The Potassium Channel Odyssey: Mechanisms of Traffic and Membrane Arrangement

 ,
, 
Abstract
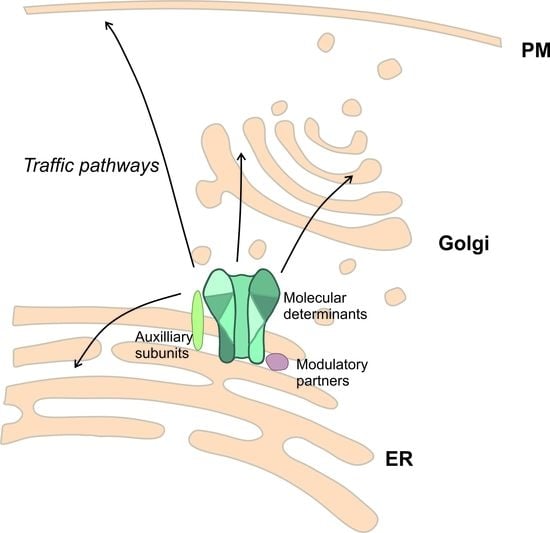
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Potassium Channel Biogenesis
3. Potassium Channel Interactions
3.1. Regulatory Subunits
3.2. Scaffolding Proteins and Other Partners
4. Subcellular Targeting of Potassium Channels
4.1. Endoplasmic Reticulum
4.2. Golgi Sorting
4.3. Plasma Membrane Arrangement
5. Post-Translational Modifications Regulating Traffic
5.1. N-Glycosylation
5.2. Phosphorylation
5.3. Redox Modifications
5.4. Lipidation
5.5. Ubiquitination
6. Mechanisms for Organelle Targeting
6.1. Lysosomes
6.2. Nucleus
6.3. Mitochondria
7. Heterotetramerization
8. Concluding Remarks
Funding
Acknowledgements
Conflicts of Interest
References
- MacKinnon, R. Potassium channels. FEBS Lett. 2003, 555, 62–65. [Google Scholar] [CrossRef]
- Gutman, G.A.; Chandy, K.G.; Adelman, J.P.; Aiyar, J.; Bayliss, D.A.; Clapham, D.E.; Covarriubias, M.; Desir, G.V.; Furuichi, K.; Ganetzky, B.; et al. International Union of Pharmacology. XLI. Compendium of voltage-gated ion channels: Potassium channels. Pharmacol. Rev. 2003, 55, 583–586. [Google Scholar] [CrossRef]
- Jeevaratnam, K.; Chadda, K.R.; Huang, C.L.; Camm, A.J. Cardiac Potassium Channels: Physiological Insights for Targeted Therapy. J. Cardiovasc. Pharmacol. Ther. 2018, 23, 119–129. [Google Scholar] [CrossRef]
- Ashcroft, F.M.; Rorsman, P. K(ATP) channels and islet hormone secretion: New insights and controversies. Nat. Rev. Endocrinol. 2013, 9, 660–669. [Google Scholar] [CrossRef]
- Comes, N.; Serrano-Albarras, A.; Capera, J.; Serrano-Novillo, C.; Condom, E.; Ramon, Y.C.S.; Ferreres, J.C.; Felipe, A. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim. Biophys. Acta 2015, 1848, 2477–2492. [Google Scholar] [CrossRef]
- Szabo, I.; Zoratti, M.; Gulbins, E. Contribution of voltage-gated potassium channels to the regulation of apoptosis. FEBS Lett. 2010, 584, 2049–2056. [Google Scholar] [CrossRef]
- Panyi, G.; Vamosi, G.; Bacso, Z.; Bagdany, M.; Bodnar, A.; Varga, Z.; Gaspar, R.; Matyus, L.; Damjanovich, S. Kv1.3 potassium channels are localized in the immunological synapse formed between cytotoxic and target cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1285–1290. [Google Scholar] [CrossRef]
- Szabo, I.; Bock, J.; Jekle, A.; Soddemann, M.; Adams, C.; Lang, F.; Zoratti, M.; Gulbins, E. A novel potassium channel in lymphocyte mitochondria. J. Biol. Chem. 2005, 280, 12790–12798. [Google Scholar] [CrossRef]
- Perez-Verdaguer, M.; Capera, J.; Ortego-Dominguez, M.; Bielanska, J.; Comes, N.; Montoro, R.J.; Camps, M.; Felipe, A. Caveolar targeting links Kv1.3 with the insulin-dependent adipocyte physiology. Cell. Mol. Life Sci. 2018, 75, 4059–4075. [Google Scholar] [CrossRef]
- Rasmussen, H.B.; Moller, M.; Knaus, H.G.; Jensen, B.S.; Olesen, S.P.; Jorgensen, N.K. Subcellular localization of the delayed rectifier K(+) channels KCNQ1 and ERG1 in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1300–H1309. [Google Scholar] [CrossRef]
- Eldstrom, J.; Van Wagoner, D.R.; Moore, E.D.; Fedida, D. Localization of Kv1.5 channels in rat and canine myocyte sarcolemma. FEBS Lett. 2006, 580, 6039–6046. [Google Scholar] [CrossRef]
- Bean, B.P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8, 451–465. [Google Scholar] [CrossRef]
- Johnston, D.; Christie, B.R.; Frick, A.; Gray, R.; Hoffman, D.A.; Schexnayder, L.K.; Watanabe, S.; Yuan, L.L. Active dendrites, potassium channels and synaptic plasticity. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2003, 358, 667–674. [Google Scholar] [CrossRef]
- Tu, L.; Wang, J.; Helm, A.; Skach, W.R.; Deutsch, C. Transmembrane biogenesis of Kv1.3. Biochemistry 2000, 39, 824–836. [Google Scholar] [CrossRef]
- Deutsch, C. Potassium channel ontogeny. Annu. Rev. Physiol. 2002, 64, 19–46. [Google Scholar] [CrossRef]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation 2006, 113, 365–373. [Google Scholar] [CrossRef]
- Furutani, M.; Trudeau, M.C.; Hagiwara, N.; Seki, A.; Gong, Q.; Zhou, Z.; Imamura, S.; Nagashima, H.; Kasanuki, H.; Takao, A.; et al. Novel mechanism associated with an inherited cardiac arrhythmia: Defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation 1999, 99, 2290–2294. [Google Scholar] [CrossRef]
- Yang, Y.; Xia, M.; Jin, Q.; Bendahhou, S.; Shi, J.; Chen, Y.; Liang, B.; Lin, J.; Liu, Y.; Liu, B.; et al. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am. J. Hum. Genet. 2004, 75, 899–905. [Google Scholar] [CrossRef]
- Humphries, E.S.; Dart, C. Neuronal and Cardiovascular Potassium Channels as Therapeutic Drug Targets: Promise and Pitfalls. J. Biomol. Screen. 2015, 20, 1055–1073. [Google Scholar] [CrossRef]
