a model for Alzheimer’s disease. Exp. Neurol. 2012, 236, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Platt, B.; Drever, B.; Koss, D.; Stoppelkamp, S.; Jyoti, A.; Plano, A.; Utan, A.; Merrick, G.; Ryan, D.; Melis, V.; et al. Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PLoS ONE 2011, 6, e27068. [Google Scholar] [CrossRef]
- Savonenko, A.; Xu, G.M.; Melnikova, T.; Morton, J.L.; Gonzales, V.; Wong, M.P.F.; Price, D.L.; Tang, F.; Markowska, A.L.; Borchelt, D.R. Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer’s disease: Relationships to β-amyloid deposition and neurotransmitter abnormalities. Neurobiol. Dis. 2005, 18, 602–617. [Google Scholar] [CrossRef] [PubMed]
- Bubu, O.M.; Brannick, M.; Mortimer, J.; Umasabor-Bubu, O.; Sebastião, Y.V.; Wen, Y.; Schwartz, S.; Borenstein, A.R.; Wu, Y.; Morgan, D.; et al. Sleep, Cognitive impairment and Alzheimer’s disease: A Systematic Review and Meta-Analysis. Sleep 2017, 40. [Google Scholar] [CrossRef]
- Lim, A.S.P.; Kowgier, M.; Yu, L.; Buchman, A.S.; Bennett, D.A. Sleep fragmentation and the risk of incident Alzheimer’s disease and cognitive decline in older persons. Sleep 2013, 36, 1027–1032. [Google Scholar] [CrossRef]
- Spira, A.P.; Gamaldo, A.A.; An, Y.; Wu, M.N.; Simonsick, E.M.; Bilgel, M.; Zhou, Y.; Wong, D.F.; Ferrucci, L.; Resnick, S.M. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 2013, 70, 1537–1543. [Google Scholar] [CrossRef]
- Palmqvist, S.; Schöll, M.; Strandberg, O.; Mattsson, N.; Stomrud, E.; Zetterberg, H.; Blennow, K.; Landau, S.; Jagust, W.; Hansson, O. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nat. Commun. 2017, 8, 1214. [Google Scholar] [CrossRef]
- Kang, J.-E.; Lim, M.M.; Bateman, R.J.; Lee, J.J.; Smyth, L.P.; Cirrito, J.R.; Fujiki, N.; Nishino, S.; Holtzman, D.M. Amyloid-beta dynamics are regulated by orexin and the sleep–wake cycle. Science 2009, 326, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Herdener, N.; Frankola, K.A.; Mughal, M.R.; Mattson, M.P. Chronic mild sleep restriction accentuates contextual memory impairments and accumulations of cortical Aβ and pTau in a mouse model of Alzheimer’s disease. Brain Res. 2013, 1529, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Zhong, R.; Liu, H.; Zhang, F.; Li, S.; Le, W. Chronic Sleep Deprivation Exacerbates Learning-Memory Disability and Alzheimer’s Disease-Like Pathologies in AβPP(swe)/PS1(ΔE9) Mice. J. Alzheimer’s Dis. 2016, 50, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Di Meco, A.; Joshi, Y.B.; Praticò, D. Sleep deprivation impairs memory, tau metabolism and synaptic integrity of a mouse model of Alzheimer’s disease with plaques and tangles. Neurobiol. Aging 2014, 35, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Yang, L.; Zeng, X.-A.; Zhang, Y.; Wang, X.; Chen, M.; Li, X.; Zhang, Y.; Zhang, M. Sleep deprivation accelerates the progression of Alzheimer’s disease by influencing Aβ-related metabolism. Neurosci. Lett. 2017, 650, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.E.; Cirrito, J.R.; Dong, H.; Csernansky, J.G.; Holtzman, D.M. Acute stress increases interstitial fluid amyloid-beta via corticotropin-releasing factor and neuronal activity. Proc. Natl. Acad. Sci. USA 2007, 104, 10673–10678. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Itoh, N.; Ohyama, S.; Kadota, K.; Oishi, K. Continuous exposure to a novel stressor based on water aversion induces abnormal circadian locomotor rhythms and sleep–wake cycles in mice. PLoS ONE 2013, 8, e55452. