Catabolism of Fibromodulin in Developmental Rudiment and Pathologic Articular Cartilage Demonstrates Novel Roles for MMP-13 and ADAMTS-4 in C-terminal Processing of SLRPs


Abstract
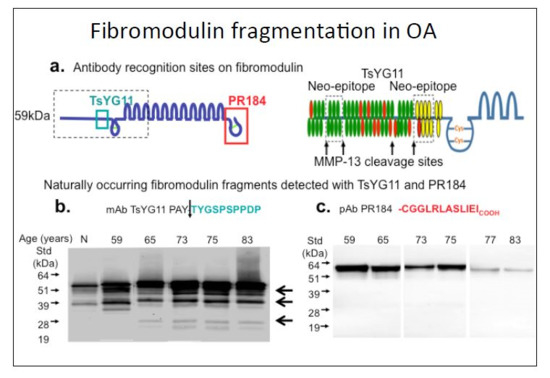
{kind=link}
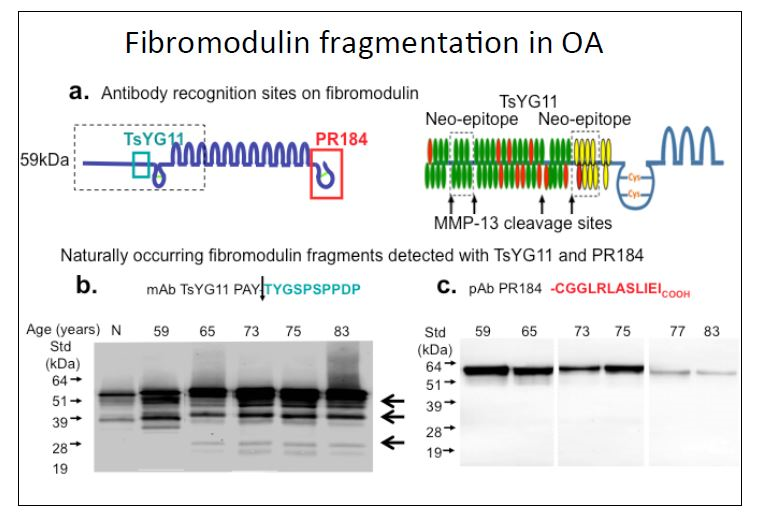
{kind=link}
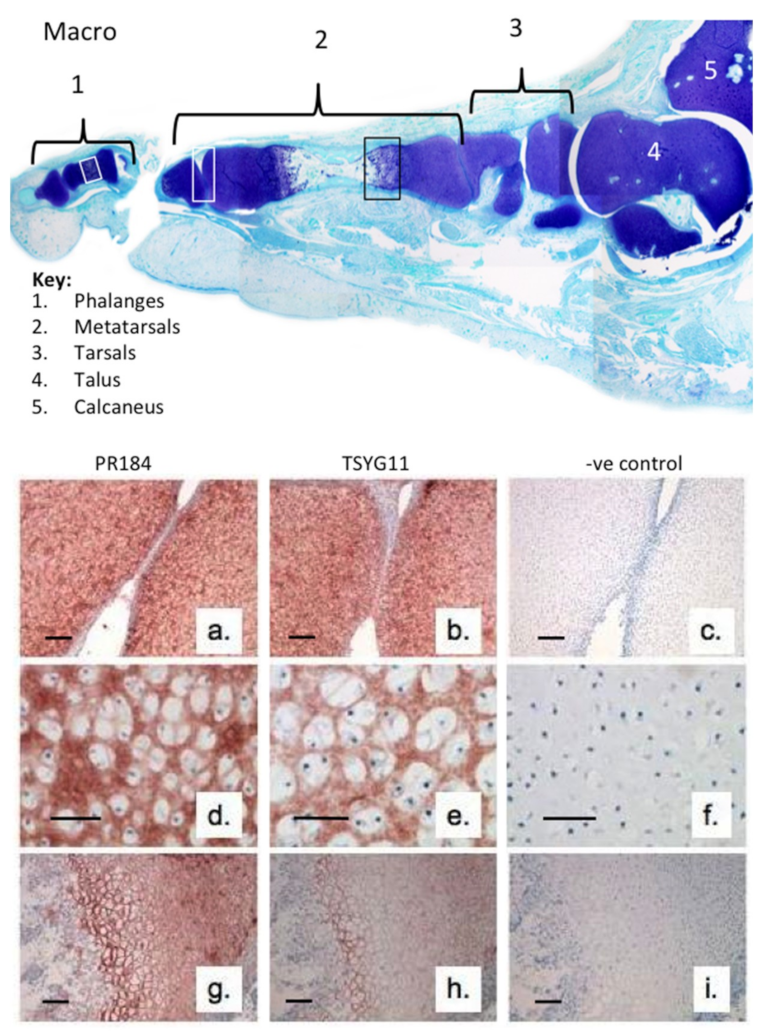
{kind=link}
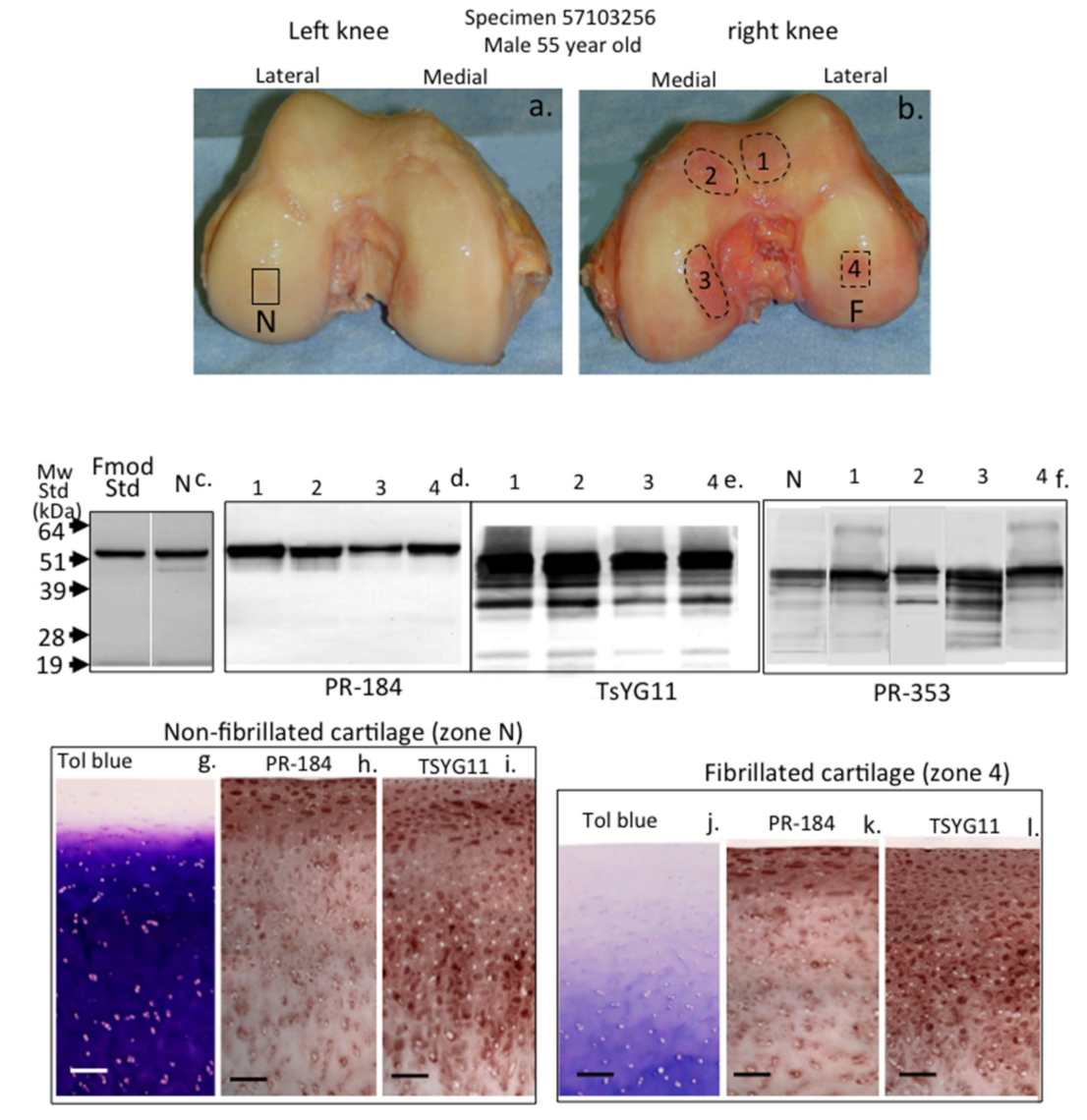
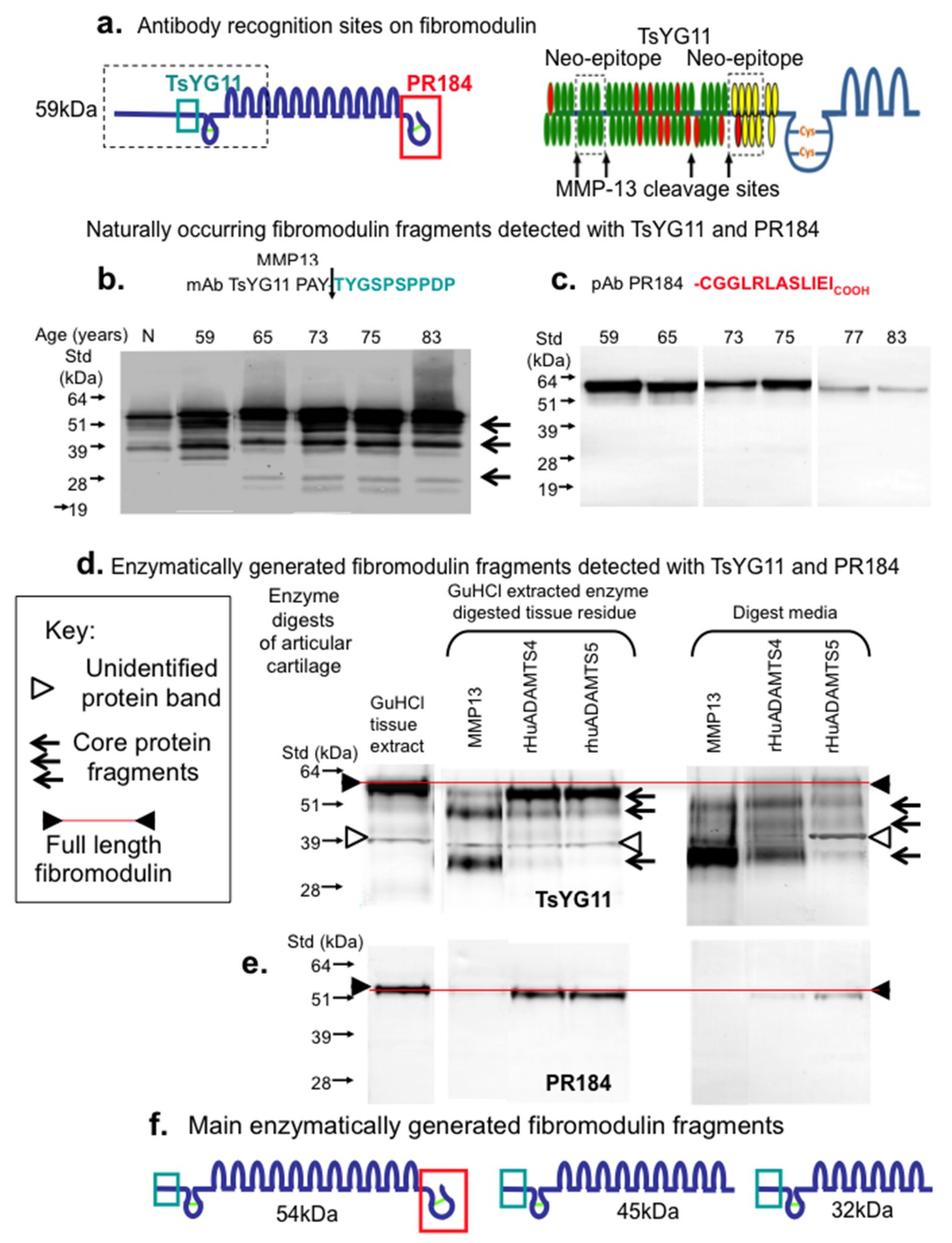{kind=link}
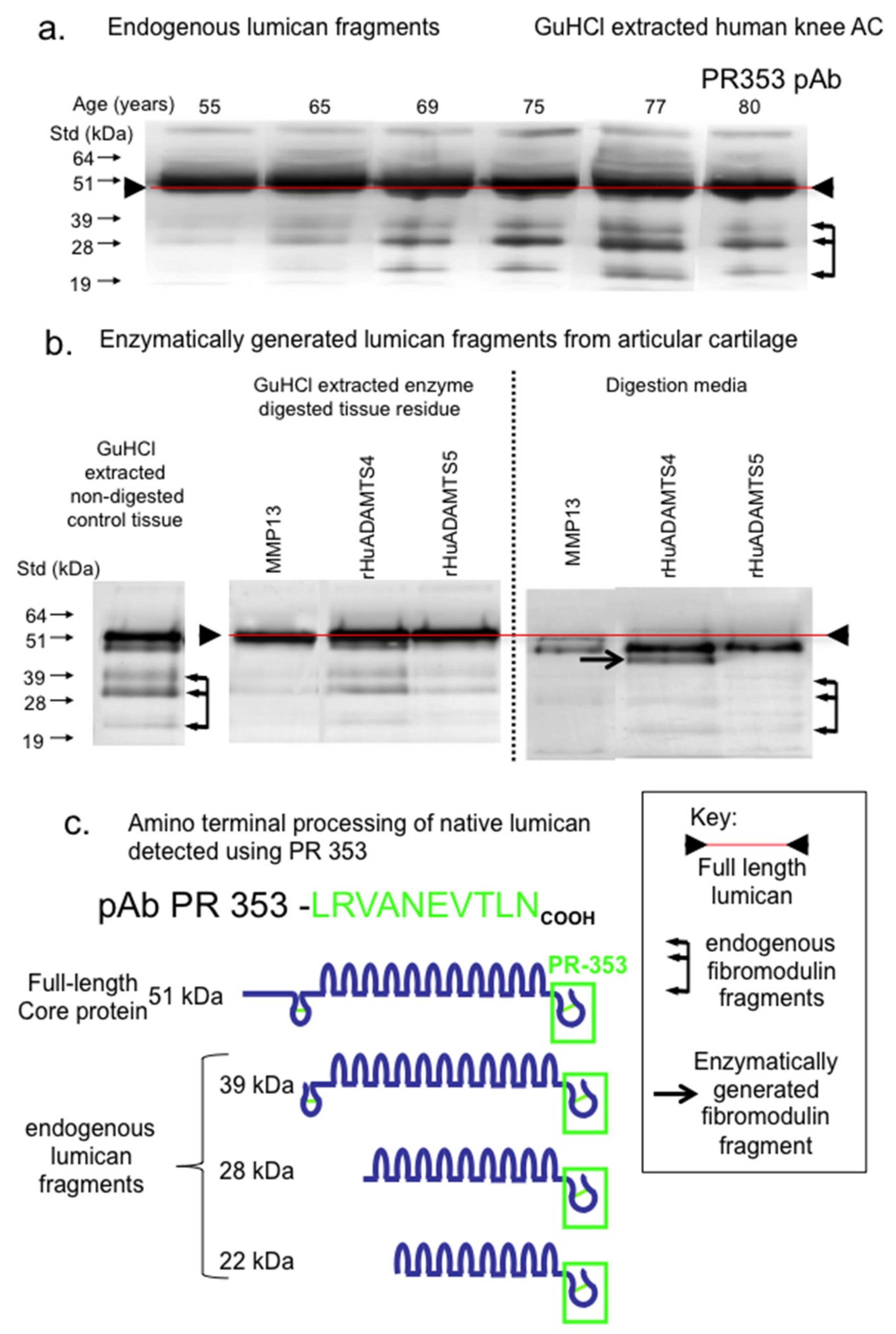{kind=link}
{kind=link}
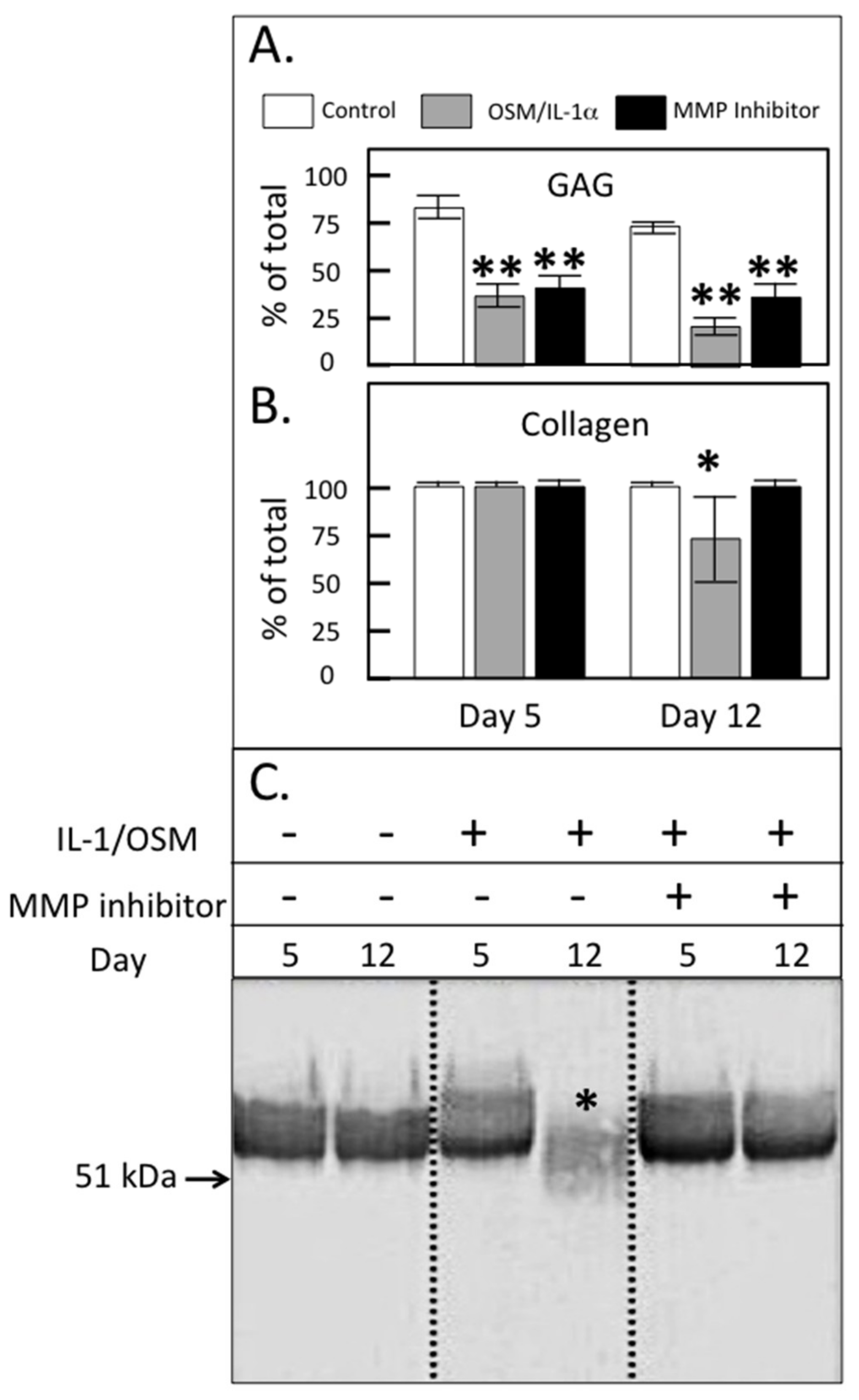{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Consumables
4.2. Tissues
4.3. Antibodies
4.4. Extraction of Tissues
4.5. Chondroitinase ABC, Keratanase-I and N-Glycanase Digestion of Tissue Extracts
4.6. SDS PAGE and Detection of SLRP Fragments by Western Blotting
4.7. Cartilage Digestion with MMP-13, ADAMTS-4 and ADAMTS-5
4.8. Ovine Cartilage Explant Cultures
4.9. Histological Processing of Specimens
4.10. Toluidine Blue Staining of Cartilage Specimens
4.11. Immunolocalization of FMOD and MMP-13-Cleaved FMOD in Cartilage Tissue Sections
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AC | articular cartilage |
| ADAMTS | A Disintegrin and Metalloproteinase with Thrombospondin motifs |
| AMPA | aminophenyl mercuric acetate |
| BGN | biglycan |
| BMI | body mass index |
| Brij-35® | Polyoxyethylene (23) lauryl ether |
| DAMPs | Damage-associated molecular pattern molecules |
| DCN | decorin |
| ECM | extracellular matrix |
| FMOD | fibromodulin |
| GuHCL | Guanidine hydrochloride |
| LRR | leucine-rich repeat |
| LUM | lumican |
| LuSDS | Lithium dodecyl sulphate |
| MOPS | 3-(N-morpholino) propanesulfonic acid |
| OA | osteoarthritis |
| OSM | oncostatin M |
| MMP | matrix metalloprotease |
| SLRP | small leucine repeat proteoglycan |
| TLR | toll-like receptor |
| PRELP | proline/arginine-rich end leucine-rich repeat protein |
| PAGE | polyacrylamide gel electrophoresis |
References
- Wallace, I.J.; Worthington, S.; Felson, D.T.; Jurmain, R.D.; Wren, K.T.; Maijanen, H.; Woods, R.J.; Lieberman, D.E. Knee osteoarthritis has doubled in prevalence since the mid-20th century. Proc. Natl. Acad. Sci. USA 2017, 114, 9332–9336. [Google Scholar] [CrossRef] [PubMed]
- Wluka, A.E.; Lombard, C.B.; Cicuttini, F.M. Tackling obesity in knee osteoarthritis. Nat. Rev. Rheumatol. 