FKBP5 Gene Expression Predicts Antidepressant Treatment Outcome in Depression

 ,
,
Abstract

1. Introduction
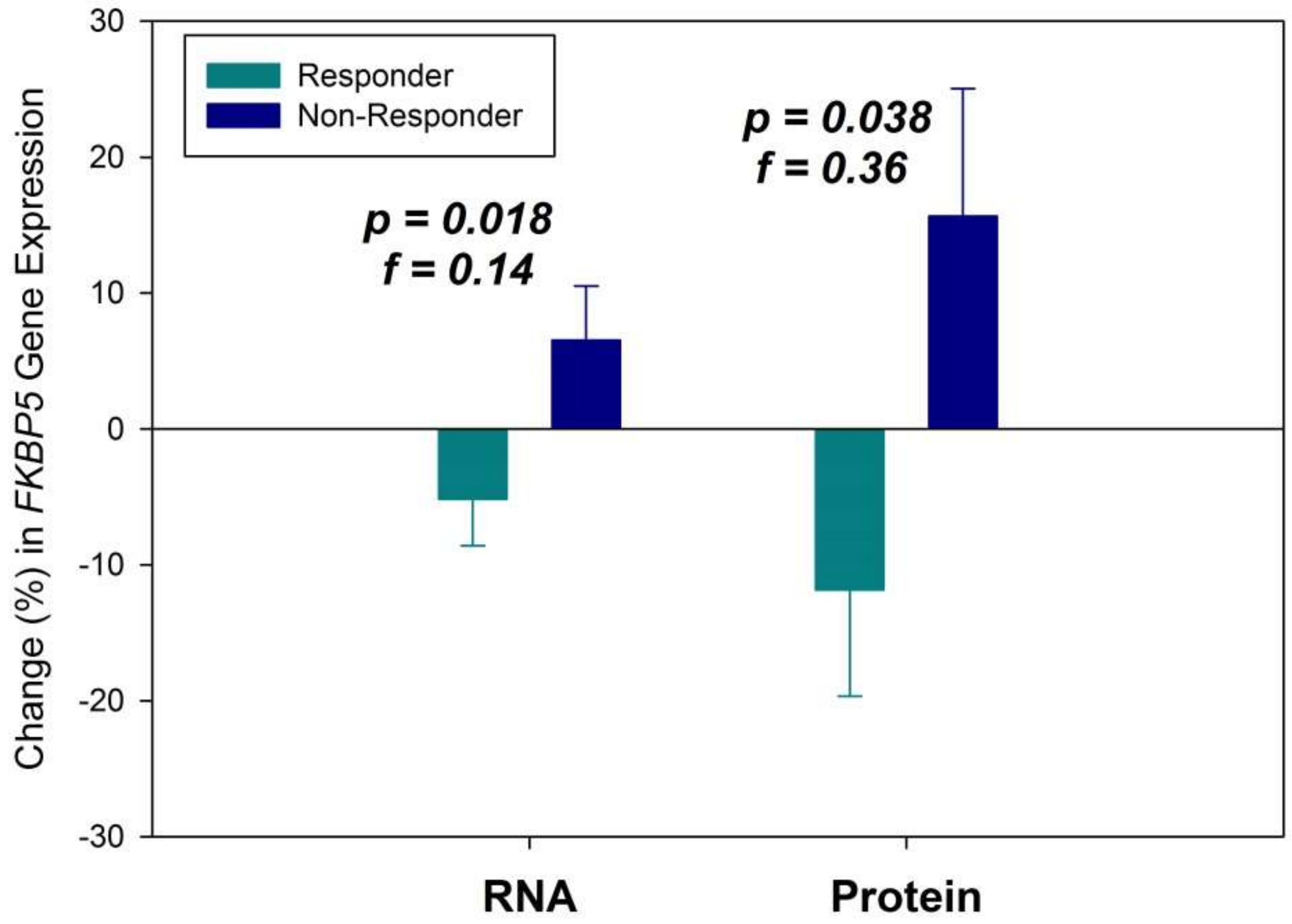
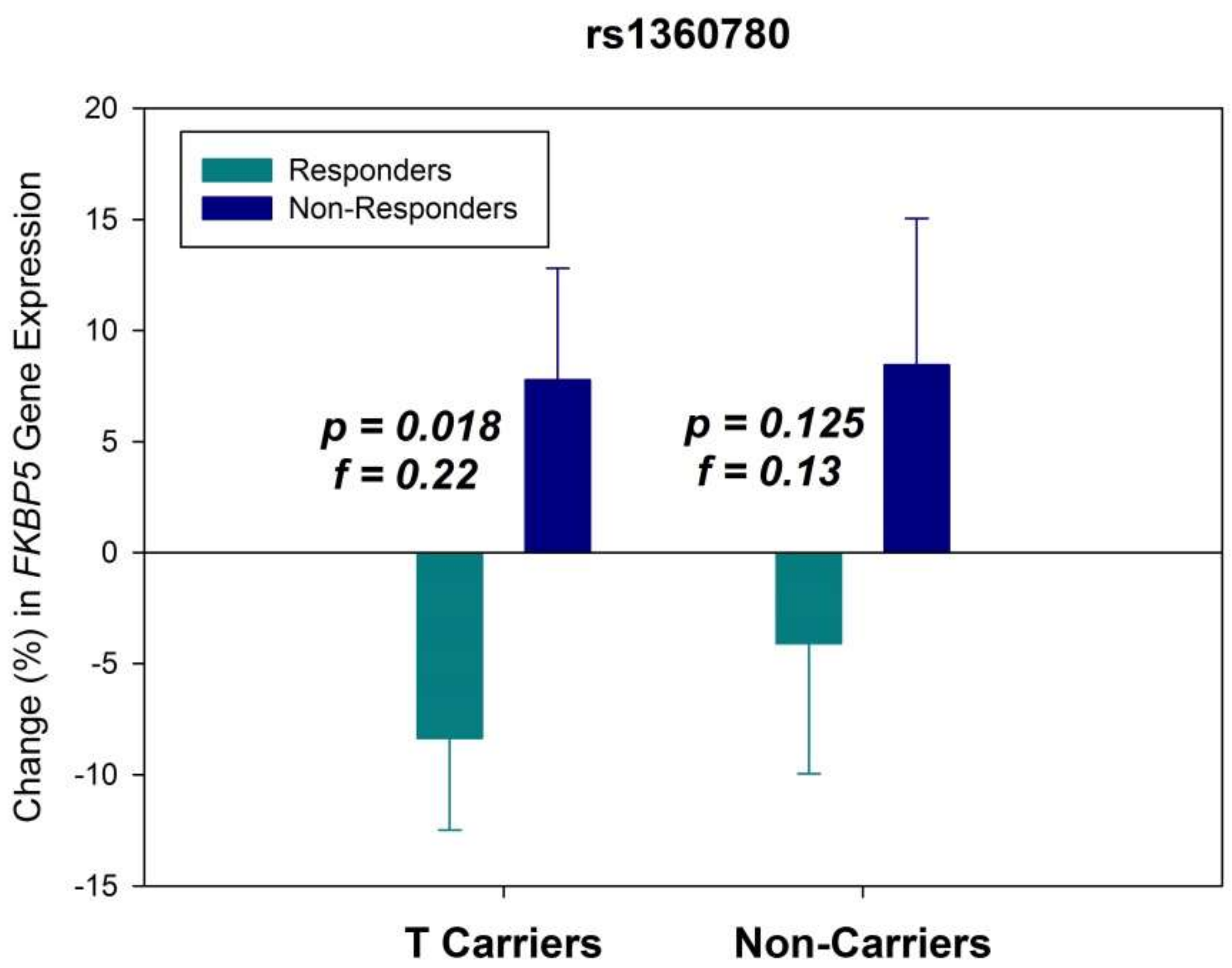
2. Results
3. Discussion
4. Materials and Methods
4.1. Sample Description and Study Protocol
4.2. Laboratory Analysis
4.3. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| CRH | Corticotropin releasing hormone |
| CpG | Cytosine-phosphate-guanine |
| Dex | Dexamethasone |
| DNA | Deoxyribonucleic acid |
| GR | Glucocorticoid receptor |
| FKBP5 | FK506-binding protein 5 gene |
| FKBP51 | FK506-binding protein 51 |
| HAMD-21 | Hamilton Depression Rating Scale, 21-items version |
| HPA | Hypothalamus–pituitary–adrenocortical |
| HSP90 | Heat-shock protein 90 |
| ICD10 | International Classification of Diseases, 10th revision |
| MARS | Munich Antidepressant Response Signature |
| OR | Odds ratio |
| RNA | Ribonucleic acid |
| RPPM | Reverse phase protein microarray |
| RT-PCR | Real-time polymerase-chain-reaction |
| SD | Standard deviation |
| SEM | Standard error of the mean |
| SNP | Single nucleotide polymorphism |
| SNRI | Selective serotonin noradrenalin reuptake inhibitor |
| SSRI | Selective serotonin reuptake inhibitor |
| TCA | Tricyclic antidepressant |
References
- Kessler, R.C.; Bromet, E.J. The epidemiology of depression across cultures. Annu. Rev. Public Health 2013, 34, 119–138. [Google Scholar] [CrossRef] [PubMed]
- Burcusa, S.L.; Iacono, W.G. Risk for recurrence in depression. Clin. Psychol. Rev. 2007, 27, 959–985. [Google Scholar] [CrossRef] [PubMed]
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef]
- Kendler, K.S.; Prescott, C.A.; Myers, J.; Neale, M.C. The structure of genetic and environmental risk factors for common psychiatric and substance use disorders in men and women. Arch. Gen. Psychiatry 2003, 60, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, M.; McInnis, M.G.; Zollner, S. Psychiatric genetics: Progress amid controversy. Nat. Rev. Genet. 2008, 9, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Hammen, C. Stress and depression. Annu. Rev. Clin. Psychol. 