- Gouas, L.; Bellocq, C.; Berthet, M.; Potet, F.; Demolombe, S.; Forhan, A.; Lescasse, R.; Simon, F.; Balkau, B.; Denjoy, I.; et al. New KCNQ1 mutations leading to haploinsufficiency in a general population; Defective trafficking of a KvLQT1 mutant. Cardiovasc. Res. 2004, 63, 60–68. [Google Scholar] [CrossRef]
- Sivaprasadarao, A.; Taneja, T.K.; Mankouri, J.; Smith, A.J. Trafficking of ATP-sensitive potassium channels in health and disease. Biochem. Soc. Transact. 2007, 35, 1055–1059. [Google Scholar] [CrossRef]
- Vaidyanathan, R.; Vega, A.L.; Song, C.; Zhou, Q.; Tan, B.H.; Berger, S.; Makielski, J.C.; Eckhardt, L.L. The interaction of caveolin 3 protein with the potassium inward rectifier channel Kir2.1: Physiology and pathology related to long qt syndrome 9 (LQT9). J. Biol. Chem. 2013, 288, 17472–17480. [Google Scholar] [CrossRef]
- Martens, J.R.; Kwak, Y.G.; Tamkun, M.M. Modulation of Kv channel alpha/beta subunit interactions. Trends Cardiovasc. Med. 1999, 9, 253–258. [Google Scholar] [CrossRef]
- Shi, G.; Nakahira, K.; Hammond, S.; Rhodes, K.J.; Schechter, L.E.; Trimmer, J.S. Beta subunits promote K+ channel surface expression through effects early in biosynthesis. Neuron 1996, 16, 843–852. [Google Scholar] [CrossRef]
- McCormack, K.; Connor, J.X.; Zhou, L.; Ho, L.L.; Ganetzky, B.; Chiu, S.Y.; Messing, A. Genetic analysis of the mammalian K+ channel beta subunit Kvbeta 2 (Kcnab2). J. Biol. Chem. 2002, 277, 13219–13228. [Google Scholar] [CrossRef]
- Roura-Ferrer, M.; Sole, L.; Oliveras, A.; Dahan, R.; Bielanska, J.; Villarroel, A.; Comes, N.; Felipe, A. Impact of KCNE subunits on KCNQ1 (Kv7.1) channel membrane surface targeting. J. Cell. Physiol. 2010, 225, 692–700. [Google Scholar] [CrossRef]
- Sole, L.; Roura-Ferrer, M.; Perez-Verdaguer, M.; Oliveras, A.; Calvo, M.; Fernandez-Fernandez, J.M.; Felipe, A. KCNE4 suppresses Kv1.3 currents by modulating trafficking, surface expression and channel gating. J. Cell Sci. 2009, 122, 3738–3748. [Google Scholar] [CrossRef]
- Flowerdew, S.E.; Burgoyne, R.D. A VAMP7/Vti1a SNARE complex distinguishes a non-conventional traffic route to the cell surface used by KChIP1 and Kv4 potassium channels. Biochem. J. 2009, 418, 529–540. [Google Scholar] [CrossRef]
- Shibata, R.; Misonou, H.; Campomanes, C.R.; Anderson, A.E.; Schrader, L.A.; Doliveira, L.C.; Carroll, K.I.; Sweatt, J.D.; Rhodes, K.J.; Trimmer, J.S. A fundamental role for KChIPs in determining the molecular properties and trafficking of Kv4.2 potassium channels. J. Biol. Chem. 2003, 278, 36445–36454. [Google Scholar] [CrossRef]
- Hasdemir, B.; Fitzgerald, D.J.; Prior, I.A.; Tepikin, A.V.; Burgoyne, R.D. Traffic of Kv4 K+ channels mediated by KChIP1 is via a novel post-ER vesicular pathway. J. Cell Biol. 2005, 171, 459–469. [Google Scholar] [CrossRef]
- Kuryshev, Y.A.; Gudz, T.I.; Brown, A.M.; Wible, B.A. KChAP as a chaperone for specific K(+) channels. Am. J. Physiol. Cell Physiol. 2000, 278, C931–C941. [Google Scholar] [CrossRef]
- Kuryshev, Y.A.; Wible, B.A.; Gudz, T.I.; Ramirez, A.N.; Brown, A.M. KChAP/Kvbeta1.2 interactions and their effects on cardiac Kv channel expression. Am. J. Physiol. Cell Physiol. 2001, 281, C290–C299. [Google Scholar] [CrossRef]
- Martinez-Marmol, R.; Styrczewska, K.; Perez-Verdaguer, M.; Vallejo-Gracia, A.; Comes, N.; Sorkin, A.; Felipe, A. Ubiquitination mediates Kv1.3 endocytosis as a mechanism for protein kinase C-dependent modulation. Sci. Rep. 2017, 7, 42395. [Google Scholar] [CrossRef]
- Folco, E.J.; Liu, G.X.; Koren, G. Caveolin-3 and SAP97 form a scaffolding protein complex that regulates the voltage-gated potassium channel Kv1.5. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H681–H690. [Google Scholar] [CrossRef]
- Davies, L.M.; Purves, G.I.; Barrett-Jolley, R.; Dart, C. Interaction with caveolin-1 modulates vascular ATP-sensitive potassium (KATP) channel activity. J. Physiol. 2010, 588, 3255–3266. [Google Scholar] [CrossRef]
- Couet, J.; Li, S.; Okamoto, T.; Ikezu, T.; Lisanti, M.P. Identification of peptide and protein ligands for the caveolin-scaffolding domain. Implications for the interaction of caveolin with caveolae-associated proteins. J. Biol. Chem. 1997, 272, 6525–6533. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Conti, L.R.; Radeke, C.M.; McGuire, L.M.; Vandenberg, C.A. A multiprotein trafficking complex composed of SAP97, CASK, Veli, and Mint1 is associated with inward rectifier Kir2 potassium channels. J. Biol. Chem. 2004, 279, 19051–19063. [Google Scholar] [CrossRef]
- Pan, C.Q.; Sudol, M.; Sheetz, M.; Low, B.C. Modularity and functional plasticity of scaffold proteins as p(l)acemakers in cell signaling. Cell. Signal. 2012, 24, 2143–2165. [Google Scholar] [CrossRef]
