Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hypoxia in Tumor Progression
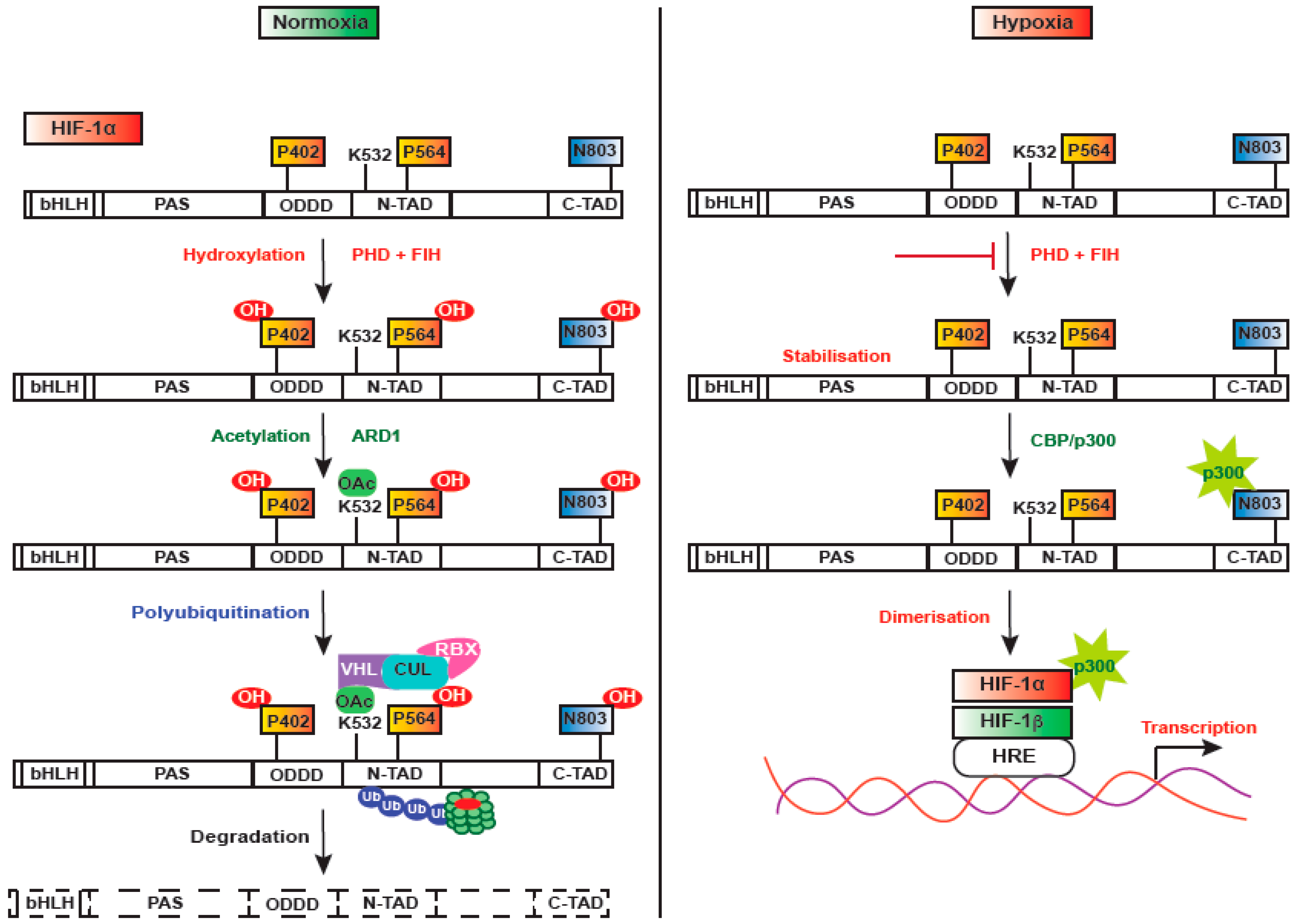
3. Regulation of HIF-1
4. Stress Responses Regulated by HIF-1 in Response to Hypoxia
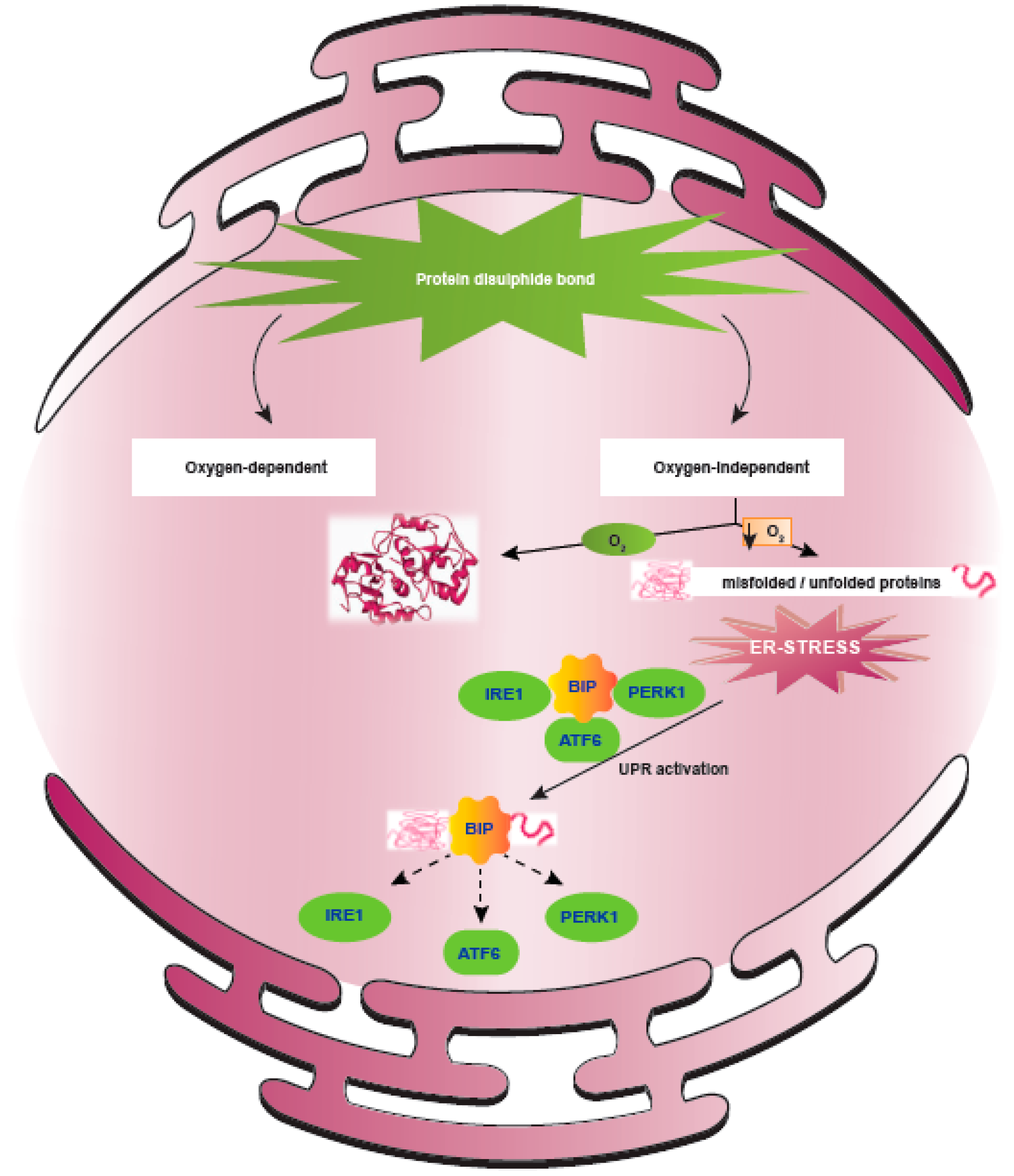
5. ER Stress Response Regulated upon Hypoxia
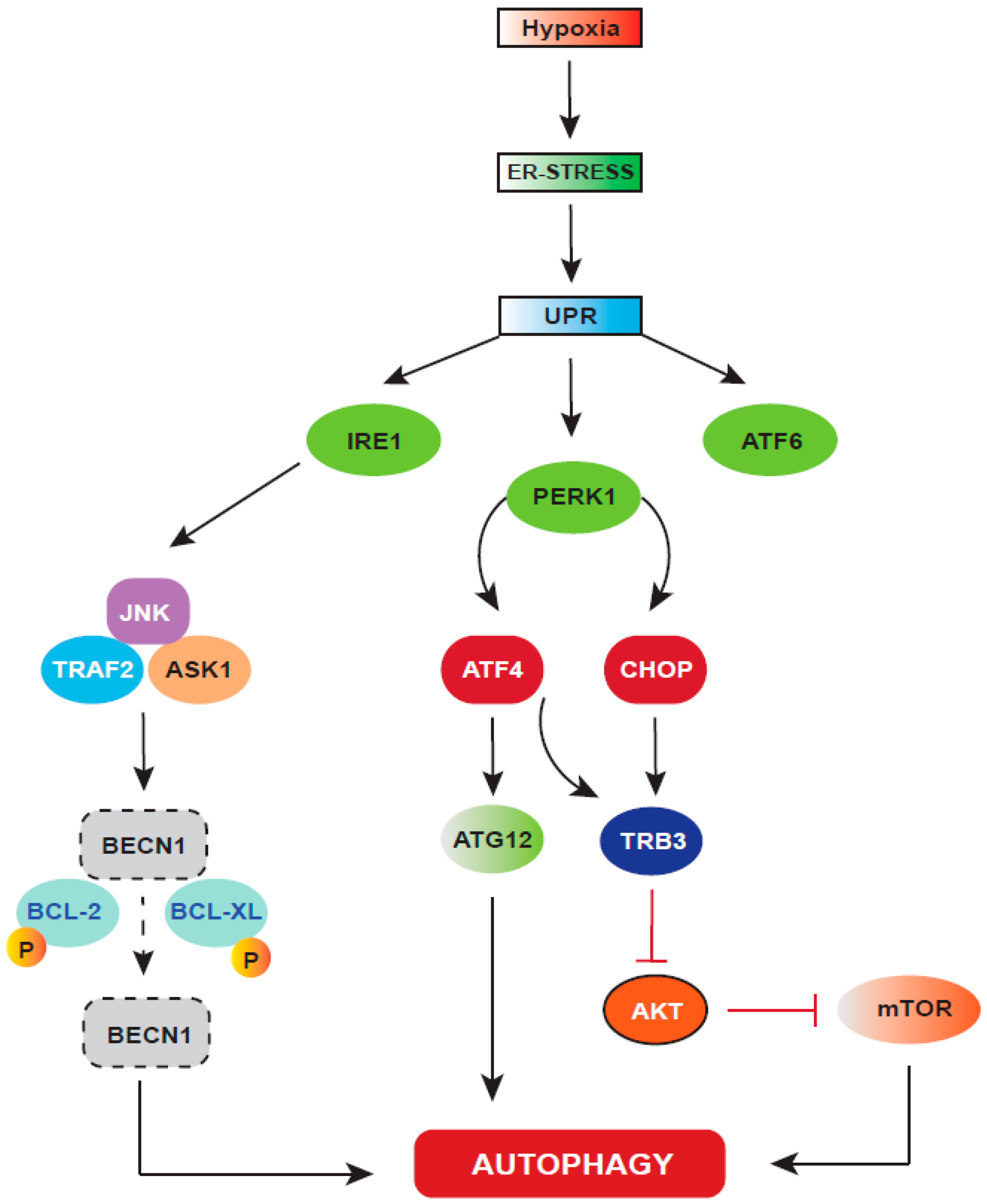
6. Autophagy: A Cellular Stress Response Regulated by HIF-1
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| IRE1 | Inositol requiring enzyme |
| PERK | Protein kinase RNA-like endoplasmic reticulum kinase |
| ATF6 | Activating transcription factor 6 |
| JNK | c-Jun N-terminal kinase |
| TRAF2 | TNF receptor-associated factor 2 |
| ASK1 | Apoptosis signal-regulating kinase 1 |
| BCL-2 | B-cell lymphoma 2 |
| BCL-XL | B-cell lymphoma-extra large |
| BECN1 | Beclin-1 |
| ATF4 | Activating transcription factor 4 |
| CHOP | C/EBP homologous protein |
| TRB3 | Tribbles homolog 3 |
| AKT | Protein kinase B (PKB) |
| mTOR | Mammalian target of rapamycin |
References
- Semenza, G.L. The hypoxic tumor microenvironment: A driving force for breast cancer progression. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Spill, F.; Reynolds, D.S.; Kamm, R.D.; Zaman, M.H. Impact of the physical microenvironment on tumor progression and metastasis. Curr. Opin. Biotechnol. 2016, 40, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Fan, X.; Houghton, J. Tumor microenvironment: The role of the tumor stroma in cancer. J. Cell. Biochem. 2007, 101, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Schito, L.; Semenza, G.L. Hypoxia-inducible factors: Master regulators of cancer progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Semenza, G.L. Oxygen homeostasis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 336–361. [Google Scholar] [CrossRef]
- Zhou, J.; Schmid, T.; Schnitzer, S.; Brüne, B. Tumor hypoxia and cancer progression. Cancer Lett. 2006, 237, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.L.; Jiang, B.-H.; Rue, E.A.; Semenza, G.L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 1995, 92, 5510–5514. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Li, P.; Ji, C. Cell death conversion under hypoxic condition in tumor development and therapy. Int. J. Mol. Sci. 2015, 16, 25536–25551. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Xie, C.; Jiang, C. The role of hypoxia-inducible factors in metabolic diseases. Nat. Rev. Endocrinol. 2018, 15, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Talks, K.L.; Turley, H.; Gatter, K.C.; Maxwell, P.H.; Pugh, C.W.; Ratcliffe, P.J.; Harris, A.L. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 2000, 157, 411–421. [Google Scholar] [CrossRef]
- Zhong, H.; De Marzo, A.M.; Laughner, E.; Lim, M.; Hilton, D.A.; Zagzag, D.; Buechler, P.; Isaacs, W.B.; Semenza, G.L.; Simons, J.W. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999, 59, 5830–5835. [Google Scholar] [PubMed]
- Ding, Z.; Yang, L.; Xie, X.; Xie, F.; Pan, F.; Li, J.; He, J.; Liang, H. Expression and significance of hypoxia-inducible factor-1 alpha and MDR1/P-glycoprotein in human colon carcinoma tissue and cells. J. Cancer Res. Clin. Oncol. 2010, 136, 1697–1707. [Google Scholar] [CrossRef]