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, E.N.; Miyazaki, K.; Maruo, K.; Yagihara, H.; Fujita, H.; Wada, K.; Nagai, Y. Chronic sleep fragmentation exacerbates amyloid β deposition in Alzheimer’s disease model mice. Neurosci. Lett. 2017, 653, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Nagai, Y.; Minakawa, E.N. Drug Development for Neurodegenerative Diseases. In Neurodegenerative Disorders as Systemic Diseases, 1st ed.; Wada, K., Ed.; Springer: Tokyo, Japan, 2015; ISBN 978-4-431-54541-5. [Google Scholar]
- Huang, Y.; Potter, R.; Sigurdson, W.; Santacruz, A.; Shih, S.; Ju, Y.-E.; Kasten, T.; Morris, J.C.; Mintun, M.; Duntley, S.; et al. Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch. Neurol. 2012, 69, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, R.M.; Farber, S.A.; Growdon, J.H.; Wurtman, R.J. Release of amyloid beta-protein precursor derivatives by electrical depolarization of rat hippocampal slices. Proc. Natl. Acad. Sci. USA 1993, 90, 5191–5193. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP Processing and Synaptic Function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Yamada, K.A.; Finn, M.B.; Sloviter, R.S.; Bales, K.R.; May, P.C.; Schoepp, D.D.; Paul, S.M.; Mennerick, S.; Holtzman, D.M. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 2005, 48, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Tanei, Z.; Hashimoto, T.; Wakabayashi, T.; Okuno, H.; Naka, Y.; Yizhar, O.; Fenno, L.E.; Fukayama, M.; Bito, H.; et al. Chronic optogenetic activation augments aβ pathology in a mouse model of Alzheimer disease. Cell Rep. 2015, 11, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Ooms, S.; Overeem, S.; Besse, K.; Rikkert, M.O.; Verbeek, M.; Claassen, J.A.H.R. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: A randomized clinical trial. JAMA Neurol. 2014, 71, 971–977. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.-E.S.; Ooms, S.J.; Sutphen, C.; Macauley, S.L.; Zangrilli, M.A.; Jerome, G.; Fagan, A.M.; Mignot, E.; Zempel, J.M.; Claassen, J.A.H.R.; et al. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 2017, 140, 2104–2111. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep drives metabolite clearance from the adult brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef]
- Lucey, B.P.; Mawuenyega, K.G.; Patterson, B.W.; Elbert, D.L.; Ovod, V.; Kasten, T.; Morris, J.C.; Bateman, R.J. Associations between β-amyloid kinetics and the β-amyloid diurnal pattern in the central nervous system. JAMA Neurol. 2017, 74, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Lucey, B.P.; Hicks, T.J.; McLeland, J.S.; Toedebusch, C.D.; Boyd, J.; Elbert, D.L.; Patterson, B.W.; Baty, J.; Morris, J.C.; Ovod, V.; et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 2018, 83, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Polanco, J.C.; Li, C.; Bodea, L.-G.; Martinez-Marmol, R.; Meunier, F.A.; Götz, J. Amyloid-β and tau complexity—towards improved biomarkers and targeted therapies. Nat. Rev. Neurol. 2018, 14, 22–39. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Wu, J.W.; Hussaini, S.A.; Bastille, I.M.; Rodriguez, G.A.; Mrejeru, A.; Rilett, K.; Sanders, D.W.; Cook, C.; Fu, H.; Boonen, R.A.C.M.; et al. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat. Neurosci. 