2013, 9, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Cross, M.; Smith, E.; Hoy, D.; Nolte, S.; Ackerman, I.; Fransen, M.; Bridgett, L.; Williams, S.; Guillemin, F.; Hill, C.L.; et al. The global burden of hip and knee osteoarthritis: Estimates from the global burden of disease 2010 study. Ann. Rheum. Dis. 2014, 73, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Lawrence, R.C.; Dieppe, P.A.; Hirsch, R.; Helmick, C.G.; Jordan, J.M.; Kington, R.S.; Lane, N.E.; Nevitt, M.C.; Zhang, Y.; et al. Osteoarthritis: New insights. Part 1: The disease and its risk factors. Ann. Intern. Med. 2000, 133, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Robinson, W.H.; Lepus, C.M.; Wang, Q.; Raghu, H.; Mao, R.; Lindstrom, T.M.; Sokolove, J. Low-grade inflammation as a key mediator of the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol 2016, 12, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Gupte, C.; Akhtar, K.; Smith, P.; Cobb, J. The Global Economic Cost of Osteoarthritis: How the UK Compares. Arthritis 2012, 2012, 698709. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef]
- Chen, S.; Oldberg, A.; Chakravarti, S.; Birk, D.E. Fibromodulin regulates collagen fibrillogenesis during peripheral corneal development. Dev. Dyn. 2010, 239, 844–854. [Google Scholar] [CrossRef]
- Chen, S.; Young, M.F.; Chakravarti, S.; Birk, D.E. Interclass small leucine-rich repeat proteoglycan interactions regulate collagen fibrillogenesis and corneal stromal assembly. Matrix Biol 2014, 35, 103–111. [Google Scholar] [CrossRef]
- Jan, A.T.; Lee, E.J.; Choi, I. Fibromodulin: A regulatory molecule maintaining cellular architecture for normal cellular function. Int. J. Biochem. Cell. Biol. 2016, 80, 66–70. [Google Scholar] [CrossRef]
- Ni, G.X.; Li, Z.; Zhou, Y.Z. The role of small leucine-rich proteoglycans in osteoarthritis pathogenesis. Osteoarthritis Cartilage 2014, 22, 896–903. [Google Scholar] [CrossRef]
- Iozzo, R.V.; Schaefer, L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol 2015, 42, 11–55. [Google Scholar] [CrossRef] [PubMed]
- Ezura, Y.; Chakravarti, S.; Oldberg, A.; Chervoneva, I.; Birk, D.E. Differential expression of lumican and fibromodulin regulate collagen fibrillogenesis in developing mouse tendons. J. Cell. Biol. 2000, 151, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Kalamajski, S.; Oldberg, A. Homologous sequence in lumican and fibromodulin leucine-rich repeat 5-7 competes for collagen binding. J. Biol. Chem. 2009, 284, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Narlid, I.; Oldberg, A. Fibromodulin and lumican bind to the same region on collagen type I fibrils. FEBS Lett. 2000, 470, 178–182. [Google Scholar] [CrossRef]
- Gill, M.R.; Oldberg, A.; Reinholt, F.P. Fibromodulin-null murine knee joints display increased incidences of osteoarthritis and alterations in tissue biochemistry. Osteoarthritis Cartilage 2002, 10, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Svensson, L.; Aszodi, A.; Reinholt, F.P.; Fassler, R.; Heinegard, D.; Oldberg, A. Fibromodulin-null mice have abnormal collagen fibrils, tissue organization, and altered lumican deposition in tendon. J. Biol. Chem. 1999, 274, 9636–9647. [Google Scholar] [CrossRef]