- Rohwer, N.; Lobitz, S.; Daskalow, K.; Jons, T.; Vieth, M.; Schlag, P.M.; Kemmner, W.; Wiedenmann, B.; Cramer, T.; Hocker, M. HIF-1alpha determines the metastatic potential of gastric cancer cells. Br. J. Cancer 2009, 100, 772–781. [Google Scholar] [CrossRef]
- Luoto, K.R.; Kumareswaran, R.; Bristow, R.G. Tumor hypoxia as a driving force in genetic instability. Genome Integr. 2013, 4, 5. [Google Scholar] [CrossRef]
- Chi, J.-T.; Wang, Z.; Nuyten, D.S.; Rodriguez, E.H.; Schaner, M.E.; Salim, A.; Wang, Y.; Kristensen, G.B.; Helland, Å.; Børresen-Dale, A.-L. Gene expression programs in response to hypoxia: Cell type specificity and prognostic significance in human cancers. PLoS Med. 2006, 3, e47. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.; Sivridis, E.; Turley, H.; Talks, K.; Pezzella, F.; Gatter, K.; Harris, A. Relation of hypoxia inducible factor 1α and 2α in operable non-small cell lung cancer to angiogenic/molecular profile of tumours and survival. Br. J. Cancer 2001, 85, 881. [Google Scholar] [CrossRef] [PubMed]
- Birner, P.; Schindl, M.; Obermair, A.; Breitenecker, G.; Oberhuber, G. Expression of hypoxia-inducible factor 1α in epithelial ovarian tumors: Its impact on prognosis and on response to chemotherapy. Clin. Cancer Res. 2001, 7, 1661–1668. [Google Scholar] [PubMed]
- Bos, R.; Van der Groep, P.; Greijer, A.E.; Shvarts, A.; Meijer, S.; Pinedo, H.M.; Semenza, G.L.; Van Diest, P.J.; Van der Wall, E. Levels of hypoxia-inducible factor-1α independently predict prognosis in patients with lymph node negative breast carcinoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 1573–1581. [Google Scholar]
- Aebersold, D.M.; Burri, P.; Beer, K.T.; Laissue, J.; Djonov, V.; Greiner, R.H.; Semenza, G.L. Expression of hypoxia-inducible factor-1α: A novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer Res. 2001, 61, 2911–2916. [Google Scholar] [PubMed]
- Zundel, W.; Schindler, C.; Haas-Kogan, D.; Koong, A.; Kaper, F.; Chen, E.; Gottschalk, A.R.; Ryan, H.E.; Johnson, R.S.; Jefferson, A.B. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000, 14, 391–396. [Google Scholar]
- Bhatia, M. Hypoxia factors suffocate leukemic stem cell initiation. J. Exp. Med. 2015, 212, 2187–2188. [Google Scholar] [CrossRef]
- Salceda, S.; Caro, J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions Its stabilization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997, 272, 22642–22647. [Google Scholar] [CrossRef]
- Liao, D.; Johnson, R.S. Hypoxia: A key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007, 26, 281–290. [Google Scholar] [CrossRef]
- Shah, T.; Krishnamachary, B.; Wildes, F.; Mironchik, Y.; Kakkad, S.M.; Jacob, D.; Artemov, D.; Bhujwalla, Z.M. HIF isoforms have divergent effects on invasion, metastasis, metabolism and formation of lipid droplets. Oncotarget 2015, 6, 28104–28119. [Google Scholar] [CrossRef]
- Luo, W.; Semenza, G.L. Pyruvate kinase M2 regulates glucose metabolism by functioning as a coactivator for hypoxia-inducible factor 1 in cancer cells. Oncotarget 2011, 2, 551–556. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-inducible factors in physiology and medicine. Cell 2012, 148, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P. The role of hypoxia-induced factors in tumor progression. Oncologist 2004, 9, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Gilany, K.; Vafakhah, M. Hypoxia: A review. J. Paramed. Sci. 2010, 1, 43–60. [Google Scholar]