2016, 19, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Cirrito, J.R.; Stewart, F.R.; Jiang, H.; Finn, M.B.; Holmes, B.B.; Binder, L.I.; Mandelkow, E.-M.; Diamond, M.I.; Lee, V.M.-Y.; et al. In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J. Neurosci. 2011, 31, 13110–13117. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Patel, T.K.; Hochgräfe, K.; Mahan, T.E.; Jiang, H.; Stewart, F.R.; Mandelkow, E.-M.; Holtzman, D.M. Analysis of in vivo turnover of tau in a mouse model of tauopathy. Mol. Neurodegen. 2015, 10, 55. [Google Scholar] [CrossRef] [PubMed]
- Barten, D.M.; Cadelina, G.W.; Hoque, N.; DeCarr, L.B.; Guss, V.L.; Yang, L.; Sankaranarayanan, S.; Wes, P.D.; Flynn, M.E.; Meredith, J.E.; et al. Tau transgenic mice as models for cerebrospinal fluid tau biomarkers. J. Alzheimer’s Dis. 2011, 24, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Wegmann, S.; Cho, H.; DeVos, S.L.; Commins, C.; Roe, A.D.; Nicholls, S.B.; Carlson, G.A.; Pitstick, R.; Nobuhara, C.K.; et al. Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat. Commun. 2015, 6, 8490. [Google Scholar] [CrossRef] [PubMed]
- Holth, J.K.; Fritschi, S.K.; Wang, C.; Pedersen, N.P.; Cirrito, J.R.; Mahan, T.E.; Finn, M.B.; Manis, M.; Geerling, J.C.; Fuller, P.M.; et al. The sleep–wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 2019, in press. [Google Scholar] [CrossRef]
- Lucey, B.P.; McCullough, A.; Landsness, E.C.; Toedebusch, C.D.; McLeland, J.S.; Zaza, A.M.; Fagan, A.M.; McCue, L.; Xiong, C.; Morris, J.C.; et al. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Mackiewicz, M.; Shockley, K.R.; Romer, M.A.; Galante, R.J.; Zimmerman, J.E.; Naidoo, N.; Baldwin, D.A.; Jensen, S.T.; Churchill, G.A.; Pack, A.I. Macromolecule biosynthesis: A key function of sleep. Physiol. Genom. 2007, 31, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.S.; Huber, J.D.; O’Callaghan, J.P.; Rosen, C.L.; Miller, D.B. A review of sleep deprivation studies evaluating the brain transcriptome. SpringerPlus 2014, 3, 728. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, N. Cellular stress/the unfolded protein response: Relevance to sleep and sleep disorders. Sleep Med. Rev. 2009, 13, 195–204. [Google Scholar] [CrossRef]
- Naidoo, N.; Giang, W.; Galante, R.J.; Pack, A.I. Sleep deprivation induces the unfolded protein response in mouse cerebral cortex. J. Neurochem. 2005, 92, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Cirelli, C.; Gutierrez, C.M.; Tononi, G. Extensive and Divergent Effects of Sleep and Wakefulness on Brain Gene Expression. Neuron 2004, 41, 35–43. [Google Scholar] [CrossRef]
- Cirelli, C.; Faraguna, U.; Tononi, G. Changes in brain gene expression after long-term sleep deprivation. J. Neurochem. 2006, 98, 1632–1645. [Google Scholar] [CrossRef]
- Naidoo, N.; Ferber, M.; Master, M.; Zhu, Y.; Pack, A.I. Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J. Neurosci. 2008, 28, 6539–6548. [Google Scholar] [CrossRef]
- Fox, M.D.; Raichle, M.E. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat. Rev. Neurosci. 2007, 8, 700–711. [Google Scholar] [CrossRef]
- Van den Heuvel, M.P.; Pol, H.E.H. Exploring the brain network: A review on resting-state fMRI functional connectivity. Eur. Neuropsychopharmacol. 2010, 20, 519–534. [Google Scholar] [CrossRef]
- Raichle, M.E.; MacLeod, A.M.; Snyder, A.Z.; Powers, W.J.; Gusnard, D.A.; Shulman, G.L. A default mode of brain function. Proc. Natl. Acad. Sci. USA 2001, 98, 676–682. [Google Scholar] [CrossRef]
