The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer

 ,
,  ,
, 


Abstract
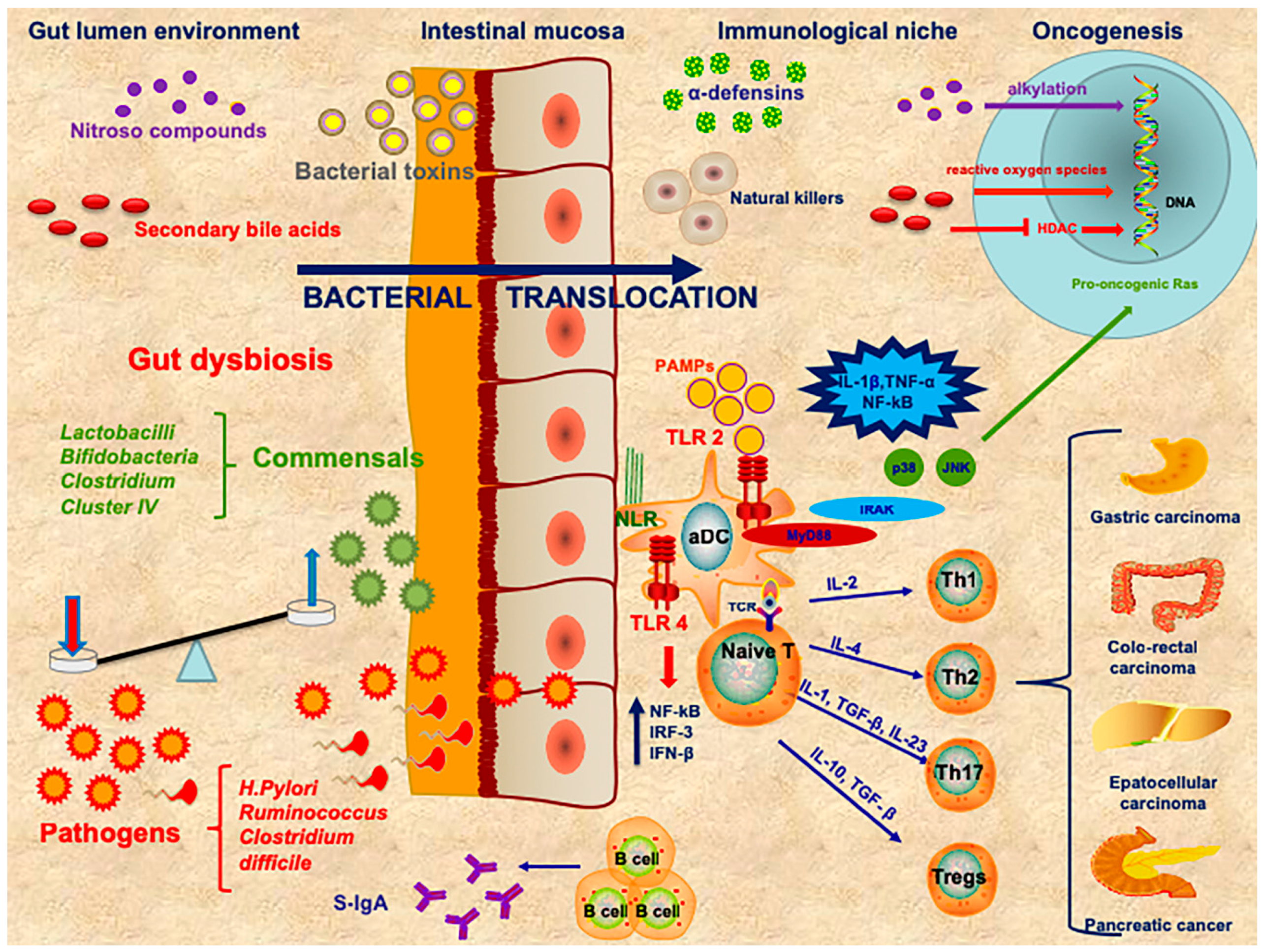
1. Introduction
2. Immune System and Cancer
3. The Role of Gut Microbiota in Cancer
4. Esophageal and Stomach Cancer
5. Colorectal Cancer
6. Hepatocellular Carcinoma
7. Pancreatic Cancer
8. The Role of Microbiota in Cancer Therapy
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cammarota, G.; Ianiro, G.; Cianci, R.; Bibbo, S.; Gasbarrini, A.; Curro, D. The involvement of gut microbiota in inflammatory bowel disease pathogenesis: Potential for therapy. Pharmacol. Ther. 2015, 149, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Lopetuso, L.R.; Petito, V.; Graziani, C.; Schiavoni, E.; Paroni Sterbini, F.; Poscia, A.; Gaetani, E.; Franceschi, F.; Cammarota, G.; Sanguinetti, M.; et al. Gut Microbiota in Health, Diverticular Disease, Irritable Bowel Syndrome, and Inflammatory Bowel Diseases: Time for Microbial Marker of Gastrointestinal Disorders. Dig. Dis. 2018, 36, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Bibbo, S.; Ianiro, G.; Dore, M.P.; Simonelli, C.; Newton, E.E.; Cammarota, G. Gut Microbiota as a Driver of Inflammation in Nonalcoholic Fatty Liver Disease. Mediat. Inflamm. 2018, 2018, 9321643. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Lonardo, A.; Byrne, C.D. Nonalcoholic fatty liver disease and chronic vascular complications of diabetes mellitus. Nat. Reviews. Endocrinol. 2018, 14, 99–114. [Google Scholar] [CrossRef]
- Pagliari, D.; Saviano, A.; Newton, E.E.; Serricchio, M.L.; Dal Lago, A.A.; Gasbarrini, A.; Cianci, R. Gut Microbiota-Immune System Crosstalk and Pancreatic Disorders. Mediat. Inflamm. 2018, 2018, 7946431. [Google Scholar] [CrossRef] [PubMed]
- Pragman, A.A.; Lyu, T.; Baller, J.A.; Gould, T.J.; Kelly, R.F.; Reilly, C.S.; Isaacson, R.E.; Wendt, C.H. The lung tissue microbiota of mild and moderate chronic obstructive pulmonary disease. Microbiome 2018, 6, 7. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.M.; Weiner, H.L. Microbiota Signaling Pathways that Influence Neurologic Disease. Neurother. J. Am. Soc. Exp. Neurother. 2018, 15, 135–145. [Google Scholar] [CrossRef]
- Abdallah, F.; Mijouin, L.; Pichon, C. Skin Immune Landscape: Inside and Outside the Organism. Mediat. Inflamm. 2017, 2017, 5095293. [Google Scholar] [CrossRef]
- York, A. Microbiome: Gut microbiota sways response to cancer immunotherapy. Nat. Reviews. Microbiol. 2018. [Google Scholar] [CrossRef]
- Mao, Q.; Jiang, F.; Yin, R.; Wang, J.; Xia, W.; Dong, G.; Ma, W.; Yang, Y.; Xu, L.; Hu, J. Interplay between the lung microbiome and lung cancer. Cancer Lett. 2018, 415, 40–48. [Google Scholar] [CrossRef]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human oral microbiome and prospective risk for pancreatic cancer: A population-based nested case-control study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Pagliari, D.; Gambassi, G.; Piccirillo, C.A.; Cianci, R. The Intricate Link among Gut “Immunological Niche,” Microbiota, and Xenobiotics in Intestinal Pathology. Mediat. Inflamm. 2017, 2017, 8390595. [Google Scholar] [CrossRef] [PubMed]
- Al-Asmakh, M.; Zadjali, F. Use of Germ-Free Animal Models in Microbiota-Related Research. J. Microbiol. Biotechnol. 2015, 25, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Geem, D.; Medina-Contreras, O.; McBride, M.; Newberry, R.D.; Koni, P.A.; Denning, T.L. Specific microbiota-induced intestinal Th17 differentiation requires MHC class II but not GALT and mesenteric lymph nodes. J. Immunol. 2014, 193, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.; Kim, N. The Effect of Microbiota on Colon Carcinogenesis. J. Cancer Prev. 2018, 23, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Cianci, R.; Pagliari, D.; Pietroni, V.; Landolfi, R.; Pandolfi, F. Tissue infiltrating lymphocytes: The role of cytokines in their growth and differentiation. J. Biol. Regul. Homeost. Agents 2010, 24, 239–249. [Google Scholar] [PubMed]