- Onnerfjord, P.; Heathfield, T.F.; Heinegard, D. Identification of tyrosine sulfation in extracellular leucine-rich repeat proteins using mass spectrometry. J. Biol. Chem. 2004, 279, 26–33. [Google Scholar] [CrossRef]
- Tillgren, V.; Onnerfjord, P.; Haglund, L.; Heinegard, D. The tyrosine sulfate-rich domains of the LRR proteins fibromodulin and osteoadherin bind motifs of basic clusters in a variety of heparin-binding proteins, including bioactive factors. J. Biol. Chem. 2009, 284, 28543–28553. [Google Scholar] [CrossRef]
- Tillgren, V.; Morgelin, M.; Onnerfjord, P.; Kalamajski, S.; Aspberg, A. The Tyrosine Sulfate Domain of Fibromodulin Binds Collagen and Enhances Fibril Formation. J. Biol. Chem. 2016, 291, 23744–23755. [Google Scholar] [CrossRef]
- Hildebrand, A.; Romaris, M.; Rasmussen, L.M.; Heinegard, D.; Twardzik, D.R.; Border, W.A.; Ruoslahti, E. Interaction of the small interstitial proteoglycans biglycan, decorin and fibromodulin with transforming growth factor beta. Biochem. J. 1994, 302, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Soo, C.; Hu, F.Y.; Zhang, X.; Wang, Y.; Beanes, S.R.; Lorenz, H.P.; Hedrick, M.H.; Mackool, R.J.; Plaas, A.; Kim, S.J.; et al. Differential expression of fibromodulin, a transforming growth factor-beta modulator, in fetal skin development and scarless repair. Am. J. Pathol. 2000, 157, 423–433. [Google Scholar] [CrossRef]
- Sjoberg, A.; Onnerfjord, P.; Morgelin, M.; Heinegard, D.; Blom, A.M. The extracellular matrix and inflammation: Fibromodulin activates the classical pathway of complement by directly binding C1q. J. Biol. Chem. 2005, 280, 32301–32308. [Google Scholar] [CrossRef] [PubMed]
- Brezillon, S.; Pietraszek, K.; Maquart, F.X.; Wegrowski, Y. Lumican effects in the control of tumour progression and their links with metalloproteinases and integrins. FEBS J. 2013, 280, 2369–2381. [Google Scholar] [CrossRef]
- Niewiarowska, J.; Brezillon, S.; Sacewicz-Hofman, I.; Bednarek, R.; Maquart, F.X.; Malinowski, M.; Wiktorska, M.; Wegrowski, Y.; Cierniewski, C.S. Lumican inhibits angiogenesis by interfering with alpha2beta1 receptor activity and downregulating MMP-14 expression. Thromb. Res. 2011, 128, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Stasiak, M.; Boncela, J.; Perreau, C.; Karamanou, K.; Chatron-Colliet, A.; Proult, I.; Przygodzka, P.; Chakravarti, S.; Maquart, F.X.; Kowalska, M.A.; et al. Lumican Inhibits SNAIL-Induced Melanoma Cell Migration Specifically by Blocking MMP-14 Activity. PLoS ONE 2016, 11, e0150226. [Google Scholar] [CrossRef] [PubMed]
- Geng, Y.; McQuillan, D.; Roughley, P.J. SLRP interaction can protect collagen fibrils from cleavage by collagenases. Matrix Biol. 2006, 25, 484–491. [Google Scholar] [CrossRef]