- Steele, D.F.; Eldstrom, J.; Fedida, D. Mechanisms of cardiac potassium channel trafficking. J. Physiol. 2007, 582, 17–26. [Google Scholar] [CrossRef]
- Ma, D.; Zerangue, N.; Lin, Y.F.; Collins, A.; Yu, M.; Jan, Y.N.; Jan, L.Y. Role of ER export signals in controlling surface potassium channel numbers. Science 2001, 291, 316–319. [Google Scholar] [CrossRef]
- Zuzarte, M.; Rinne, S.; Schlichthorl, G.; Schubert, A.; Daut, J.; Preisig-Muller, R. A di-acidic sequence motif enhances the surface expression of the potassium channel TASK-3. Traffic 2007, 8, 1093–1100. [Google Scholar] [CrossRef]
- Martinez-Marmol, R.; Perez-Verdaguer, M.; Roig, S.R.; Vallejo-Gracia, A.; Gotsi, P.; Serrano-Albarras, A.; Bahamonde, M.I.; Ferrer-Montiel, A.; Fernandez-Ballester, G.; Comes, N.; et al. A non-canonical di-acidic signal at the C-terminus of Kv1.3 determines anterograde trafficking and surface expression. J. Cell Sci. 2013, 126, 5681–5691. [Google Scholar] [CrossRef]
- Spear, J.M.; Koborssy, D.A.; Schwartz, A.B.; Johnson, A.J.; Audhya, A.; Fadool, D.A.; Stagg, S.M. Kv1.3 contains an alternative C-terminal ER exit motif and is recruited into COPII vesicles by Sec24a. BMC Biochem. 2015, 16, 16. [Google Scholar] [CrossRef]
- Chen, L.; Jeffries, O.; Rowe, I.C.; Liang, Z.; Knaus, H.G.; Ruth, P.; Shipston, M.J. Membrane trafficking of large conductance calcium-activated potassium channels is regulated by alternative splicing of a transplantable, acidic trafficking motif in the RCK1-RCK2 linker. J. Biol. Chem. 2010, 285, 23265–23275. [Google Scholar] [CrossRef]
- Levitan, E.S.; Takimoto, K. Surface expression of Kv1 voltage-gated K+ channels is governed by a C-terminal motif. Trends Cardiovasc. Med. 2000, 10, 317–320. [Google Scholar] [CrossRef]
- Akhavan, A.; Atanasiu, R.; Noguchi, T.; Han, W.; Holder, N.; Shrier, A. Identification of the cyclic-nucleotide-binding domain as a conserved determinant of ion-channel cell-surface localization. J. Cell Sci. 2005, 118, 2803–2812. [Google Scholar] [CrossRef]
- Michelsen, K.; Schmid, V.; Metz, J.; Heusser, K.; Liebel, U.; Schwede, T.; Spang, A.; Schwappach, B. Novel cargo-binding site in the beta and delta subunits of coatomer. J. Cell Biol. 2007, 179, 209–217. [Google Scholar] [CrossRef]
- Zerangue, N.; Schwappach, B.; Jan, Y.N.; Jan, L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 1999, 22, 537–548. [Google Scholar] [CrossRef]
- Kupershmidt, S.; Yang, T.; Chanthaphaychith, S.; Wang, Z.; Towbin, J.A.; Roden, D.M. Defective human Ether-a-go-go-related gene trafficking linked to an endoplasmic reticulum retention signal in the C terminus. J. Biol. Chem. 2002, 277, 27442–27448. [Google Scholar] [CrossRef]
- Zuzarte, M.; Heusser, K.; Renigunta, V.; Schlichthorl, G.; Rinne, S.; Wischmeyer, E.; Daut, J.; Schwappach, B.; Preisig-Muller, R. Intracellular traffic of the K+ channels TASK-1 and TASK-3: Role of N- and C-terminal sorting signals and interaction with 14-3-3 proteins. J. Physiol. 2009, 587, 929–952. [Google Scholar] [CrossRef]
- Manganas, L.N.; Wang, Q.; Scannevin, R.H.; Antonucci, D.E.; Rhodes, K.J.; Trimmer, J.S. Identification of a trafficking determinant localized to the Kv1 potassium channel pore. Proc. Natl. Acad. Sci. USA 2001, 98, 14055–14059. [Google Scholar] [CrossRef]
- Gomez-Posada, J.C.; Etxeberria, A.; Roura-Ferrer, M.; Areso, P.; Masin, M.; Murrell-Lagnado, R.D.; Villarroel, A. A pore residue of the KCNQ3 potassium M-channel subunit controls surface expression. J. Neurosci. 2010, 30, 9316–9323. [Google Scholar] [CrossRef]
- Zarei, M.M.; Eghbali, M.; Alioua, A.; Song, M.; Knaus, H.G.; Stefani, E.; Toro, L. An endoplasmic reticulum trafficking signal prevents surface expression of a voltage- and Ca2+-activated K+ channel splice variant. Proc. Natl. Acad. Sci. USA 2004, 101, 10072–10077. [Google Scholar] [CrossRef]
- Sole, L.; Roig, S.R.; Vallejo-Gracia, A.; Serrano-Albarras, A.; Martinez-Marmol, R.; Tamkun, M.M.; Felipe, A. The C-terminal domain of Kv1.3 regulates functional interactions with the KCNE4 subunit. J. Cell Sci. 2016, 129, 4265–4277. [Google Scholar] [CrossRef]
- Li, H.; Guo, W.; Mellor, R.L.; Nerbonne, J.M. KChIP2 modulates the cell surface expression of Kv 1.5-encoded K(+) channels. J. Mol. Cell. Cardiol. 2005, 39, 121–132. [Google Scholar] [CrossRef]
- Etxeberria, A.; Aivar, P.; Rodriguez-Alfaro, J.A.; Alaimo, A.; Villace, P.; Gomez-Posada, J.C.; Areso, P.; Villarroel, A. Calmodulin regulates the trafficking of KCNQ2 potassium channels. FASEB J. 2008, 22, 1135–1143. [Google Scholar] [CrossRef]
- Alaimo, A.; Gomez-Posada, J.C.; Aivar, P.; Etxeberria, A.; Rodriguez-Alfaro, J.A.; Areso, P.; Villarroel, A. Calmodulin activation limits the rate of KCNQ2 K+ channel exit from the endoplasmic reticulum. J. Biol. Chem. 2009, 284, 20668–20675. [Google Scholar] [CrossRef]