- Buckner, R.L.; Andrews-Hanna, J.R.; Schacter, D.L. The brain’s default network. Ann. N. Y. Acad. Sci. 2008, 1124, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, T.; Elvsåshagen, T.; Alnæs, D.; Zak, N.; Pedersen, P.Ø.; Norbom, L.B.; Quraishi, S.H.; Tagliazucchi, E.; Laufs, H.; Bjørnerud, A.; et al. The brain functional connectome is robustly altered by lack of sleep. NeuroImage 2016, 127, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Horovitz, S.G.; Braun, A.R.; Carr, W.S.; Picchioni, D.; Balkin, T.J.; Fukunaga, M.; Duyn, J.H. Decoupling of the brain’s default mode network during deep sleep. Proc. Natl. Acad. Sci. USA 2009, 106, 11376–11381. [Google Scholar] [CrossRef] [PubMed]
- Fjell, A.M.; McEvoy, L.; Holland, D.; Dale, A.M.; Walhovd, K.B. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog. Neurobiol. 2014, 117, 20–40. [Google Scholar] [CrossRef] [PubMed]
- Brier, M.R.; Thomas, J.B.; Ances, B.M. Network dysfunction in Alzheimer’s disease: Refining the disconnection hypothesis. Brain Connect. 2014, 4, 299–311. [Google Scholar] [CrossRef]
- Sheline, Y.I.; Morris, J.C.; Snyder, A.Z.; Price, J.L.; Yan, Z.; D’Angelo, G.; Liu, C.; Dixit, S.; Benzinger, T.; Fagan, A.; et al. APOE4 allele disrupts resting state fMRI connectivity in the absence of amyloid plaques or decreased CSF Aβ42. J. Neurosci. 2010, 30, 17035–17040. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.C.; Diamond, M.I.; Duff, K.E.; Hyman, B.T. Mechanisms of protein seeding in neurodegenerative diseases. JAMA Neurol. 2013, 70, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gennatas, E.D.; Kramer, J.H.; Miller, B.L.; Seeley, W.W. Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron 2012, 73, 1216–1227. [Google Scholar] [CrossRef]
- Ingiosi, A.M.; Opp, M.R.; Krueger, J.M. Sleep and immune function: Glial contributions and consequences of aging. Curr. Opin. Neurobiol. 2013, 23, 806–811. [Google Scholar] [CrossRef]
- Hurtado-Alvarado, G.; Pavón, L.; Castillo-García, S.A.; Hernández, M.E.; Domínguez-Salazar, E.; Velázquez-Moctezuma, J.; Gómez-González, B. Sleep loss as a factor to induce cellular and molecular inflammatory variations. Clin. Dev. Immunol. 2013, 2013, 801341. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Bellesi, M.; de Vivo, L.; Chini, M.; Gilli, F.; Tononi, G.; Cirelli, C. Sleep loss promotes astrocytic phagocytosis and microglial activation in mouse cerebral cortex. J. Neurosci. 2017, 37, 5263–5273. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Alvarado, G.; Domínguez-Salazar, E.; Pavon, L.; Velazquez-Moctezuma, J.; Gomez-Gonzalez, B. Blood-brain barrier disruption induced by chronic sleep loss: Low-grade inflammation may be the link. J. Immunol. Res. 2016, 2016, e4576012. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer’s disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Villafuerte, G.; Miguel-Puga, A.; Murillo Rodríguez, E.; Machado, S.; Manjarrez, E.; Arias-Carrión, O. Sleep deprivation and oxidative stress in animal models: A systematic review. Oxid. Med. Cell. Longev. 2015, 2015, 234952. [Google Scholar] [CrossRef]
- Sharma, R.A.; Varga, A.W.; Bubu, O.M.; Pirraglia, E.; Kam, K.; Parekh, A.; Wohlleber, M.; Miller, M.D.; Andrade, A.; Lewis, C.; et al. Obstructive sleep apnea severity affects amyloid burden in cognitively normal elderly. A longitudinal study. Am. J. Respir. Crit. Care. Med. 2017, 197, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Kashiwagi, M.; Yasuda, K.; Ando, R.; Kanuka, M.; Sakai, K.; Itohara, S. Cells of a common developmental origin regulate REM/non-REM sleep and wakefulness in mice. Science 2015, aad1023. [Google Scholar] [CrossRef]