- Cs-Szabo, G.; Melching, L.I.; Roughley, P.J.; Glant, T.T. Changes in messenger RNA and protein levels of proteoglycans and link protein in human osteoarthritic cartilage samples. Arthritis Rheum. 1997, 40, 1037–1045. [Google Scholar] [CrossRef]
- Melrose, J.; Fuller, E.S.; Roughley, P.J.; Smith, M.M.; Kerr, B.; Hughes, C.E.; Caterson, B.; Little, C.B. Fragmentation of decorin, biglycan, lumican and keratocan is elevated in degenerate human meniscus, knee and hip articular cartilages compared with age-matched macroscopically normal and control tissues. Arthritis Res. Ther. 2008, 10, R79. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Brittain, I.J.; Laska, D.A.; Mitchell, P.G.; Sumer, E.U.; Karsdal, M.A.; Duffin, K.L. Characterization of metalloprotease cleavage products of human articular cartilage. Arthritis Rheum. 2008, 58, 2420–2431. [Google Scholar] [CrossRef]
- Danfelter, M.; Onnerfjord, P.; Heinegard, D. Fragmentation of proteins in cartilage treated with interleukin-1: Specific cleavage of type IX collagen by matrix metalloproteinase 13 releases the NC4 domain. J. Biol. Chem. 2007, 282, 36933–36941. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, M.; Enghild, J.J.; Gendron, C.; Hughes, C.; Caterson, B.; Itoh, Y.; Nagase, H. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J. Biol. Chem. 2004, 279, 10109–10119. [Google Scholar] [CrossRef]
- Flannery, C.R. MMPs and ADAMTSs: Functional studies. Front. Biosci. 2006, 11, 544–569. [Google Scholar] [CrossRef]
- Flannery, C.R.; Zeng, W.; Corcoran, C.; Collins-Racie, L.A.; Chockalingam, P.S.; Hebert, T.; Mackie, S.A.; McDonagh, T.; Crawford, T.K.; Tomkinson, K.N.; et al. Autocatalytic cleavage of ADAMTS-4 (Aggrecanase-1) reveals multiple glycosaminoglycan-binding sites. J. Biol. Chem. 2002, 277, 42775–42780. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.T.; Moradi, B.; Smith, M.M.; Jackson, C.J.; Little, C.B. Activation of matrix metalloproteinases 2, 9, and 13 by activated protein C in human osteoarthritic cartilage chondrocytes. Arthritis Rheum. 2014, 66, 1525–1536. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.T.; Moradi, B.; Zaki, S.; Smith, M.M.; McCracken, S.; Smith, S.M.; Jackson, C.J.; Little, C.B. Depletion of protease-activated receptor 2 but not protease-activated receptor 1 may confer protection against osteoarthritis in mice through extracartilaginous mechanisms. Arthritis Rheum. 2014, 66, 3337–3348. [Google Scholar] [CrossRef]
- Jackson, M.T.; Smith, M.M.; Smith, S.M.; Jackson, C.J.; Xue, M.; Little, C.B. Activation of cartilage matrix metalloproteinases by activated protein C. Arthritis Rheum. 2009, 60, 780–791. [Google Scholar] [CrossRef]