- Pagliari, D.; Piccirillo, C.A.; Larbi, A.; Cianci, R. The Interactions between Innate Immunity and Microbiota in Gastrointestinal Diseases. J. Immunol. Res. 2015, 2015, 898297. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, G.L.V.; Leite, A.Z.; Higuchi, B.S.; Gonzaga, M.I.; Mariano, V.S. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 2017, 152, 1–12. [Google Scholar] [CrossRef]
- Weiss, G.A.; Hennet, T. Mechanisms and consequences of intestinal dysbiosis. Cell. Mol. Life Sci. 2017, 74, 2959–2977. [Google Scholar] [CrossRef]
- Zeng, M.Y.; Inohara, N.; Nunez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017, 10, 18–26. [Google Scholar] [CrossRef]
- Mondot, S.; de Wouters, T.; Dore, J.; Lepage, P. The human gut microbiome and its dysfunctions. Dig. Dis. 2013, 31, 278–285. [Google Scholar] [CrossRef]
- Tetel, M.J.; de Vries, G.J.; Melcangi, R.C.; Panzica, G.; O’Mahony, S.M. Steroids, Stress, and the Gut Microbiome-Brain Axis. J. Neuroendocrinol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Reddavide, R.; Rotolo, O.; Caruso, M.G.; Stasi, E.; Notarnicola, M.; Miraglia, C.; Nouvenne, A.; Meschi, T.; De’ Angelis, G.L.; Di Mario, F.; et al. The role of diet in the prevention and treatment of Inflammatory Bowel Diseases. Acta Bio-Med. Atenei Parm. 2018, 89, 60–75. [Google Scholar] [CrossRef]
- Satitsuksanoa, P.; Jansen, K.; Globinska, A.; van de Veen, W.; Akdis, M. Regulatory Immune Mechanisms in Tolerance to Food Allergy. Front. Immunol. 2018, 9, 2939. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A., Jr. The immune system evolved to discriminate infectious nonself from noninfectious self. Immunol. Today 1992, 13, 11–16. [Google Scholar] [CrossRef]
- Kobe, B.; Deisenhofer, J. A structural basis of the interactions between leucine-rich repeats and protein ligands. Nature 1995, 374, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [PubMed]
- Menendez, A.; Willing, B.P.; Montero, M.; Wlodarska, M.; So, C.C.; Bhinder, G.; Vallance, B.A.; Finlay, B.B. Bacterial stimulation of the TLR-MyD88 pathway modulates the homeostatic expression of ileal Paneth cell alpha-defensins. J. Innate Immun. 2013, 5, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Schnare, M.; Barton, G.M.; Holt, A.C.; Takeda, K.; Akira, S.; Medzhitov, R. Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2001, 2, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Rakoff-Nahoum, S.; Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007, 317, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Apidianakis, Y.; Pitsouli, C.; Perrimon, N.; Rahme, L. Synergy between bacterial infection and genetic predisposition in intestinal dysplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 20883–20888. [Google Scholar] [CrossRef] [PubMed]
- Dzutsev, A.; Goldszmid, R.S.; Viaud, S.; Zitvogel, L.; Trinchieri, G. The role of the microbiota in inflammation, carcinogenesis, and cancer therapy. Eur. J. Immunol. 2015, 45, 17–31. [Google Scholar] [CrossRef] [PubMed]
- von Frieling, J.; Fink, C.; Hamm, J.; Klischies, K.; Forster, M.; Bosch, T.C.G.; Roeder, T.; Rosenstiel, P.; Sommer, F. Grow With the Challenge—Microbial Effects on Epithelial Proliferation, Carcinogenesis, and Cancer Therapy. Front. Microbiol. 2018, 9, 2020. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Niccolai, E.; Ricci, F.; Russo, E.; Nannini, G.; Emmi, G.; Taddei, A.; Ringressi, M.N.; Melli, F.; Miloeva, M.; Cianchi, F.; et al. The Different Functional Distribution of “Not Effector” T Cells (Treg/Tnull) in Colorectal Cancer. Front. Immunol. 2017, 8, 1900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wen, B.; Anton, O.M.; Yao, Z.; Dubois, S.; Ju, W.; Sato, N.; DiLillo, D.J.; Bamford, R.N.; Ravetch, J.V. IL-15 enhanced antibody-dependent cellular cytotoxicity mediated by NK cells and macrophages. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Cianci, R.; Pagliari, D.; Landolfi, R.; Cammarota, G. Cellular mediators of inflammation: Tregs and TH17 cells in gastrointestinal diseases. Mediat. Inflamm. 2009, 2009, 132028. [Google Scholar] [CrossRef]