- Ivan, M.; Kondo, K.; Yang, H.; Kim, W.; Valiando, J.; Ohh, M.; Salic, A.; Asara, J.M.; Lane, W.S.; Kaelin, W.G., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science 2001, 292, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Jokilehto, T.; Jaakkola, P.M. The role of HIF prolyl hydroxylases in tumour growth. J. Cell. Mol. Med. 2010, 14, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Ohh, M.; Park, C.W.; Ivan, M.; Hoffman, M.A.; Kim, T.-Y.; Huang, L.E.; Pavletich, N.; Chau, V.; Kaelin, W.G. Ubiquitination of hypoxia-inducible factor requires direct binding to the β-domain of the von Hippel–Lindau protein. Nat. Cell Biol. 2000, 2, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Groulx, I.; Lee, S. Oxygen-dependent ubiquitination and degradation of hypoxia-inducible factor requires nuclear-cytoplasmic trafficking of the von Hippel-Lindau tumor suppressor protein. Mol. Cell. Biol. 2002, 22, 5319–5336. [Google Scholar] [CrossRef] [PubMed]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Singh, A.R.; Durden, D.L. MDM2 regulates hypoxic HIF1α stability in an E3 ligase proteasome and PTEN-PI-3 kinase-AKT dependent manner. J. Biol. Chem. 2014, 289, 22785–227897. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α pathway: Role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef]
- Gradin, K.; McGuire, J.; Wenger, R.H.; Kvietikova, I.; Toftgård, R.; Tora, L.; Gassmann, M.; Poellinger, L. Functional interference between hypoxia and dioxin signal transduction pathways: Competition for recruitment of the Arnt transcription factor. Mol. Cell. Biol. 1996, 16, 5221–5231. [Google Scholar] [CrossRef] [PubMed]
- Land, S.C.; Tee, A.R. Hypoxia inducible factor 1α is regulated by the mammalian target of rapamycin (mTOR) via an mTOR-signalling motif. J. Biol. Chem. 2007, 282, 20534–20543. [Google Scholar] [CrossRef] [PubMed]
- Mcneill, L.A.; Hewitson, K.S.; Claridge, T.D.; Seibel, J.F.; Horsfall, L.E.; Schofield, C.J. Hypoxia-inducible factor asparaginyl hydroxylase (FIH-1) catalyses hydroxylation at the β-carbon of asparagine-803. Biochem. J. 2002, 367, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Lando, D.; Peet, D.J.; Gorman, J.J.; Whelan, D.A.; Whitelaw, M.L.; Bruick, R.K. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002, 16, 1466–1471. [Google Scholar] [CrossRef] [PubMed]
- Dann, C.E.; Bruick, R.K.; Deisenhofer, J. Structure of factor-inhibiting hypoxia-inducible factor 1: An asparaginyl hydroxylase involved in the hypoxic response pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 15351–15356. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Weljie, A.M.; Jirik, F.R. Hypoxia-induced metabolic shifts in cancer cells: Moving beyond the Warburg effect. Int. J. Biochem. Cell Biol. 2011, 43, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.E.; Simon, M.C. Hypoxia-inducible factors: Crosstalk between inflammation and metabolism. Semin. Cell Dev. Biol. 2012, 23, 389–394. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef]