- D’Angelo, M.; Yan, Z.; Nooreyazdan, M.; Pacifici, M.; Sarment, D.S.; Billings, P.C.; Leboy, P.S. MMP-13 is induced during chondrocyte hypertrophy. J. Cell. Biochem. 2000, 77, 678–693. [Google Scholar] [CrossRef]
- Favero, M.; Belluzzi, E.; Trisolino, G.; Goldring, M.B.; Goldring, S.R.; Cigolotti, A.; Pozzuoli, A.; Ruggieri, P.; Ramonda, R.; Grigolo, B.; et al. Inflammatory molecules produced by meniscus and synovium in early and end-stage osteoarthritis: A coculture study. J. Cell Physiol. 2018. [Google Scholar] [CrossRef]
- Favero, M.; Ramonda, R.; Goldring, M.B.; Goldring, S.R.; Punzi, L. Early knee osteoarthritis. RMD Open 2015, 1, e000062. [Google Scholar] [CrossRef] [PubMed]
- Belluzzi, E.; El Hadi, H.; Granzotto, M.; Rossato, M.; Ramonda, R.; Macchi, V.; De Caro, R.; Vettor, R.; Favero, M. Systemic and Local Adipose Tissue in Knee Osteoarthritis. J. Cell. Physiol. 2017, 232, 1971–1978. [Google Scholar] [CrossRef]
- Chang, J.; Liao, Z.; Lu, M.; Meng, T.; Han, W.; Ding, C. Systemic and local adipose tissue in knee osteoarthritis. Osteoarthritis Cartilage 2018, 26, 864–871. [Google Scholar] [CrossRef] [PubMed]
- Favero, M.; El-Hadi, H.; Belluzzi, E.; Granzotto, M.; Porzionato, A.; Sarasin, G.; Rambaldo, A.; Iacobellis, C.; Cigolotti, A.; Fontanella, C.G.; et al. Infrapatellar fat pad features in osteoarthritis: A histopathological and molecular study. Rheumatology (Oxford) 2017, 56, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Goldring, S.R. Alterations in periarticular bone and cross talk between subchondral bone and articular cartilage in osteoarthritis. Ther. Adv. Musculoskelet. Dis. 2012, 4, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Trzeciak, T.; Owecki, M.; Pucher, A.; Kaczmarczyk, J. The role of adipocytokines in the pathogenesis of knee joint osteoarthritis. Int. Orthop. 2015, 39, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef]
- Fuller, E.; Little, C.B.; Melrose, J. Interleukin-1alpha induces focal degradation of biglycan and tissue degeneration in an in-vitro ovine meniscal model. Exp. Mol. Pathol. 2016, 101, 214–220. [Google Scholar] [CrossRef]
- Fuller, E.S.; Smith, M.M.; Little, C.B.; Melrose, J. Zonal differences in meniscus matrix turnover and cytokine response. Osteoarthritis Cartilage 2012, 20, 49–59. [Google Scholar] [CrossRef]
- Melrose, J.; Fuller, E.S.; Little, C.B. The biology of meniscal pathology in osteoarthritis and its contribution to joint disease: Beyond simple mechanics. Connect. Tissue Res. 2017, 58, 282–294. [Google Scholar] [CrossRef]
- Madry, H.; Kon, E.; Condello, V.; Peretti, G.M.; Steinwachs, M.; Seil, R.; Berruto, M.; Engebretsen, L.; Filardo, G.; Angele, P. Early osteoarthritis of the knee. Knee Surg. Sports Traumatol. Arthrosc. 2016, 24, 1753–1762. [Google Scholar] [CrossRef] [PubMed]