- Liu, W.; Devaux, J.J. Calmodulin orchestrates the heteromeric assembly and the trafficking of KCNQ2/3 (Kv7.2/3) channels in neurons. Mol. Cell. Neurosci. 2014, 58, 40–52. [Google Scholar] [CrossRef]
- Cavaretta, J.P.; Sherer, K.R.; Lee, K.Y.; Kim, E.H.; Issema, R.S.; Chung, H.J. Polarized axonal surface expression of neuronal KCNQ potassium channels is regulated by calmodulin interaction with KCNQ2 subunit. PLoS ONE 2014, 9, e103655. [Google Scholar] [CrossRef]
- Joiner, W.J.; Khanna, R.; Schlichter, L.C.; Kaczmarek, L.K. Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J. Biol. Chem. 2001, 276, 37980–37985. [Google Scholar] [CrossRef]
- Balut, C.M.; Hamilton, K.L.; Devor, D.C. Trafficking of intermediate (KCa3.1) and small (KCa2.x) conductance, Ca(2+)-activated K(+) channels: A novel target for medicinal chemistry efforts? ChemMedChem 2012, 7, 1741–1755. [Google Scholar] [CrossRef]
- Hund, T.J.; Mohler, P.J. Differential roles for SUR subunits in KATP channel membrane targeting and regulation. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H33–H35. [Google Scholar] [CrossRef]
- Li, X.; Ortega, B.; Kim, B.; Welling, P.A. A Common Signal Patch Drives AP-1 Protein-dependent Golgi Export of Inwardly Rectifying Potassium Channels. J. Biol. Chem. 2016, 291, 14963–14972. [Google Scholar] [CrossRef]
- Jensen, C.S.; Watanabe, S.; Rasmussen, H.B.; Schmitt, N.; Olesen, S.P.; Frost, N.A.; Blanpied, T.A.; Misonou, H. Specific sorting and post-Golgi trafficking of dendritic potassium channels in living neurons. J. Biol. Chem. 2014, 289, 10566–10581. [Google Scholar] [CrossRef]
- Lim, S.T.; Antonucci, D.E.; Scannevin, R.H.; Trimmer, J.S. A novel targeting signal for proximal clustering of the Kv2.1 K+ channel in hippocampal neurons. Neuron 2000, 25, 385–397. [Google Scholar] [CrossRef]
- Rivera, J.F.; Ahmad, S.; Quick, M.W.; Liman, E.R.; Arnold, D.B. An evolutionarily conserved dileucine motif in Shal K+ channels mediates dendritic targeting. Nat. Neurosci. 2003, 6, 243–250. [Google Scholar] [CrossRef]
- Perez-Verdaguer, M.; Capera, J.; Martinez-Marmol, R.; Camps, M.; Comes, N.; Tamkun, M.M.; Felipe, A. Caveolin interaction governs Kv1.3 lipid raft targeting. Sci. Rep. 2016, 6, 22453. [Google Scholar] [CrossRef]
- Bundis, F.; Neagoe, I.; Schwappach, B.; Steinmeyer, K. Involvement of Golgin-160 in cell surface transport of renal ROMK channel: Co-expression of Golgin-160 increases ROMK currents. Cell. Physiol. Biochem. 2006, 17, 1–12. [Google Scholar] [CrossRef]
- Taneja, T.K.; Ma, D.; Kim, B.Y.; Welling, P.A. Golgin-97 Targets Ectopically Expressed Inward Rectifying Potassium Channel, Kir2.1, to the trans-Golgi Network in COS-7 Cells. Front. Physiol. 2018, 9, 1070. [Google Scholar] [CrossRef]
- Stockklausner, C.; Klocker, N. Surface expression of inward rectifier potassium channels is controlled by selective Golgi export. J. Biol. Chem. 2003, 278, 17000–17005. [Google Scholar] [CrossRef]
- Pan, Z.; Kao, T.; Horvath, Z.; Lemos, J.; Sul, J.Y.; Cranstoun, S.D.; Bennett, V.; Scherer, S.S.; Cooper, E.C. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J. Neurosci. 2006, 26, 2599–2613. [Google Scholar] [CrossRef]
- Rasmussen, H.B.; Frokjaer-Jensen, C.; Jensen, C.S.; Jensen, H.S.; Jorgensen, N.K.; Misonou, H.; Trimmer, J.S.; Olesen, S.P.; Schmitt, N. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J. Cell Sci. 2007, 120, 953–963. [Google Scholar] [CrossRef]
- Hajdu, P.; Martin, G.V.; Chimote, A.A.; Szilagyi, O.; Takimoto, K.; Conforti, L. The C-terminus SH3-binding domain of Kv1.3 is required for the actin-mediated immobilization of the channel via cortactin. Mol. Biol. Cell 2015, 26, 1640–1651. [Google Scholar] [CrossRef]
- Burke, N.A.; Takimoto, K.; Li, D.; Han, W.; Watkins, S.C.; Levitan, E.S. Distinct structural requirements for clustering and immobilization of K+ channels by PSD-95. J. Gener. Physiol. 1999, 113, 71–80. [Google Scholar] [CrossRef]
- Martens, J.R.; O’Connell, K.; Tamkun, M. Targeting of ion channels to membrane microdomains: Localization of KV channels to lipid rafts. Trends Pharmacol. Sci. 2004, 25, 16–21. [Google Scholar] [CrossRef]
- Romanenko, V.G.; Fang, Y.; Byfield, F.; Travis, A.J.; Vandenberg, C.A.; Rothblat, G.H.; Levitan, I. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys. J. 2004, 87, 3850–3861. [Google Scholar] [CrossRef]
- Campomanes, C.R.; Carroll, K.I.; Manganas, L.N.; Hershberger, M.E.; Gong, B.; Antonucci, D.E.; Rhodes, K.J.; Trimmer, J.S. Kv beta subunit oxidoreductase activity and Kv1 potassium channel trafficking. J. Biol. Chem. 2002, 277, 8298–8305. [Google Scholar] [CrossRef]