2005, 1, 293–319. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Newport, D.J.; Mletzko, T.; Miller, A.H.; Nemeroff, C.B. The link between childhood trauma and depression: Insights from HPA axis studies in humans. Psychoneuroendocrinology 2008, 33, 693–710. [Google Scholar] [PubMed]
- Kendler, K.S.; Karkowski, L.M.; Prescott, C.A. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry 1999, 156, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000, 23, 477–501. [Google Scholar] [CrossRef]
- Heuser, I.J.; Yassouridis, A.; Holsboer, F. The combined dexamethasone/CRH test: A refined laboratory test for psychiatric disorders. J. Psychiatr. Res. 1994, 28, 341–356. [Google Scholar] [CrossRef]
- Ising, M.; Künzel, H.E.; Binder, E.B.; Nickel, T.; Modell, S.; Holsboer, F. The combined dexamethasone/CRH test as a potential surrogate marker in depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2005, 29, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Ising, M.; Horstmann, S.; Kloiber, S.; Lucae, S.; Binder, E.B.; Kern, N.; Kunzel, H.E.; Pfennig, A.; Uhr, M.; Holsboer, F. Combined dexamethasone/corticotropin releasing hormone test predicts treatment response in major depression-a potential biomarker? Biol. Psychiatry 2007, 62, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Appelhof, B.C.; Huyser, J.; Verweij, M.; Brouwer, J.P.; van Dyck, R.; Fliers, E.; Hoogendijk, W.J.G.; Tijssen, J.G.P.; Wiersinga, W.M.; Schene, A.H. Glucocorticoids and Relapse of Major Depression (Dexamethasone/Corticotropin-Releasing Hormone Test in Relation to Relapse of Major Depression). Biol. Psychiatry 2006, 59, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Zobel, A.W.; Nickel, T.; Sonntag, A.; Uhr, M.; Holsboer, F.; Ising, M. Cortisol response in the combined dexamethasone/CRH test as predictor of relapse in patients with remitted depression: A prospective study. J. Psychiatr. Res. 2001, 35, 83–94. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Joels, M.; Holsboer, F. Stress and the brain: From adaptation to disease. Nat. Rev. Neurosci. 2005, 6, 463–475. [Google Scholar] [CrossRef] [PubMed]