- Malempati, C.; Jacobs, C.A.; Lattermann, C. The Early Osteoarthritic Knee: Implications for Cartilage Repair. Clin. Sports Med. 2017, 36, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, H.; Okazaki, K.; Takayama, Y.; Osaki, K.; Matsuo, Y.; Honda, H.; Iwamoto, Y. Detection of early cartilage deterioration associated with meniscal tear using T1rho mapping magnetic resonance imaging. BMC Musculoskelet. Disord. 2015, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Winalski, C.S.; Chu, C.R. Early articular cartilage MRI T2 changes after anterior cruciate ligament reconstruction correlate with later changes in T2 and cartilage thickness. J. Orthop. Res. 2017, 35, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Liu-Bryan, R. Synovium and the innate inflammatory network in osteoarthritis progression. Curr. Rheum. Rep. 2013, 15, 323. [Google Scholar] [CrossRef] [PubMed]
- Merline, R.; Schaefer, R.M.; Schaefer, L. The matricellular functions of small leucine-rich proteoglycans (SLRPs). J. Cell. Commun. Signal. 2009, 3, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.H.; Rai, V.; Dilisio, M.F.; Agrawal, D.K. Damage-associated molecular patterns in the pathogenesis of osteoarthritis: Potentially novel therapeutic targets. Mol. Cell. Biochem. 2017, 434, 171–179. [Google Scholar] [CrossRef]
- Miller, R.E.; Belmadani, A.; Ishihara, S.; Tran, P.B.; Ren, D.; Miller, R.J.; Malfait, A.M. Damage-associated molecular patterns generated in osteoarthritis directly excite murine nociceptive neurons through Toll-like receptor 4. Arthritis Rheum. 2015, 67, 2933–2943. [Google Scholar] [CrossRef]
- Ricard-Blum, S.; Ballut, L. Matricryptins derived from collagens and proteoglycans. Front. Biosci. (Landmark Ed.) 2011, 16, 674–697. [Google Scholar] [CrossRef]
- Moreth, K.; Iozzo, R.V.; Schaefer, L. Small leucine-rich proteoglycans orchestrate receptor crosstalk during inflammation. Cell Cycle 2012, 11, 2084–2091. [Google Scholar] [CrossRef]
- Nastase, M.V.; Janicova, A.; Roedig, H.; Hsieh, L.T.; Wygrecka, M.; Schaefer, L. Small Leucine-Rich Proteoglycans in Renal Inflammation: Two Sides of the Coin. J. Histochem. Cytochem. 2018, 66, 261–272. [Google Scholar] [CrossRef]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef]
- Stanton, H.; Melrose, J.; Little, C.B.; Fosang, A.J. Proteoglycan degradation by the ADAMTS family of proteinases. Biochim. Biophys. Acta 2011, 1812, 1616–1629. [Google Scholar] [CrossRef]
- Pietraszek, K.; Chatron-Colliet, A.; Brezillon, S.; Perreau, C.; Jakubiak-Augustyn, A.; Krotkiewski, H.; Maquart, F.X.; Wegrowski, Y. Lumican: A new inhibitor of matrix metalloproteinase-14 activity. FEBS Lett. 2014, 588, 4319–4324. [Google Scholar] [CrossRef]