- Nystoriak, M.A.; Zhang, D.; Jagatheesan, G.; Bhatnagar, A. Heteromeric complexes of aldo-keto reductase auxiliary KVbeta subunits (AKR6A) regulate sarcolemmal localization of KV1.5 in coronary arterial myocytes. Chem. Biol. Interact. 2017, 276, 210–217. [Google Scholar] [CrossRef]
- Lefoulon, C.; Waghmare, S.; Karnik, R.; Blatt, M.R. Gating control and K(+) uptake by the KAT1 K(+) channel leaveraged through membrane anchoring of the trafficking protein SYP121. Plant Cell Environ. 2018, 41, 2668–2677. [Google Scholar] [CrossRef]
- Kilisch, M.; Lytovchenko, O.; Schwappach, B.; Renigunta, V.; Daut, J. The role of protein-protein interactions in the intracellular traffic of the potassium channels TASK-1 and TASK-3. Pflugers Archiv 2015, 467, 1105–1120. [Google Scholar] [CrossRef]
- Michaelevski, I.; Chikvashvili, D.; Tsuk, S.; Singer-Lahat, D.; Kang, Y.; Linial, M.; Gaisano, H.Y.; Fili, O.; Lotan, I. Direct interaction of target SNAREs with the Kv2.1 channel. Modal regulation of channel activation and inactivation gating. J. Biol. Chem. 2003, 278, 34320–34330. [Google Scholar] [CrossRef]
- Nehring, R.B.; Wischmeyer, E.; Doring, F.; Veh, R.W.; Sheng, M.; Karschin, A. Neuronal inwardly rectifying K(+) channels differentially couple to PDZ proteins of the PSD-95/SAP90 family. J. Neurosci. 2000, 20, 156–162. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Mailliard, W.; Wingerd, K.; Clegg, D.; Vandenberg, C. Inward rectifier potassium channel Kir2.2 is associated with synapse-associated protein SAP97. J. Cell Sci. 2001, 114, 987–998. [Google Scholar]
- Leonoudakis, D.; Conti, L.R.; Anderson, S.; Radeke, C.M.; McGuire, L.M.; Adams, M.E.; Froehner, S.C.; Yates, J.R., 3rd; Vandenberg, C.A. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J. Biol. Chem. 2004, 279, 22331–22346. [Google Scholar] [CrossRef]
- Saheki, Y.; De Camilli, P. Endoplasmic Reticulum-Plasma Membrane Contact Sites. Annu. Rev. Biochem. 2017, 86, 659–684. [Google Scholar] [CrossRef]
- Rabouille, C. Pathways of Unconventional Protein Secretion. Trends Cell Biol. 2017, 27, 230–240. [Google Scholar] [CrossRef]
- Yoo, J.S.; Moyer, B.D.; Bannykh, S.; Yoo, H.M.; Riordan, J.R.; Balch, W.E. Non-conventional trafficking of the cystic fibrosis transmembrane conductance regulator through the early secretory pathway. J. Biol. Chem. 2002, 277, 11401–11409. [Google Scholar] [CrossRef]
- Fox, P.D.; Haberkorn, C.J.; Akin, E.J.; Seel, P.J.; Krapf, D.; Tamkun, M.M. Induction of stable ER-plasma-membrane junctions by Kv2.1 potassium channels. J. Cell Sci. 2015, 128, 2096–2105. [Google Scholar] [CrossRef]
- Deutsch, E.; Weigel, A.V.; Akin, E.J.; Fox, P.; Hansen, G.; Haberkorn, C.J.; Loftus, R.; Krapf, D.; Tamkun, M.M. Kv2.1 cell surface clusters are insertion platforms for ion channel delivery to the plasma membrane. Mol. Biol. Cell 2012, 23, 2917–2929. [Google Scholar] [CrossRef]
- Takimoto, K.; Yang, E.K.; Conforti, L. Palmitoylation of KChIP splicing variants is required for efficient cell surface expression of Kv4.3 channels. J. Biol. Chem. 2002, 277, 26904–26911. [Google Scholar] [CrossRef]
- Wong, W.; Schlichter, L.C. Differential recruitment of Kv1.4 and Kv4.2 to lipid rafts by PSD-95. J. Biol. Chem. 2004, 279, 444–452. [Google Scholar] [CrossRef]
- Bao, L.; Hadjiolova, K.; Coetzee, W.A.; Rindler, M.J. Endosomal KATP channels as a reservoir after myocardial ischemia: A role for SUR2 subunits. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H262–H270. [Google Scholar] [CrossRef]
- Gao, Y.; Balut, C.M.; Bailey, M.A.; Patino-Lopez, G.; Shaw, S.; Devor, D.C. Recycling of the Ca2+-activated K+ channel, KCa2.3, is dependent upon RME-1, Rab35/EPI64C, and an N-terminal domain. J. Biol. Chem. 2010, 285, 17938–17953. [Google Scholar] [CrossRef]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci. Rep. 2015, 5, 8926. [Google Scholar] [CrossRef]
- Watanabe, I.; Zhu, J.; Recio-Pinto, E.; Thornhill, W.B. Glycosylation affects the protein stability and cell surface expression of Kv1.4 but Not Kv1.1 potassium channels. A pore region determinant dictates the effect of glycosylation on trafficking. J. Biol. Chem. 2004, 279, 8879–8885. [Google Scholar] [CrossRef]
- Thayer, D.A.; Yang, S.B.; Jan, Y.N.; Jan, L.Y. N-linked glycosylation of Kv1.2 voltage-gated potassium channel facilitates cell surface expression and enhances the stability of internalized channels. J. Physiol. 2016, 594, 6701–6713. [Google Scholar] [CrossRef]
- Zhu, J.; Yan, J.; Thornhill, W.B. N-glycosylation promotes the cell surface expression of Kv1.3 potassium channels. FEBS J. 2012, 279, 2632–2644. [Google Scholar] [CrossRef]
- Noma, K.; Kimura, K.; Minatohara, K.; Nakashima, H.; Nagao, Y.; Mizoguchi, A.; Fujiyoshi, Y. Triple N-glycosylation in the long S5-P loop regulates the activation and trafficking of the Kv12.2 potassium channel. J. Biol. Chem. 2009, 284, 33139–33150. [Google Scholar] [CrossRef]