- Lim, J.-H.; Lee, Y.-M.; Chun, Y.-S.; Chen, J.; Kim, J.-E.; Park, J.-W. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1α. Mol. Cell 2010, 38, 864–878. [Google Scholar] [CrossRef]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, E.; Vander Heiden, M.G.; Thompson, C.B. Bcl-xL prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell. Biol. 2000, 20, 5680–5689. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.; Maltepe, E.; Goldwasser, E.; Mathieu, C.; Simon, M.; Schumacker, P. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Kazak, L.; Jedrychowski, M.P.; Lu, G.Z.; Erickson, B.K.; Szpyt, J.; Pierce, K.A.; Laznik-Bogoslavski, D.; Vetrivelan, R.; Clish, C.B. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 2016, 532, 112. [Google Scholar] [CrossRef] [PubMed]
- Görlach, A.; Dimova, E.Y.; Petry, A.; Martínez-Ruiz, A.; Hernansanz-Agustín, P.; Rolo, A.P.; Palmeira, C.M.; Kietzmann, T. Reactive oxygen species, nutrition, hypoxia and diseases: Problems solved? Redox Biol. 2015, 6, 372–385. [Google Scholar] [CrossRef]
- Finley, L.W.; Carracedo, A.; Lee, J.; Souza, A.; Egia, A.; Zhang, J.; Teruya-Feldstein, J.; Moreira, P.I.; Cardoso, S.M.; Clish, C.B. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell 2011, 19, 416–428. [Google Scholar] [CrossRef]
- Taguchi, K.; Yamamoto, M. The KEAP1-NRF2 System in Cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Y.; Feun, L.G.; Thongkum, A.; Tu, C.-H.; Chen, S.-M.; Wangpaichitr, M.; Wu, C.; Kuo, M.T.; Savaraj, N. Autophagic mechanism in anti-cancer immunity: Its pros and cons for cancer therapy. Int. J. Mol. Sci. 2017, 18, 1297. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Ryter, S.W.; Levine, B. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Y.; Kim, E.; Beemiller, P.; Wang, C.-Y.; Swanson, J.; You, M.; Guan, K.-L. Bnip3 mediates the hypoxia-induced inhibition on mammalian target of rapamycin by interacting with Rheb. J. Biol. Chem. 2007, 282, 35803–35813. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bosch-Marce, M.; Shimoda, L.A.; Tan, Y.S.; Baek, J.H.; Wesley, J.B.; Gonzalez, F.J.; Semenza, G.L. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J. Biol. Chem. 2008, 283, 10892–10903. [Google Scholar] [CrossRef] [PubMed]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Sofer, A.; Lei, K.; Johannessen, C.M.; Ellisen, L.W. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol. Cell. Biol. 2005, 25, 5834–5845. [Google Scholar] [CrossRef]
- Whitney, M.L.; Jefferson, L.S.; Kimball, S.R. ATF4 is necessary and sufficient for ER stress-induced upregulation of REDD1 expression. Biochem. Biophys. Res. Commun. 2009, 379, 451–455. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yang, Z.-Q.; Zhang, K. Endoplasmic reticulum stress response in cancer: Molecular mechanism and therapeutic potential. Am. J. Transl. Res. 2010, 2, 65–74. [Google Scholar] [PubMed]
- Gething, M.-J.; Sambrook, J. Protein folding in the cell. Nature 1992, 355, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.R.; Gething, M.-J.; Sambrook, J. The cellular response to unfolded proteins: Intercompartmental signaling. Curr. Opin. Biotechnol. 1994, 5, 540–545. [Google Scholar] [CrossRef]