- Shahmarvand, N.; Nagy, A.; Shahryari, J.; Ohgami, R.S. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018, 109, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Hurtado, C.G.; Wan, F.; Housseau, F.; Sears, C.L. Roles for Interleukin 17 and Adaptive Immunity in Pathogenesis of Colorectal Cancer. Gastroenterology 2018. [Google Scholar] [CrossRef] [PubMed]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Microbiome, inflammation, and cancer. Cancer J. 2014, 20, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Cianci, R.; Pagliari, D.; Casciano, F.; Bagala, C.; Astone, A.; Landolfi, R.; Barone, C. The immune response to tumors as a tool toward immunotherapy. Clin. Dev. Immunol. 2011, 2011, 894704. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Wei, S.; Szeliga, W.; Vatan, L.; Zou, W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 2009, 114, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 2018, 175, 1780–1795. [Google Scholar] [CrossRef] [PubMed]
- McCaw, T.R.; Li, M.; Starenki, D.; Cooper, S.J. The expression of MHC class II molecules on murine breast tumors delays T-cell exhaustion, expands the T-cell repertoire, and slows tumor growth. Cancer Immunol. Immunother. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Godec, J.; Wolski, D.; Adland, E.; Yates, K.; Pauken, K.E.; Cosgrove, C.; Ledderose, C.; Junger, W.G.; Robson, S.C.; et al. CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog. 2015, 11, e1005177. [Google Scholar] [CrossRef]
- Bhatt, A.P.; Redinbo, M.R.; Bultman, S.J. The role of the microbiome in cancer development and therapy. A Cancer J. Clin. 2017, 67, 326–344. [Google Scholar] [CrossRef]
- Vannucci, L.; Stepankova, R.; Kozakova, H.; Fiserova, A.; Rossmann, P.; Tlaskalova-Hogenova, H. Colorectal carcinogenesis in germ-free and conventionally reared rats: Different intestinal environments affect the systemic immunity. Int. J. Oncol. 2008, 32, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.; Li, J.V.; Lahti, L. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Fang, L.; Lee, M.H. Dysbiosis of gut microbiota in promoting the development of colorectal cancer. Gastroenterol. Rep. 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Brownawell, A.M.; Caers, W.; Gibson, G.R.; Kendall, C.W.; Lewis, K.D.; Ringel, Y.; Slavin, J.L. Prebiotics and the health benefits of fiber: Current regulatory status, future research, and goals. J. Nutr. 2012, 142, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Youssef, O.; Lahti, L.; Kokkola, A.; Karla, T.; Tikkanen, M.; Ehsan, H.; Carpelan-Holmstrom, M.; Koskensalo, S.; Bohling, T.; Rautelin, H.; et al. Stool Microbiota Composition Differs in Patients with Stomach, Colon, and Rectal Neoplasms. Dig. Dis. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, K.J.; Cobiac, L.; Le Leu, R.K.; Van der Hoek, M.B.; Michael, M.Z. Histone deacetylase inhibition in colorectal cancer cells reveals competing roles for members of the oncogenic miR-17-92 cluster. Mol. Carcinog. 2013, 52, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Wu, S.; Yan, C.; Zhao, C.; Jin, H.; Yan, N.; Xu, J.; Wu, Y.; Li, C.; Shao, Q.; et al. Butyrate upregulates the TLR4 expression and the phosphorylation of MAPKs and NK-kappaB in colon cancer cell in vitro. Oncol. Lett. 2018, 16, 4439–4447. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Inflammation-Modulating Effect of Butyrate in the Prevention of Colon Cancer by Dietary Fiber. Clin. Colorectal Cancer 2018, 17, e541–e544. [Google Scholar] [CrossRef]