- Criado-Marrero, M.; Rein, T.; Binder, E.B.; Porter, J.T.; Koren, J., III.; Blair, L.J. Hsp90 and FKBP51: Complex regulators of psychiatric diseases. Philos. Trans. R. Soc. Lond B Biol. Sci. 2018, 373, 20160532. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Morishima, Y.; Murphy, M.; Harrell, M. Chaperoning of glucocorticoid receptors. In Handbook of Experimental Pharmacology; Starke, K., Gaestel, M., Eds.; Springer: Heidelberg, Germany, 2006; Volume 172, pp. 111–138. [Google Scholar]
- Denny, W.B.; Valentine, D.L.; Reynolds, P.D.; Smith, D.F.; Scammell, J.G. Squirrel monkey immunophilin FKBP51 is a potent inhibitor of glucocorticoid receptor binding. Endocrinology, 2000; 141, 4107–4113. [Google Scholar] [CrossRef]
- Galigniana, N.M.; Ballmer, L.T.; Toneatto, J.; Erlejman, A.G.; Lagadari, M.; Galigniana, M.D. Regulation of the glucocorticoid response to stress-related disorders by the Hsp90-binding immunophilin FKBP51. J. Neurochem. 2012, 122, 4–18. [Google Scholar] [CrossRef]
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34 (Suppl. 1), S186–S195. [Google Scholar] [CrossRef]
- Fabbri, C.; Hosak, L.; Mossner, R.; Giegling, I.; Mandelli, L.; Bellivier, F.; Claes, S.; Collier, D.A.; Corrales, A.; Delisi, L.E.; et al. Consensus paper of the WFSBP Task Force on Genetics: Genetics, epigenetics and gene expression markers of major depressive disorder and antidepressant response. World J. Biol. Psychiatry 2017, 18, 5–28. [Google Scholar] [CrossRef]
- Wang, Q.; Shelton, R.C.; Dwivedi, Y. Interaction between early-life stress and FKBP5 gene variants in major depressive disorder and post-traumatic stress disorder: A systematic review and meta-analysis. J. Affect. Disord. 2018, 225, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, P.; Bruckl, T.; Nocon, A.; Pfister, H.; Binder, E.B.; Uhr, M.; Lieb, R.; Moffitt, T.E.; Caspi, A.; Holsboer, F.; et al. Interaction of FKBP5 gene variants and adverse life events in predicting depression onset: Results from a 10-year prospective community study. Am. J. Psychiatry 