- Sidrauski, C.; Chapman, R.; Walter, P. The unfolded protein response: An intracellular signalling pathway with many surprising features. Trends Cell Biol. 1998, 8, 245–249. [Google Scholar] [CrossRef]
- Mori, K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell 2000, 101, 451–454. [Google Scholar] [CrossRef]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef]
- Koritzinsky, M.; Levitin, F.; van den Beucken, T.; Rumantir, R.A.; Harding, N.J.; Chu, K.C.; Boutros, P.C.; Braakman, I.; Wouters, B.G. Two phases of disulfide bond formation have differing requirements for oxygen. J. Cell Biol. 2013, 203, 615–627. [Google Scholar] [CrossRef]
- Li, X.; Zhang, K.; Li, Z. Unfolded protein response in cancer: The physician’s perspective. J. Hematol. Oncol. 2011, 4, 8. [Google Scholar] [CrossRef]
- Sano, R.; Reed, J.C. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2013, 1833, 3460–3470. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Gagliardi, M.; Fimia, G.M.; Piacentini, M. Endoplasmic reticulum stress, unfolded protein response, and cancer cell fate. Front. Oncol. 2017, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Lovat, P.; Armstrong, J.; Fimia, G.; Hill, D.; Birch-Machin, M.; Redfern, C.; Piacentini, M. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: The role of stress proteins ERdj5 and ERp57. Br. J. Cancer 2007, 96, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Lovat, P.E.; Corazzari, M.; Armstrong, J.L.; Martin, S.; Pagliarini, V.; Hill, D.; Brown, A.M.; Piacentini, M.; Birch-Machin, M.A.; Redfern, C.P. Increasing melanoma cell death using inhibitors of protein disulfide isomerases to abrogate survival responses to endoplasmic reticulum stress. Cancer Res. 2008, 68, 5363–5369. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sankar, S.; Neamati, N. Protein disulfide isomerase: A promising target for cancer therapy. Drug Discov. Today 2014, 19, 222–240. [Google Scholar] [CrossRef] [PubMed]
- Won, J.K.; Yu, S.J.; Hwang, C.Y.; Cho, S.H.; Park, S.M.; Kim, K.; Choi, W.M.; Cho, H.; Cho, E.J.; Lee, J.H.; et al. Protein disulfide isomerase inhibition synergistically enhances the efficacy of sorafenib for hepatocellular carcinoma. Hepatology 2017, 66, 855–868. [Google Scholar] [CrossRef]
- Kozutsumi, Y.; Segal, M.; Normington, K.; Gething, M.-J.; Sambrook, J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 1988, 332, 462–464. [Google Scholar] [CrossRef]
- Lee, A.S. GRP78 induction in cancer: Therapeutic and prognostic implications. Cancer Res. 2007, 67, 3496–3499. [Google Scholar] [CrossRef]
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381. [Google Scholar] [CrossRef]
- Shapiro, D.J.; Livezey, M.; Yu, L.; Zheng, X.; Andruska, N. Anticipatory UPR activation: A protective pathway and target in cancer. Trends Endocrinol. Metab. 2016, 27, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Z. Glucose regulated protein 78: A critical link between tumor microenvironment and cancer hallmarks. Biochim. Biophys. Acta (BBA) Rev. Cancer 2012, 1826, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O. Activation of the ATF6, XBP1 and grp78 genes in human hepatocellular carcinoma: A possible involvement of the ER stress pathway in hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Song, M.S.; Park, Y.K.; Lee, J.-H.; Park, K. Induction of glucose-regulated protein 78 by chronic hypoxia in human gastric tumor cells through a protein kinase C-ε/ERK/AP-1 signaling cascade. Cancer Res. 2001, 61, 8322–8330. [Google Scholar] [PubMed]