- Napp, J.; Monje, F.; Stuhmer, W.; Pardo, L.A. Glycosylation of Eag1 (Kv10.1) potassium channels: Intracellular trafficking and functional consequences. J. Biol. Chem. 2005, 280, 29506–29512. [Google Scholar] [CrossRef]
- Mant, A.; Williams, S.; Roncoroni, L.; Lowry, E.; Johnson, D.; O’Kelly, I. N-glycosylation-dependent control of functional expression of background potassium channels K2P3.1 and K2P9.1. J. Biol. Chem. 2013, 288, 3251–3264. [Google Scholar] [CrossRef]
- Petrecca, K.; Atanasiu, R.; Akhavan, A.; Shrier, A. N-linked glycosylation sites determine HERG channel surface membrane expression. J. Physiol. 1999, 515, 41–48. [Google Scholar] [CrossRef]
- Gong, Q.; Anderson, C.L.; January, C.T.; Zhou, Z. Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H77–H84. [Google Scholar] [CrossRef]
- Brooks, N.L.; Corey, M.J.; Schwalbe, R.A. Characterization of N-glycosylation consensus sequences in the Kv3.1 channel. FEBS J. 2006, 273, 3287–3300. [Google Scholar] [CrossRef]
- Hall, M.K.; Weidner, D.A.; Bernetski, C.J.; Schwalbe, R.A. N-Linked glycan site occupancy impacts the distribution of a potassium channel in the cell body and outgrowths of neuronal-derived cells. Biochim. Biophys. Acta 2014, 1840, 595–604. [Google Scholar] [CrossRef]
- Hall, M.K.; Weidner, D.A.; Chen, J.; Bernetski, C.J.; Schwalbe, R.A. Glycan structures contain information for the spatial arrangement of glycoproteins in the plasma membrane. PLoS ONE 2013, 8, e75013. [Google Scholar] [CrossRef]
- Bas, T.; Gao, G.Y.; Lvov, A.; Chandrasekhar, K.D.; Gilmore, R.; Kobertz, W.R. Post-translational N-glycosylation of type I transmembrane KCNE1 peptides: Implications for membrane protein biogenesis and disease. J. Biol. Chem. 2011, 286, 28150–28159. [Google Scholar] [CrossRef]
- Yoo, D.; Fang, L.; Mason, A.; Kim, B.Y.; Welling, P.A. A phosphorylation-dependent export structure in ROMK (Kir 1.1) channel overrides an endoplasmic reticulum localization signal. J. Biol. Chem. 2005, 280, 35281–35289. [Google Scholar] [CrossRef]
- Martinez-Marmol, R.; Comes, N.; Styrczewska, K.; Perez-Verdaguer, M.; Vicente, R.; Pujadas, L.; Soriano, E.; Sorkin, A.; Felipe, A. Unconventional EGF-induced ERK1/2-mediated Kv1.3 endocytosis. Cell. Mol. Life Sci. 2016, 73, 1515–1528. [Google Scholar] [CrossRef]
- Hammond, R.S.; Lin, L.; Sidorov, M.S.; Wikenheiser, A.M.; Hoffman, D.A. Protein kinase a mediates activity-dependent Kv4.2 channel trafficking. J. Neurosci. 2008, 28, 7513–7519. [Google Scholar] [CrossRef]
- Lin, L.; Sun, W.; Wikenheiser, A.M.; Kung, F.; Hoffman, D.A. KChIP4a regulates Kv4.2 channel trafficking through PKA phosphorylation. Mol. Cell. Neurosci. 2010, 43, 315–325. [Google Scholar] [CrossRef]
- Yang, J.W.; Vacher, H.; Park, K.S.; Clark, E.; Trimmer, J.S. Trafficking-dependent phosphorylation of Kv1.2 regulates voltage-gated potassium channel cell surface expression. Proc. Natl. Acad. Sci. USA 2007, 104, 20055–20060. [Google Scholar] [CrossRef]
- Nesti, E.; Everill, B.; Morielli, A.D. Endocytosis as a mechanism for tyrosine kinase-dependent suppression of a voltage-gated potassium channel. Mol. Biol. Cell 2004, 15, 4073–4088. [Google Scholar] [CrossRef]
- Misonou, H.; Mohapatra, D.P.; Park, E.W.; Leung, V.; Zhen, D.; Misonou, K.; Anderson, A.E.; Trimmer, J.S. Regulation of ion channel localization and phosphorylation by neuronal activity. Nat. Neurosci. 2004, 7, 711–718. [Google Scholar] [CrossRef]
- Fernandez-Orth, J.; Ehling, P.; Ruck, T.; Pankratz, S.; Hofmann, M.S.; Landgraf, P.; Dieterich, D.C.; Smalla, K.H.; Kahne, T.; Seebohm, G.; et al. 14-3-3 Proteins regulate K2P 5.1 surface expression on T lymphocytes. Traffic 2017, 18, 29–43. [Google Scholar] [CrossRef]
- Bogeski, I.; Niemeyer, B.A. Redox regulation of ion channels. Antioxid. Redox Signal. 2014, 21, 859–862. [Google Scholar] [CrossRef]
- Svoboda, L.K.; Reddie, K.G.; Zhang, L.; Vesely, E.D.; Williams, E.S.; Schumacher, S.M.; O’Connell, R.P.; Shaw, R.; Day, S.M.; Anumonwo, J.M.; et al. Redox-sensitive sulfenic acid modification regulates surface expression of the cardiovascular voltage-gated potassium channel Kv1.5. Circ. Res. 2012, 111, 842–853. [Google Scholar] [CrossRef]
- Chimote, A.A.; Kuras, Z.; Conforti, L. Disruption of kv1.3 channel forward vesicular trafficking by hypoxia in human T lymphocytes. J. Biol. Chem. 2012, 287, 2055–2067. [Google Scholar] [CrossRef]
- Jindal, H.K.; Folco, E.J.; Liu, G.X.; Koren, G. Posttranslational modification of voltage-dependent potassium channel Kv1.5: COOH-terminal palmitoylation modulates its biological properties. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2012–H2021. [Google Scholar] [CrossRef]