- Tylichova, Z.; Strakova, N.; Vondracek, J.; Vaculova, A.H.; Kozubik, A.; Hofmanova, J. Activation of autophagy and PPARgamma protect colon cancer cells against apoptosis induced by interactive effects of butyrate and DHA in a cell type-dependent manner: The role of cell differentiation. J. Nutr. Biochem. 2017, 39, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Chu, H. Host gene-microbiome interactions: Molecular mechanisms in inflammatory bowel disease. Genome Med. 2017, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Rea, D.; Coppola, G.; Palma, G.; Barbieri, A.; Luciano, A.; Del Prete, P.; Rossetti, S.; Berretta, M.; Facchini, G.; Perdona, S.; et al. Microbiota effects on cancer: From risks to therapies. Oncotarget 2018, 9, 17915–17927. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Tilg, H.; Gasbarrini, A. Antibiotics as deep modulators of gut microbiota: Between good and evil. Gut 2016, 65, 1906–1915. [Google Scholar] [CrossRef] [PubMed]
- Boursi, B.; Mamtani, R.; Haynes, K.; Yang, Y.X. Recurrent antibiotic exposure may promote cancer formation—Another step in understanding the role of the human microbiota? Eur. J. Cancer 2015, 51, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chaudhary, N.; Baghdadi, J.; Pei, Z. Microbiome in reflux disorders and esophageal adenocarcinoma. Cancer J. 2014, 20, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Vogiatzi, P.; Cassone, M.; Luzzi, I.; Lucchetti, C.; Otvos, L., Jr.; Giordano, A. Helicobacter pylori as a class I carcinogen: Physiopathology and management strategies. J. Cell. Biochem. 2007, 102, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Begum, S.; Sano, T.; Endo, H.; Kawamata, H.; Urakami, Y. Mucosal change of the stomach with low-grade mucosa-associated lymphoid tissue lymphoma after eradication of Helicobacter pylori: Follow-up study of 48 cases. J. Med Investig. 2000, 47, 36–46. [Google Scholar]
- Cammarota, G.; Fedeli, P.; Bianchi, A.; Cianci, R.; Martino, A.; Fedeli, G.; Gasbarrini, G. Regression of EI2-stage low-grade gastric MALT-lymphoma after H. pylori eradication. Hepato-Gastroenterol. 2005, 52, 975–977. [Google Scholar]
- Wang, L.; Zhou, J.; Xin, Y.; Geng, C.; Tian, Z.; Yu, X.; Dong, Q. Bacterial overgrowth and diversification of microbiota in gastric cancer. Eur. J. Gastroenterol. Hepatol. 2016, 28, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.; Wong, S.H.; Chen, Z.; Sung, J.J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef] [PubMed]
- Rubenstein, J.H.; Inadomi, J.M.; Scheiman, J.; Schoenfeld, P.; Appelman, H.; Zhang, M.; Metko, V.; Kao, J.Y. Association between Helicobacter pylori and Barrett’s esophagus, erosive esophagitis, and gastroesophageal reflux symptoms. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2014, 12, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kono, K.; Kawaida, H.; Takahashi, A.; Sugai, H.; Mimura, K.; Miyagawa, N.; Omata, H.; Fujii, H. CD4(+)CD25high regulatory T cells increase with tumor stage in patients with gastric and esophageal cancers. Cancer Immunol. Immunother. Cii 2006, 55, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, F.; Kono, K.; Takahashi, A.; Kawaida, H.; Sugai, H.; Fujii, H. Increased populations of regulatory T cells in peripheral blood and tumor-infiltrating lymphocytes in patients with gastric and esophageal cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 4404–4408. [Google Scholar]
- Chen, J.; Domingue, J.C.; Sears, C.L. Microbiota dysbiosis in select human cancers: Evidence of association and causality. Semin. Immunol. 2017, 32, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y. The Role of the Gut Microbiome in Colorectal Cancer. Clin. Colon Rectal Surg. 2018, 31, 192–198. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [PubMed]