- Zorzi, E.; Bonvini, P. Inducible hsp70 in the regulation of cancer cell survival: Analysis of chaperone induction, expression and activity. Cancers 2011, 3, 3921–3956. [Google Scholar] [CrossRef] [PubMed]
- Kern, J.; Untergasser, G.; Zenzmaier, C.; Sarg, B.; Gastl, G.; Gunsilius, E.; Steurer, M. GRP-78 secreted by tumor cells blocks the antiangiogenic activity of bortezomib. Blood 2009, 114, 3960–3967. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.R.; Eckhardt, B.L.; Cao, Y.; Pasqualini, R.; Argani, P.; Arap, W.; Ramsay, R.G.; Anderson, R.L. Inhibition of Established Micrometastases by Targeted Drug Delivery via Cell Surface-Associated GRP78. Clin. Cancer Res. 2013, 19, 2107–2116. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.; Ni, M.; Li, J.; Xiong, S.; Ye, W.; Virrey, J.J.; Mao, C.; Ye, R.; Wang, M.; Pen, L. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008, 68, 498–505. [Google Scholar] [CrossRef]
- Fu, Y.; Li, J.; Lee, A.S. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation–induced apoptosis. Cancer Res. 2007, 67, 3734–3740. [Google Scholar] [CrossRef]
- Lee, H.K.; Xiang, C.; Cazacu, S.; Finniss, S.; Kazimirsky, G.; Lemke, N.; Lehman, N.L.; Rempel, S.A.; Mikkelsen, T.; Brodie, C. GRP78 is overexpressed in glioblastomas and regulates glioma cell growth and apoptosis. Neuro Oncol. 2008, 10, 236–243. [Google Scholar] [CrossRef]
- Lee, E.; Nichols, P.; Spicer, D.; Groshen, S.; Mimi, C.Y.; Lee, A.S. GRP78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006, 66, 7849–7853. [Google Scholar] [CrossRef] [PubMed]
- Daneshmand, S.; Quek, M.L.; Lin, E.; Lee, C.; Cote, R.J.; Hawes, D.; Cai, J.; Groshen, S.; Lieskovsky, G.; Skinner, D.G. Glucose-regulated protein GRP78 is up-regulated in prostate cancer and correlates with recurrence and survival. Hum. Pathol. 2007, 38, 1547–1552. [Google Scholar] [CrossRef]
- Zheng, H.-C.; Takahashi, H.; Li, X.-H.; Hara, T.; Masuda, S.; Guan, Y.-F.; Takano, Y. Overexpression of GRP78 and GRP94 are markers for aggressive behavior and poor prognosis in gastric carcinomas. Hum. Pathol. 2008, 39, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Pootrakul, L.; Datar, R.H.; Shi, S.-R.; Cai, J.; Hawes, D.; Groshen, S.G.; Lee, A.S.; Cote, R.J. Expression of stress response protein Grp78 is associated with the development of castration-resistant prostate cancer. Clin. Cancer Res. 2006, 12, 5987–5993. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.; Bultynck, G.; Rizzuto, R.; Decuypere, J.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1α. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef]
- Liu, C.Y.; Schröder, M.; Kaufman, R.J. Ligand-independent dimerization activates the stress response kinases IRE1 and PERK in the lumen of the endoplasmic reticulum. J. Biol. Chem. 2000, 275, 24881–24885. [Google Scholar] [CrossRef]