- Trauer, J.M.; Qian, M.Y.; Doyle, J.S.; Rajaratnam, S.M.W.; Cunnington, D. Cognitive behavioral therapy for chronic insomnia: A systematic review and meta-analysis. Ann. Intern. Med. 2015, 163, 191–204. [Google Scholar] [CrossRef]
- Van Straten, A.; van der Zweerde, T.; Kleiboer, A.; Cuijpers, P.; Morin, C.M.; Lancee, J. Cognitive and behavioral therapies in the treatment of insomnia: A meta-analysis. Sleep. Med. Rev. 2018, 38, 3–16. [Google Scholar] [CrossRef]
- Kay-Stacey, M.; Attarian, H. Advances in the management of chronic insomnia. BMJ 2016, 354, i2123. [Google Scholar] [CrossRef]
- Wilson, S.; Nutt, D.; Alford, C.; Argyropoulos, S.; Baldwin, D.; Bateson, A.; Britton, T.; Crowe, C.; Dijk, D.-J.; Espie, C.; et al. British Association for Psychopharmacology consensus statement on evidence-based treatment of insomnia, parasomnias and circadian rhythm disorders. J. Psychopharmacol. 2010, 24, 1577–1601. [Google Scholar] [CrossRef] [PubMed]
- Abad, V.C.; Guilleminault, C. Insomnia in elderly patients: Recommendations for pharmacological management. Drugs Aging. 2018, 35, 791–817. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhao, X.; Veasey, S.C. Neural consequences of chronic short sleep: Reversible or lasting? Front. Neurol. 2017, 8, 235. [Google Scholar] [CrossRef] [PubMed]
- Van Dongen, H.P.A.; Maislin, G.; Mullington, J.M.; Dinges, D.F. The cumulative cost of additional wakefulness: Dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep 2003, 26, 117–126. [Google Scholar] [CrossRef]
- Belenky, G.; Wesensten, N.J.; Thorne, D.R.; Thomas, M.L.; Sing, H.C.; Redmond, D.P.; Russo, M.B.; Balkin, T.J. Patterns of performance degradation and restoration during sleep restriction and subsequent recovery: A sleep dose-response study. J. Sleep Res. 2003, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Pejovic, S.; Basta, M.; Vgontzas, A.N.; Kritikou, I.; Shaffer, M.L.; Tsaoussoglou, M.; Stiffler, D.; Stefanakis, Z.; Bixler, E.O.; Chrousos, G.P. Effects of recovery sleep after one work week of mild sleep restriction on interleukin-6 and cortisol secretion and daytime sleepiness and performance. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E890–E896. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minakawa, E.N.; Wada, K.; Nagai, Y. Sleep Disturbance as a Potential Modifiable Risk Factor for Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 803. https://doi.org/10.3390/ijms20040803
Minakawa EN, Wada K, Nagai Y. Sleep Disturbance as a Potential Modifiable Risk Factor for Alzheimer’s Disease. International Journal of Molecular Sciences. 2019; 20(4):803. https://doi.org/10.3390/ijms20040803
Chicago/Turabian StyleMinakawa, Eiko N., Keiji Wada, and Yoshitaka Nagai. 2019. "Sleep Disturbance as a Potential Modifiable Risk Factor for Alzheimer’s Disease" International Journal of Molecular Sciences 20, no. 4: 803. https://doi.org/10.3390/ijms20040803
APA StyleMinakawa, E. N., Wada, K., & Nagai, Y. (2019). Sleep Disturbance as a Potential Modifiable Risk Factor for Alzheimer’s Disease. International Journal of Molecular Sciences, 20(4), 803. https://doi.org/10.3390/ijms20040803