- Gagnière, J.; Raisch, J.; Veziant, J.; Barnich, N.; Bonnet, R.; Buc, E.; Bringer, M.-A.; Pezet, D.; Bonnet, M. Gut microbiota imbalance and colorectal cancer. World J. Gastroenterol. 2016, 22, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.S.; Recco, R.A.; Catalano, M.T.; Edberg, S.C.; Casey, J.I.; Steigbigel, N.H. Association of Streptococcus bovis with carcinoma of the colon. N. Engl. J. Med. 1977, 297, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Zumkeller, N.; Brenner, H.; Zwahlen, M.; Rothenbacher, D. Helicobacter pylori infection and colorectal cancer risk: A meta-analysis. Helicobacter 2006, 11, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Housseau, F.; Sears, C.L. Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/−) mice: A human commensal-based murine model of colon carcinogenesis. Cell Cycle 2010, 9, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Balamurugan, R.; Rajendiran, E.; George, S.; Samuel, G.V.; Ramakrishna, B.S. Real-time polymerase chain reaction quantification of specific butyrate-producing bacteria, Desulfovibrio and Enterococcus faecalis in the feces of patients with colorectal cancer. J. Gastroenterol. Hepatol. 2008, 23, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Kwong, T.N.Y.; Wang, X.; Nakatsu, G.; Chow, T.C.; Tipoe, T.; Dai, R.Z.W.; Tsoi, K.K.K.; Wong, M.C.S.; Tse, G.; Chan, M.T.V.; et al. Association Between Bacteremia From Specific Microbes and Subsequent Diagnosis of Colorectal Cancer. Gastroenterology 2018, 155, 383–390.e388. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.A.; Khan, Z.; Malik, A.; Kalam, M.A.; Cash, P.; Ashraf, M.T.; Alshamsan, A. Colorectal cancer-inflammatory bowel disease nexus and felony of Escherichia coli. Life Sci. 2017, 180, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Sears, C.L. Enterotoxigenic Bacteroides fragilis: A rogue among symbiotes. Clin. Microbiol. Rev. 2009, 22, 349–369. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Rhee, K.J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Boleij, A.; Tjalsma, H. The itinerary of Streptococcus gallolyticus infection in patients with colonic malignant disease. Lancet Infect. Dis. 2013, 13, 719–724. [Google Scholar] [CrossRef]
- Zhou, Y.; He, H.; Xu, H.; Li, Y.; Li, Z.; Du, Y.; He, J.; Zhou, Y.; Wang, H.; Nie, Y. Association of oncogenic bacteria with colorectal cancer in South China. Oncotarget 2016, 7, 80794–80802. [Google Scholar] [CrossRef]
- Tjalsma, H.; Boleij, A.; Marchesi, J.R.; Dutilh, B.E. A bacterial driver-passenger model for colorectal cancer: Beyond the usual suspects. Nat. Rev. Microbiol. 2012, 10, 575–582. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, C.V.; Taddei, A.; Amedei, A. The controversial role of Enterococcus faecalis in colorectal cancer. Ther. Adv. Gastroenterol. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Adolph, T.E.; Grander, C.; Moschen, A.R.; Tilg, H. Liver-Microbiome Axis in Health and Disease. Trends Immunol. 2018, 39, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Akram, N.; Imran, M.; Noreen, M.; Ahmed, F.; Atif, M.; Fatima, Z.; Bilal Waqar, A. Oncogenic Role of Tumor Viruses in Humans. Viral Immunol. 2017, 30, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Rongrui, L.; Na, H.; Zongfang, L.; Fanpu, J.; Shiwen, J. Epigenetic mechanism involved in the HBV/HCV-related hepatocellular carcinoma tumorigenesis. Curr. Pharm. Des. 2014, 20, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.S.; Cho, Y.G.; Kang, B.S.; Lester, I.M.; Hahn, Y.S. The HCV core protein acts as a positive regulator of fas-mediated apoptosis in a human lymphoblastoid T cell line. Virology 2000, 276, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Feng, Y.; Theve, E.J.; Raczynski, A.R.; Fiala, J.L.; Doernte, A.L.; Williams, M.; McFaline, J.L.; Essigmann, J.M.; Schauer, D.B.; et al. Gut microbes define liver cancer risk in mice exposed to chemical and viral transgenic hepatocarcinogens. Gut 2010, 59, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Schwabe, R.F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 527–539. [Google Scholar] [CrossRef]
- Mima, K.; Nakagawa, S.; Sawayama, H.; Ishimoto, T.; Imai, K.; Iwatsuki, M.; Hashimoto, D.; Baba, Y.; Yamashita, Y.I.; Yoshida, N.; et al. The microbiome and hepatobiliary-pancreatic cancers. Cancer Lett. 2017, 402, 9–15. [Google Scholar] [CrossRef]