- Jeffries, O.; Geiger, N.; Rowe, I.C.; Tian, L.; McClafferty, H.; Chen, L.; Bi, D.; Knaus, H.G.; Ruth, P.; Shipston, M.J. Palmitoylation of the S0-S1 linker regulates cell surface expression of voltage- and calcium-activated potassium (BK) channels. J. Biol. Chem. 2010, 285, 33307–33314. [Google Scholar] [CrossRef]
- Tian, L.; McClafferty, H.; Knaus, H.G.; Ruth, P.; Shipston, M.J. Distinct acyl protein transferases and thioesterases control surface expression of calcium-activated potassium channels. J. Biol. Chem. 2012, 287, 14718–14725. [Google Scholar] [CrossRef]
- Alioua, A.; Li, M.; Wu, Y.; Stefani, E.; Toro, L. Unconventional myristoylation of large-conductance Ca(2)(+)-activated K(+) channel (Slo1) via serine/threonine residues regulates channel surface expression. Proc. Natl. Acad. Sci. USA 2011, 108, 10744–10749. [Google Scholar] [CrossRef]
- Lin, D.H.; Sterling, H.; Wang, Z.; Babilonia, E.; Yang, B.; Dong, K.; Hebert, S.C.; Giebisch, G.; Wang, W.H. ROMK1 channel activity is regulated by monoubiquitination. Proc. Natl. Acad. Sci. USA 2005, 102, 4306–4311. [Google Scholar] [CrossRef]
- Albesa, M.; Grilo, L.S.; Gavillet, B.; Abriel, H. Nedd4-2-dependent ubiquitylation and regulation of the cardiac potassium channel hERG1. J. Mol. Cell. Cardiol. 2011, 51, 90–98. [Google Scholar] [CrossRef]
- Jespersen, T.; Membrez, M.; Nicolas, C.S.; Pitard, B.; Staub, O.; Olesen, S.P.; Baro, I.; Abriel, H. The KCNQ1 potassium channel is down-regulated by ubiquitylating enzymes of the Nedd4/Nedd4-like family. Cardiovasc. Res. 2007, 74, 64–74. [Google Scholar] [CrossRef]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. BK Channels Alleviate Lysosomal Storage Diseases by Providing Positive Feedback Regulation of Lysosomal Ca2+ Release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef]
- Cang, C.; Aranda, K.; Seo, Y.J.; Gasnier, B.; Ren, D. TMEM175 Is an Organelle K(+) Channel Regulating Lysosomal Function. Cell 2015, 162, 1101–1112. [Google Scholar] [CrossRef]
- Quesada, I.; Rovira, J.M.; Martin, F.; Roche, E.; Nadal, A.; Soria, B. Nuclear KATP channels trigger nuclear Ca(2+) transients that modulate nuclear function. Proc. Natl. Acad. Sci. USA 2002, 99, 9544–9549. [Google Scholar] [CrossRef]
- Stonehouse, A.H.; Grubb, B.D.; Pringle, J.H.; Norman, R.I.; Stanfield, P.R.; Brammar, W.J. Nuclear immunostaining in rat neuronal cells using two anti-Kir2.2 ion channel polyclonal antibodies. J. Mol. Neurosci. 2003, 20, 189–194. [Google Scholar] [CrossRef]
- Chachi, L.; Shikotra, A.; Duffy, S.M.; Tliba, O.; Brightling, C.; Bradding, P.; Amrani, Y. Functional KCa3.1 channels regulate steroid insensitivity in bronchial smooth muscle cells. J. Immunol. 2013, 191, 2624–2636. [Google Scholar] [CrossRef]
- Li, B.; Jie, W.; Huang, L.; Wei, P.; Li, S.; Luo, Z.; Friedman, A.K.; Meredith, A.L.; Han, M.H.; Zhu, X.H.; et al. Nuclear BK channels regulate gene expression via the control of nuclear calcium signaling. Nat. Neurosci. 2014, 17, 1055–1063. [Google Scholar] [CrossRef]
- Jang, S.H.; Byun, J.K.; Jeon, W.I.; Choi, S.Y.; Park, J.; Lee, B.H.; Yang, J.E.; Park, J.B.; O’Grady, S.M.; Kim, D.Y.; et al. Nuclear localization and functional characteristics of voltage-gated potassium channel Kv1.3. J. Biol. Chem. 2015, 290, 12547–12557. [Google Scholar] [CrossRef]
- Katta, S.S.; Smoyer, C.J.; Jaspersen, S.L. Destination: Inner nuclear membrane. Trends Cell Biol. 2014, 24, 221–229. [Google Scholar] [CrossRef]
- Chen, Y.; Sanchez, A.; Rubio, M.E.; Kohl, T.; Pardo, L.A.; Stuhmer, W. Functional K(v)10.1 channels localize to the inner nuclear membrane. PLoS ONE 2011, 6, e19257. [Google Scholar] [CrossRef]
- Soltysinska, E.; Bentzen, B.H.; Barthmes, M.; Hattel, H.; Thrush, A.B.; Harper, M.E.; Qvortrup, K.; Larsen, F.J.; Schiffer, T.A.; Losa-Reyna, J.; et al. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS ONE 2014, 9, e103402. [Google Scholar] [CrossRef]
- Szabo, I.; Bock, J.; Grassme, H.; Soddemann, M.; Wilker, B.; Lang, F.; Zoratti, M.; Gulbins, E. Mitochondrial potassium channel Kv1.3 mediates Bax-induced apoptosis in lymphocytes. Proc. Natl. Acad. Sci. USA 2008, 105, 14861–14866. [Google Scholar] [CrossRef]
- Yu, K.Y.; Wang, Y.P.; Wang, L.H.; Jian, Y.; Zhao, X.D.; Chen, J.W.; Murao, K.; Zhu, W.; Dong, L.; Wang, G.Q.; et al. Mitochondrial KATP channel involvement in angiotensin II-induced autophagy in vascular smooth muscle cells. Basic Res. Cardiol. 2014, 109, 416. [Google Scholar] [CrossRef]
- Singh, H.; Lu, R.; Bopassa, J.C.; Meredith, A.L.; Stefani, E.; Toro, L. MitoBK(Ca) is encoded by the Kcnma1 gene, and a splicing sequence defines its mitochondrial location. Proc. Natl. Acad. Sci. USA 2013, 110, 10836–10841. [Google Scholar] [CrossRef]