- Scheuner, D.; Song, B.; McEwen, E.; Liu, C.; Laybutt, R.; Gillespie, P.; Saunders, T.; Bonner-Weir, S.; Kaufman, R.J. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell 2001, 7, 1165–1176. [Google Scholar] [CrossRef]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Yaman, I.; Fernandez, J.; Liu, H.; Caprara, M.; Komar, A.A.; Koromilas, A.E.; Zhou, L.; Snider, M.D.; Scheuner, D.; Kaufman, R.J. The zipper model of translational control: A small upstream ORF is the switch that controls structural remodeling of an mRNA leader. Cell 2003, 113, 519–531. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Akha, A.A.S.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; LaVail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Patel, R.; Wang, F.; Lee, K.; Kumar, K.; Wu, J.; Nilsson, A.; Karin, M.; Kaufman, R.J. dsRNA-dependent protein kinase phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2 mediates apoptosis. J. Biol. Chem. 2006, 281, 21458–21468. [Google Scholar] [CrossRef] [PubMed]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef] [PubMed]
- Ranganathan, A.C.; Zhang, L.; Adam, A.P.; Aguirre-Ghiso, J.A. Functional coupling of p38-induced up-regulation of BiP and activation of RNA-dependent protein kinase–like endoplasmic reticulum kinase to drug resistance of dormant carcinoma cells. Cancer Res. 2006, 66, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Fels, D.R.; Koumenis, C. The PERK/eIF2alpha/ATF4 module of the UPR in hypoxia resistance and tumor growth. Cancer Biol. Ther. 2006, 5, 723–728. [Google Scholar] [CrossRef]
- Gupta, S.; Deepti, A.; Deegan, S.; Lisbona, F.; Hetz, C.; Samali, A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1α-XBP1 signaling through a physical interaction. PLoS Biol. 2010, 8, e1000410. [Google Scholar] [CrossRef]
- Lee, K.; Tirasophon, W.; Shen, X.; Michalak, M.; Prywes, R.; Okada, T.; Yoshida, H.; Mori, K.; Kaufman, R.J. IRE1-mediated unconventional mRNA splicing and S2P-mediated ATF6 cleavage merge to regulate XBP1 in signaling the unfolded protein response. Genes Dev. 2002, 16, 452–466. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.-H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Niwa, M.; Koong, A.C. Targeting the IRE1alpha-XBP1 branch of the unfolded protein response in human diseases. Semin. Cancer Biol. 2015, 33, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Scriven, P.; Coulson, S.; Haines, R.; Balasubramanian, S.; Cross, S.; Wyld, L. Activation and clinical significance of the unfolded protein response in breast cancer. Br. J. Cancer 2009, 101, 1692–1698. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Wu, W.; Valkovska, N.; Jäger, C.; Hong, X.; Nitsche, U.; Friess, H.; Esposito, I.; Erkan, M.; Kleeff, J. A common genetic variation of melanoma inhibitory activity-2 labels a subtype of pancreatic adenocarcinoma with high endoplasmic reticulum stress levels. Sci. Rep. 2015, 5, 8109. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Lerner, A.G.; Walle, L.V.; Upton, J.-P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1α kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [PubMed]