- Krüttgen, A.; Horz, H.-P.; Weber-Heynemann, J.; Vucur, M.; Trautwein, C.; Haase, G.; Luedde, T.; Roderburg, C. Study on the association of helicobacter species with viral hepatitis-induced hepatocellular carcinoma. Gut Microbes 2012, 3, 228–233. [Google Scholar] [CrossRef]
- Sandler, N.G.; Koh, C.; Roque, A.; Eccleston, J.L.; Siegel, R.B.; Demino, M.; Kleiner, D.E.; Deeks, S.G.; Liang, T.J.; Heller, T.; et al. Host response to translocated microbial products predicts outcomes of patients with HBV or HCV infection. Gastroenterology 2011, 141, 1220–1230. [Google Scholar] [CrossRef] [PubMed]
- Grąt, M.; Wronka, K.M.; Krasnodębski, M.; Masior, Ł.; Lewandowski, Z.; Kosińska, I.; Grąt, K.; Stypułkowski, J.; Rejowski, S.; Wasilewicz, M.; et al. Profile of Gut Microbiota Associated With the Presence of Hepatocellular Cancer in Patients With Liver Cirrhosis. Transplant. Proc. 2016, 48, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Diehl, A.M. Nonalcoholic Fatty Liver Disease and the Gut Microbiome. Clin. Liver Dis. 2016, 20, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Brandi, G.; De Lorenzo, S.; Candela, M.; Pantaleo, M.A.; Bellentani, S.; Tovoli, F.; Saccoccio, G.; Biasco, G. Microbiota, NASH, HCC and the potential role of probiotics. Carcinogenesis 2017, 38, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Han, D.; Xu, R.; Li, S.; Wu, H.; Qu, C.; Wang, F.; Wang, X.; Zhao, Y. A Model of Metabolic Syndrome and Related Diseases with Intestinal Endotoxemia in Rats Fed a High Fat and High Sucrose Diet. PLoS ONE 2014, 9, e115148. [Google Scholar] [CrossRef] [PubMed]
- Lowe, P.P.; Gyongyosi, B.; Satishchandran, A.; Iracheta-Vellve, A.; Ambade, A.; Kodys, K.; Catalano, D.; Ward, D.V.; Szabo, G. Alcohol-related changes in the intestinal microbiome influence neutrophil infiltration, inflammation and steatosis in early alcoholic hepatitis in mice. PLoS ONE 2017, 12, e0174544. [Google Scholar] [CrossRef]
- Hartmann, P.; Seebauer, C.T.; Schnabl, B. Alcoholic liver disease: The gut microbiome and liver cross talk. Alcohol. Clin. Exp. Res. 2015, 39, 763–775. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.X.; Huang, C.L.; Luo, S.Z.; Zhang, X.M.; Zeng, Y.; Lu, Y.Y. Characterization of the duodenal bacterial microbiota in patients with pancreatic head cancer vs. healthy controls. Pancreatol. Off. J. Int. Assoc. Pancreatol. 2018, 18, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, H.; Bording-Jorgensen, M.; Dijk, S.; Wine, E. The Complex Interplay between Chronic Inflammation, the Microbiome, and Cancer: Understanding Disease Progression and What We Can Do to Prevent It. Cancers 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef]
- Huang, H.; Daniluk, J.; Liu, Y.; Chu, J.; Li, Z.; Ji, B.; Logsdon, C.D. Oncogenic K-Ras requires activation for enhanced activity. Oncogene 2014, 33, 532–535. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Bai, C.; Brown, T.D.; Hood, L.E.; Tian, Q. Human Gut Microbiota and Gastrointestinal Cancer. Genom. Proteom. Bioinform. 2018, 16, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Chen, Y.T.; Wang, R.; Chen, X.Z. Helicobacter pylori infection, atrophic gastritis, and pancreatic cancer risk: A meta-analysis of prospective epidemiologic studies. Medicine 2017, 96, e7811. [Google Scholar] [CrossRef] [PubMed]
- Ertz-Archambault, N.; Keim, P.; Von Hoff, D. Microbiome and pancreatic cancer: A comprehensive topic review of literature. World J. Gastroenterol. 2017, 23, 1899–1908. [Google Scholar] [CrossRef]
- Koh, G.Y.; Kane, A.; Lee, K.; Xu, Q.; Wu, X.; Roper, J.; Mason, J.B.; Crott, J.W. Parabacteroides distasonis attenuates toll-like receptor 4 signaling and Akt activation and blocks colon tumor formation in high-fat diet-fed azoxymethane-treated mice. Int. J. Cancer 2018. [Google Scholar] [CrossRef]