- Pietraszek, K.; Brezillon, S.; Perreau, C.; Malicka-Blaszkiewicz, M.; Maquart, F.X.; Wegrowski, Y. Lumican -derived peptides inhibit melanoma cell growth and migration. PLoS ONE 2013, 8, e76232. [Google Scholar] [CrossRef]
- Li, Y.; Aoki, T.; Mori, Y.; Ahmad, M.; Miyamori, H.; Takino, T.; Sato, H. Cleavage of lumican by membrane-type matrix metalloproteinase-1 abrogates this proteoglycan-mediated suppression of tumor cell colony formation in soft agar. Cancer Res. 2004, 64, 7058–7064. [Google Scholar] [CrossRef]
- Heathfield, T.F.; Onnerfjord, P.; Dahlberg, L.; Heinegard, D. Cleavage of fibromodulin in cartilage explants involves removal of the N-terminal tyrosine sulfate-rich region by proteolysis at a site that is sensitive to matrix metalloproteinase-13. J. Biol. Chem. 2004, 279, 6286–6295. [Google Scholar] [CrossRef]
- Young, A.A.; Smith, M.M.; Smith, S.M.; Cake, M.A.; Ghosh, P.; Read, R.A.; Melrose, J.; Sonnabend, D.H.; Roughley, P.J.; Little, C.B. Regional assessment of articular cartilage gene expression and small proteoglycan metabolism in an animal model of osteoarthritis. Arthritis Res. Ther. 2005, 7, R852–R861. [Google Scholar] [CrossRef]
- Hedlund, H.; Mengarelli-Widholm, S.; Heinegard, D.; Reinholt, F.P.; Svensson, O. Fibromodulin distribution and association with collagen. Matrix Biol. 1994, 14, 227–232. [Google Scholar] [CrossRef]
- Cs-Szabo, G.; Roughley, P.J.; Plaas, A.H.; Glant, T.T. Large and small proteoglycans of osteoarthritic and rheumatoid articular cartilage. Arthritis Rheum. 1995, 38, 660–668. [Google Scholar] [CrossRef]
- Roughley, P.J.; White, R.J.; Cs-Szabo, G.; Mort, J.S. Changes with age in the structure of fibromodulin in human articular cartilage. Osteoarthritis Cartilage 1996, 4, 153–161. [Google Scholar] [CrossRef]
- Melrose, J.; Fuller, E.S.; Roughley, P.J.; Smith, M.M.; Kerr, B.; Hughes, C.E.; Caterson, B.; Little, C.B. Fragmentation of Decorin, Biglycan, Lumican and Keratocan is elevated in Degenerate Human Meniscus, Knee and Hip Articular Cartilages compared to Age-matched Macroscopically Normal and Control Tissues. Arthritis Res. Ther. 2008, 10, R79. [Google Scholar]
- Sztrolovics, R.; White, R.J.; Poole, A.R.; Mort, J.S.; Roughley, P.J. Resistance of small leucine-rich repeat proteoglycans to proteolytic degradation during interleukin-1-stimulated cartilage catabolism. Biochem. J. 1999, 339, 571–577. [Google Scholar] [CrossRef]
- Gendron, C.; Kashiwagi, M.; Lim, N.H.; Enghild, J.J.; Thogersen, I.B.; Hughes, C.; Caterson, B.; Nagase, H. Proteolytic activities of human ADAMTS-5: Comparative studies with ADAMTS-4. J. Biol. Chem. 2007, 282, 18294–18306. [Google Scholar] [CrossRef]