- Foster, D.B.; Ho, A.S.; Rucker, J.; Garlid, A.O.; Chen, L.; Sidor, A.; Garlid, K.D.; O’Rourke, B. Mitochondrial ROMK channel is a molecular component of mitoK(ATP). Circ. Res. 2012, 111, 446–454. [Google Scholar] [CrossRef]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabo, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef]
- Dolga, A.M.; Netter, M.F.; Perocchi, F.; Doti, N.; Meissner, L.; Tobaben, S.; Grohm, J.; Zischka, H.; Plesnila, N.; Decher, N.; et al. Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J. Biol. Chem. 2013, 288, 10792–10804. [Google Scholar] [CrossRef]
- Testai, L.; Barrese, V.; Soldovieri, M.V.; Ambrosino, P.; Martelli, A.; Vinciguerra, I.; Miceli, F.; Greenwood, I.A.; Curtis, M.J.; Breschi, M.C.; et al. Expression and function of Kv7.4 channels in rat cardiac mitochondria: Possible targets for cardioprotection. Cardiovasc. Res. 2016, 110, 40–50. [Google Scholar] [CrossRef]
- Yao, J.; McHedlishvili, D.; McIntire, W.E.; Guagliardo, N.A.; Erisir, A.; Coburn, C.A.; Santarelli, V.P.; Bayliss, D.A.; Barrett, P.Q. Functional TASK-3-Like Channels in Mitochondria of Aldosterone-Producing Zona Glomerulosa Cells. Hypertension 2017, 70, 347–356. [Google Scholar] [CrossRef]
- Smith, C.O.; Wang, Y.T.; Nadtochiy, S.M.; Miller, J.H.; Jonas, E.A.; Dirksen, R.T.; Nehrke, K.; Brookes, P.S. Cardiac metabolic effects of KNa1.2 channel deletion and evidence for its mitochondrial localization. FASEB J. 2018. [Google Scholar] [CrossRef]
- Villarejo, A.; Buren, S.; Larsson, S.; Dejardin, A.; Monne, M.; Rudhe, C.; Karlsson, J.; Jansson, S.; Lerouge, P.; Rolland, N.; et al. Evidence for a protein transported through the secretory pathway en route to the higher plant chloroplast. Nat. Cell Biol. 2005, 7, 1224–1231. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Lin, T.Y.; Chiou, S.H.; Chow, K.C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE 2012, 7, e33657. [Google Scholar] [CrossRef]
- Von Charpuis, C.; Meckel, T.; Moroni, A.; Thiel, G. The sorting of a small potassium channel in mammalian cells can be shifted between mitochondria and plasma membrane. Cell Calcium 2015, 58, 114–121. [Google Scholar] [CrossRef]
- Vicente, R.; Villalonga, N.; Calvo, M.; Escalada, A.; Solsona, C.; Soler, C.; Tamkun, M.M.; Felipe, A. Kv1.5 association modifies Kv1.3 traffic and membrane localization. J. Biol. Chem. 2008, 283, 8756–8764. [Google Scholar] [CrossRef]
- Manganas, L.N.; Trimmer, J.S. Subunit composition determines Kv1 potassium channel surface expression. J. Biol. Chem. 2000, 275, 29685–29693. [Google Scholar] [CrossRef]
- Zhu, J.; Watanabe, I.; Gomez, B.; Thornhill, W.B. Heteromeric Kv1 potassium channel expression: Amino acid determinants involved in processing and trafficking to the cell surface. J. Biol. Chem. 2003, 278, 25558–25567. [Google Scholar] [CrossRef]
- Ponce-Balbuena, D.; Guerrero-Serna, G.; Valdivia, C.R.; Caballero, R.; Diez-Guerra, F.J.; Jimenez-Vazquez, E.N.; Ramirez, R.J.; Monteiro da Rocha, A.; Herron, T.J.; Campbell, K.F.; et al. Cardiac Kir2.1 and NaV1.5 Channels Traffic Together to the Sarcolemma to Control Excitability. Circ. Res. 2018, 122, 1501–1516. [Google Scholar] [CrossRef]
- Utrilla, R.G.; Nieto-Marin, P.; Alfayate, S.; Tinaquero, D.; Matamoros, M.; Perez-Hernandez, M.; Sacristan, S.; Ondo, L.; de Andres, R.; Diez-Guerra, F.J.; et al. Kir2.1-Nav1.5 Channel Complexes Are Differently Regulated than Kir2.1 and Nav1.5 Channels Alone. Front. Physiol. 2017, 8, 903. [Google Scholar] [CrossRef] [PubMed]
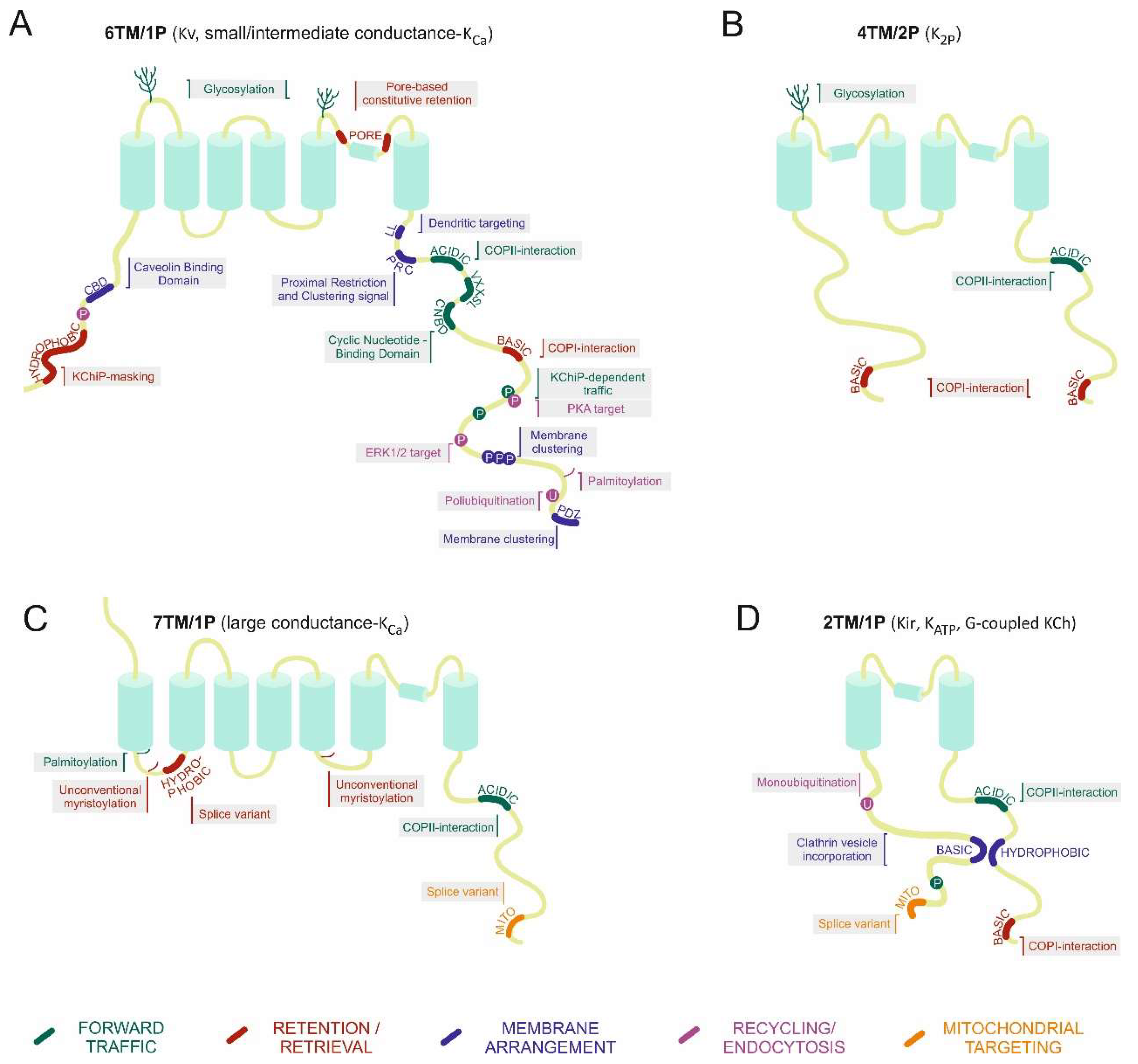
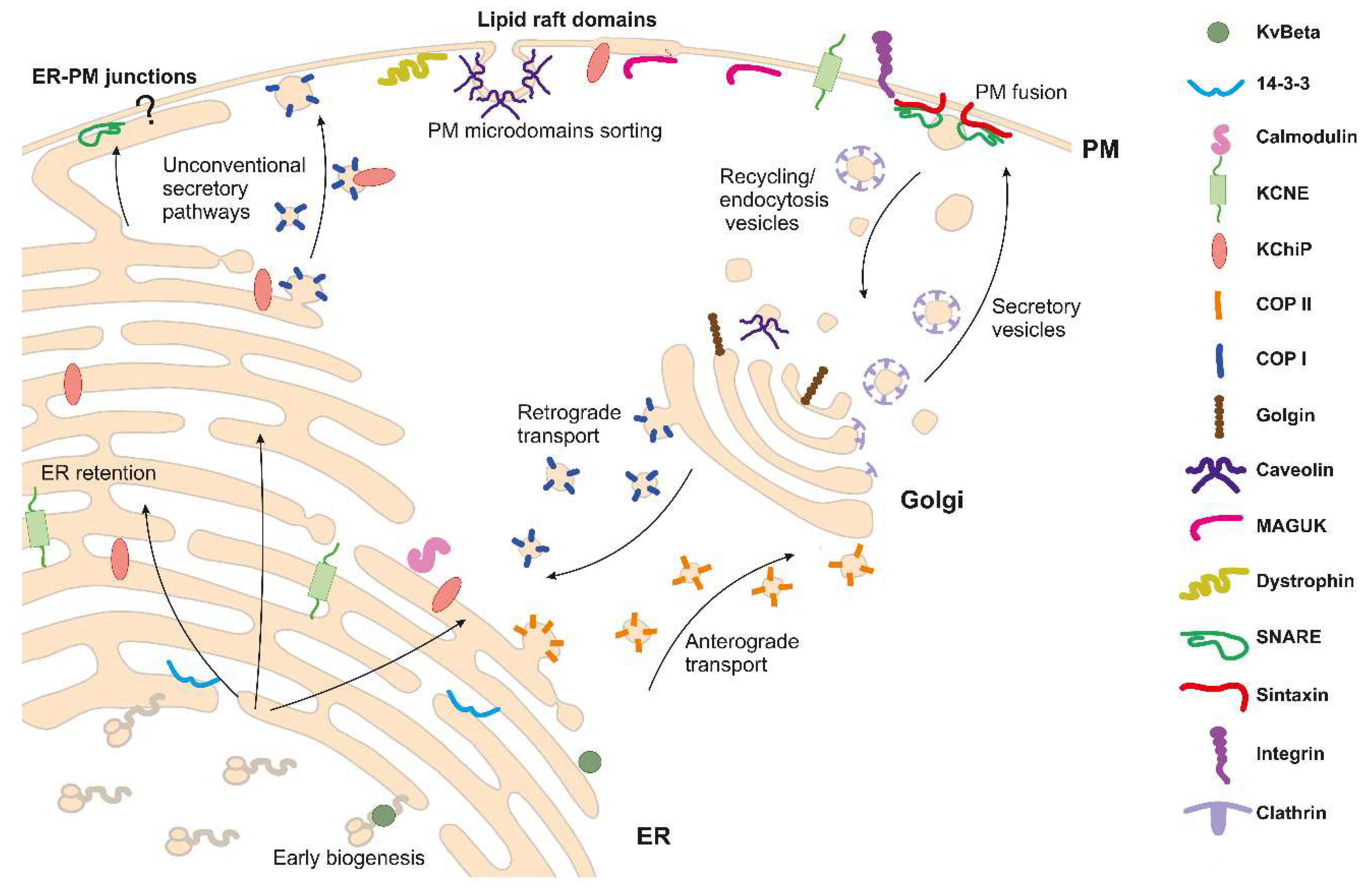
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capera, J.; Serrano-Novillo, C.; Navarro-Pérez, M.; Cassinelli, S.; Felipe, A. The Potassium Channel Odyssey: Mechanisms of Traffic and Membrane Arrangement. Int. J. Mol. Sci. 2019, 20, 734. https://doi.org/10.3390/ijms20030734
Capera J, Serrano-Novillo C, Navarro-Pérez M, Cassinelli S, Felipe A. The Potassium Channel Odyssey: Mechanisms of Traffic and Membrane Arrangement. International Journal of Molecular Sciences. 2019; 20(3):734. https://doi.org/10.3390/ijms20030734
Chicago/Turabian StyleCapera, Jesusa, Clara Serrano-Novillo, María Navarro-Pérez, Silvia Cassinelli, and Antonio Felipe. 2019. "The Potassium Channel Odyssey: Mechanisms of Traffic and Membrane Arrangement" International Journal of Molecular Sciences 20, no. 3: 734. https://doi.org/10.3390/ijms20030734
APA StyleCapera, J., Serrano-Novillo, C., Navarro-Pérez, M., Cassinelli, S., & Felipe, A. (2019). The Potassium Channel Odyssey: Mechanisms of Traffic and Membrane Arrangement. International Journal of Molecular Sciences, 20(3), 734. https://doi.org/10.3390/ijms20030734