- Geller, L.T.; Straussman, R. Intratumoral bacteria may elicit chemoresistance by metabolizing anticancer agents. Mol. Cell. Oncol. 2018, 5, e1405139. [Google Scholar] [CrossRef]
- Michaud, D.S.; Izard, J.; Wilhelm-Benartzi, C.S.; You, D.-H.; Grote, V.A.; Tjønneland, A.; Dahm, C.C.; Overvad, K.; Jenab, M.; Fedirko, V.; et al. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut 2013, 62, 1764–1770. [Google Scholar] [CrossRef] [PubMed]
- Geller, L.T.; Barzily-Rokni, M. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Vetizou, M.; Trinchieri, G. Anti-PD1 in the wonder-gut-land. Cell Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Wilson, I.D.; Teare, J.; Marchesi, J.R.; Nicholson, J.K.; Kinross, J.M. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 356. [Google Scholar] [CrossRef] [PubMed]
- Wardill, H.R.; Bowen, J.M. Chemotherapy-induced mucosal barrier dysfunction: An updated review on the role of intestinal tight junctions. Curr. Opin. Supportive Palliat. Care 2013, 7, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Trinchieri, G. Microbiota: A key orchestrator of cancer therapy. Nat. Rev. Cancer 2017, 17, 271. [Google Scholar] [CrossRef] [PubMed]
- Lehouritis, P.; Cummins, J.; Stanton, M.; Murphy, C.T.; McCarthy, F.O.; Reid, G.; Urbaniak, C.; Byrne, W.L.; Tangney, M. Local bacteria affect the efficacy of chemotherapeutic drugs. Sci. Rep. 2015, 5, 14554. [Google Scholar] [CrossRef] [PubMed]
- Daillere, R.; Vetizou, M.; Waldschmitt, N.; Yamazaki, T.; Isnard, C.; Poirier-Colame, V.; Duong, C.P.M.; Flament, C.; Lepage, P.; Roberti, M.P.; et al. Enterococcus hirae and Barnesiella intestinihominis Facilitate Cyclophosphamide-Induced Therapeutic Immunomodulatory Effects. Immunity 2016, 45, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Guyton, K.; Alverdy, J.C. The gut microbiota and gastrointestinal surgery. Nat. Rev. Gastroenterol. Hepatol. 2016, 14, 43. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Dummer, R. The 2017 complete overhaul of adjuvant therapies for high-risk melanoma and its consequences for staging and management of melanoma patients. Eur. J. Cancer 2017, 86, 101–105. [Google Scholar] [CrossRef]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Farkona, S.; Diamandis, E.P.; Blasutig, I.M. Cancer immunotherapy: The beginning of the end of cancer? Bmc Med. 2016, 14, 73. [Google Scholar] [CrossRef] [PubMed]
- Carbonnel, F.; Soularue, E.; Coutzac, C.; Chaput, N.; Mateus, C.; Lepage, P.; Robert, C. Inflammatory bowel disease and cancer response due to anti-CTLA-4: Is it in the flora? Semin. Immunopathol. 2017, 39, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.; Tenore, G.C.; Novellino, E. Probiotic species in the modulation of the anticancer immune response. Semin. Cancer Biol. 2017, 46, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Dubin, K.; Callahan, M.K.; Ren, B.; Khanin, R.; Viale, A.; Ling, L.; No, D.; Gobourne, A.; Littmann, E.; Huttenhower, C.; et al. Intestinal microbiome analyses identify melanoma patients at risk for checkpoint-blockade-induced colitis. Nat. Commun. 2016, 7, 10391. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Man Lei, Y.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Amedei, A.; Boem, F. I’ve Gut A Feeling: Microbiota Impacting the Conceptual and Experimental Perspectives of Personalized Medicine. Int. J. Mol. Sci. 2018, 19, 3756. [Google Scholar] [CrossRef] [PubMed]
- Castro, C.; Peleteiro, B.; Lunet, N. Modifiable factors and esophageal cancer: A systematic review of published meta-analyses. J. Gastroenterol. 2018, 53, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Ladinsky, M.S. Gut microbiota utilize immunoglobulin A for mucosal colonization. Science 2018, 360, 795–800. [Google Scholar] [CrossRef] [PubMed]