- Ishiwata-Kimata, Y.; Promlek, T.; Kohno, K.; Kimata, Y. B i P-bound and nonclustered mode of I re1 evokes a weak but sustained unfolded protein response. Genes Cells 2013, 18, 288–301. [Google Scholar] [CrossRef]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Davé, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Wu, J.; Rutkowski, D.T.; Dubois, M.; Swathirajan, J.; Saunders, T.; Wang, J.; Song, B.; Yau, G.D.-Y.; Kaufman, R.J. ATF6α optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev. Cell 2007, 13, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Binet, F.; Sapieha, P. ER stress and angiogenesis. Cell Metab. 2015, 22, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Vandewynckel, Y.-P.; Laukens, D.; Bogaerts, E.; Paridaens, A.; Van den Bussche, A.; Verhelst, X.; Van Steenkiste, C.; Descamps, B.; Vanhove, C.; Libbrecht, L. Modulation of the unfolded protein response impedes tumor cell adaptation to proteotoxic stress: A PERK for hepatocellular carcinoma therapy. Hepatol. Int. 2015, 9, 93–104. [Google Scholar] [CrossRef]
- Liu, J.; Xiao, M.; Li, J.; Wang, D.; He, Y.; He, J.; Gao, F.; Mai, L.; Li, Y.; Liang, Y. Activation of UPR signaling pathway is associated with the malignant progression and poor prognosis in prostate cancer. Prostate 2017, 77, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Salazar, M.; Velasco, G.; Agostinis, P. Linking ER stress to autophagy: Potential implications for cancer therapy. Int. J. Cell Biol. 2010, 2010, 930509. [Google Scholar] [CrossRef] [PubMed]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef] [PubMed]
- Young, M.; Takahashi, Y.; Khan, O. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef]
- Deegan, S.; Saveljeva, S.; Logue, S.E.; Pakos-Zebrucka, K.; Gupta, S.; Vandenabeele, P.; Bertrand, M.J.; Samali, A. Deficiency in the mitochondrial apoptotic pathway reveals the toxic potential of autophagy under ER stress conditions. Autophagy 2014, 10, 1921–1936. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Messah, C.; Han, J.; LaVail, M.M.; Kaufman, R.J.; Lin, J.H. Translational and posttranslational regulation of XIAP by eIF2α and ATF4 promotes ER stress–induced cell death during the unfolded protein response. Mol. Biol. Cell 2014, 25, 1411–1420. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chipurupalli, S.; Kannan, E.; Tergaonkar, V.; D’Andrea, R.; Robinson, N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. Int. J. Mol. Sci. 2019, 20, 749. https://doi.org/10.3390/ijms20030749
Chipurupalli S, Kannan E, Tergaonkar V, D’Andrea R, Robinson N. Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. International Journal of Molecular Sciences. 2019; 20(3):749. https://doi.org/10.3390/ijms20030749
Chicago/Turabian StyleChipurupalli, Sandhya, Elango Kannan, Vinay Tergaonkar, Richard D’Andrea, and Nirmal Robinson. 2019. "Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer" International Journal of Molecular Sciences 20, no. 3: 749. https://doi.org/10.3390/ijms20030749
APA StyleChipurupalli, S., Kannan, E., Tergaonkar, V., D’Andrea, R., & Robinson, N. (2019). Hypoxia Induced ER Stress Response as an Adaptive Mechanism in Cancer. International Journal of Molecular Sciences, 20(3), 749. https://doi.org/10.3390/ijms20030749