- Fushimi, K.; Troeberg, L.; Nakamura, H.; Lim, N.H.; Nagase, H. Functional differences of the catalytic and non-catalytic domains in human ADAMTS-4 and ADAMTS-5 in aggrecanolytic activity. J. Biol. Chem. 2008, 283, 6706–6716. [Google Scholar] [CrossRef]
- Sztrolovics, R.; Alini, M.; Mort, J.S.; Roughley, P.J. Age-related changes in fibromodulin and lumican in human intervertebral discs. Spine 1999, 24, 1765–1771. [Google Scholar] [CrossRef]
- Ge, G.; Seo, N.S.; Liang, X.; Hopkins, D.R.; Hook, M.; Greenspan, D.S. Bone morphogenetic protein-1/tolloid-related metalloproteinases process osteoglycin and enhance its ability to regulate collagen fibrillogenesis. J. Biol. Chem. 2004, 279, 41626–41633. [Google Scholar] [CrossRef]
- Melrose, J.; Smith, S.M.; Fuller, E.S.; Young, A.A.; Roughley, P.J.; Dart, A.; Little, C.B. Biglycan and fibromodulin fragmentation correlates with temporal and spatial annular remodelling in experimentally injured ovine intervertebral discs. Eur. Spine J. 2007, 16, 2193–2205. [Google Scholar] [CrossRef]
- Little, C.B.; Hughes, C.E.; Curtis, C.L.; Janusz, M.J.; Bohne, R.; Wang-Weigand, S.; Taiwo, Y.O.; Mitchell, P.G.; Otterness, I.G.; Flannery, C.R.; et al. Matrix metalloproteinases are involved in C-terminal and interglobular domain processing of cartilage aggrecan in late stage cartilage degradation. Matrix Biol. 2002, 21, 271–288. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, C.C.; Flannery, C.R.; Little, C.B.; Melrose, J. Catabolism of Fibromodulin in Developmental Rudiment and Pathologic Articular Cartilage Demonstrates Novel Roles for MMP-13 and ADAMTS-4 in C-terminal Processing of SLRPs. Int. J. Mol. Sci. 2019, 20, 579. https://doi.org/10.3390/ijms20030579
Shu CC, Flannery CR, Little CB, Melrose J. Catabolism of Fibromodulin in Developmental Rudiment and Pathologic Articular Cartilage Demonstrates Novel Roles for MMP-13 and ADAMTS-4 in C-terminal Processing of SLRPs. International Journal of Molecular Sciences. 2019; 20(3):579. https://doi.org/10.3390/ijms20030579
Chicago/Turabian StyleShu, Cindy C, Carl R Flannery, Christopher B Little, and James Melrose. 2019. "Catabolism of Fibromodulin in Developmental Rudiment and Pathologic Articular Cartilage Demonstrates Novel Roles for MMP-13 and ADAMTS-4 in C-terminal Processing of SLRPs" International Journal of Molecular Sciences 20, no. 3: 579. https://doi.org/10.3390/ijms20030579
APA StyleShu, C. C., Flannery, C. R., Little, C. B., & Melrose, J. (2019). Catabolism of Fibromodulin in Developmental Rudiment and Pathologic Articular Cartilage Demonstrates Novel Roles for MMP-13 and ADAMTS-4 in C-terminal Processing of SLRPs. International Journal of Molecular Sciences, 20(3), 579. https://doi.org/10.3390/ijms20030579