- Browne, H.P.; Neville, B.A.; Forster, S.C.; Lawley, T.D. Transmission of the gut microbiota: Spreading of health. Nat. Rev. Microbiol. 2017, 15, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Gamage, S.M.K.; Dissabandara, L.; Lam, A.K.; Gopalan, V. The role of heme iron molecules derived from red and processed meat in the pathogenesis of colorectal carcinoma. Crit. Rev. Oncol. Hematol. 2018, 126, 121–128. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Site | Effect | Mechanism | References | |
|---|---|---|---|---|
| Neisseria elongate | Oral | ↓↓↓ pancreatic tumor | Promotes oral homeostasis. | [121] |
| Streptococcus mitis | Oral | ↓↓↓ pancreatic tumor | Promotes oral homeostasis. | [121] |
| Porphyromonas gingivalis (strain ATCC 53978) | Oral | ↑↑↑ pancreatic tumor | Promotes oral dysbiosis and inflammation. | [121] |
| Helicobacter pylori | Stomach, liver, intestine | ↑↑↑ gastric liver pancreatic colorectal tumor; ↓↓↓ esophageal tumor | Immune-modulating effect through Th17 pathway; promoting factor for dysbiosis; not clear protective properties in esophageal tumor. | [67,80,97,111,142] |
| Helicobacter hepaticus | Liver | ↑↑↑ liver tumor | Directly damages DNA, through WNT and NF-κB signaling pathways in tumor cells; suppresses intra-tumor immunity in aflatoxin- and hepatitis C virus-induced HCC. | [97,100,101] |
| Streptococcus bovis | Intestine | ↑↑↑ colorectal tumor | Immune-modulating effect; symbiotic relation with tumor cells. | [79,89] |
| Bacteroidesfragilis | Intestine | ↑↑↑ progression colorectal tumor | Immune-modulating effect through TH17 pathway; promotion of WNT, NF-κB and STS-3 pathways; direct effect of BFT toxin. | [87,88,134,143] |
| Enterococcus faecalis | Intestine | ↑↑↑ colorectal tumor | Inflammatory effect through ROS production; increases risk of epithelial damage | [82,92] |
| Clostridium septicum | Intestine | ↑↑↑ colorectal tumor | Inflammatory effect; increases risk of infectious complications. | [83] |
| Fusobacterium spp. | Intestine | ↓↓↓ colorectal tumor; ↑↑↑ esophageal tumor. | Immune-modulating effect. Esophageal dysbiosis marker. | [66,84,144] |
| Escherichia coli | Intestine, pancreas | ↑↑↑ colorectal and liver tumor; ↓↓ pancreatic tumor | Direct epithelial invasion; production of nitrous compounds through eme-metabolism; promotes dysbiosis. | [85,86,102,145] |
| Lactobacillum spp. | Gastro intestinal apparatus | ↓↓↓↓ malignancies | Promotes gut homeostasis; anti-inflammatory effects. | [54,55,56,76,110] |
| Bifidobacter spp. | Gastro intestinal apparatus | ↓↓↓↓ malignancies; ↓↓ immunotherapy side-effects | Promotes gut homeostasis through competition with pathogens; anti-inflammatory effects. | [54,55,77,110] |
| Clostridium cluster IV | Gastro intestinal apparatus | ↓↓↓↓ malignancies | Promotes gut homeostasis; anti-inflammatory effects. | [55] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cianci, R.; Franza, L.; Schinzari, G.; Rossi, E.; Ianiro, G.; Tortora, G.; Gasbarrini, A.; Gambassi, G.; Cammarota, G. The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer. Int. J. Mol. Sci. 2019, 20, 501. https://doi.org/10.3390/ijms20030501
Cianci R, Franza L, Schinzari G, Rossi E, Ianiro G, Tortora G, Gasbarrini A, Gambassi G, Cammarota G. The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer. International Journal of Molecular Sciences. 2019; 20(3):501. https://doi.org/10.3390/ijms20030501
Chicago/Turabian StyleCianci, Rossella, Laura Franza, Giovanni Schinzari, Ernesto Rossi, Gianluca Ianiro, Giampaolo Tortora, Antonio Gasbarrini, Giovanni Gambassi, and Giovanni Cammarota. 2019. "The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer" International Journal of Molecular Sciences 20, no. 3: 501. https://doi.org/10.3390/ijms20030501
APA StyleCianci, R., Franza, L., Schinzari, G., Rossi, E., Ianiro, G., Tortora, G., Gasbarrini, A., Gambassi, G., & Cammarota, G. (2019). The Interplay between Immunity and Microbiota at Intestinal Immunological Niche: The Case of Cancer. International Journal of Molecular Sciences, 20(3), 501. https://doi.org/10.3390/ijms20030501