2011, 168, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Binder, E.B.; Bradley, R.G.; Liu, W.; Epstein, M.P.; Deveau, T.C.; Mercer, K.B.; Tang, Y.; Gillespie, C.F.; Heim, C.M.; Nemeroff, C.B.; et al. Association of FKBP5 polymorphisms and childhood abuse with risk of posttraumatic stress disorder symptoms in adults. JAMA 2008, 299, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, A.; Gennarelli, M.; Uher, R.; Breen, G.; Farmer, A.; Aitchison, K.J.; Craig, I.W.; Anacker, C.; Zunsztain, P.A.; McGuffin, P.; et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: Differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 2013, 38, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Banach, E.; Szczepankiewicz, A.; Leszczynska-Rodziewicz, A.; Pawlak, J.; Dmitrzak-Weglarz, M.; Zaremba, D.; Twarowska-Hauser, J. Venlafaxine and sertraline does not affect the expression of genes regulating stress response in female MDD patients. Psychiatr. Pol. 2017, 51, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Niitsu, T.; Fabbri, C.; Bentini, F.; Serretti, A. Pharmacogenetics in major depression: A comprehensive meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 45, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J. Statistical Power Analysis for the Behavioral Sciences, 2nd ed.; Lawrence Erlbaum Associates Inc.: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Vermeer, H.; Hendriks-Stegeman, B.I.; van der, B.B.; van Buul-Offers, S.C.; Jansen, M. Glucocorticoid-induced increase in lymphocytic FKBP51 messenger ribonucleic acid expression: A potential marker for glucocorticoid sensitivity, potency, and bioavailability. J. Clin. Endocrinol. Metab 2003, 88, 277–284. [Google Scholar] [CrossRef]
- Binder, E.B.; Salyakina, D.; Lichtner, P.; Wochnik, G.; Ising, M.; Pütz, B.; Papiol, S.; Seaman, S.; Lucae, S.; Kohli, M.; et al. Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet. 2004, 36, 1319–1325. [Google Scholar] [CrossRef]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B.; et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef]
- Gassen, N.C.; Hartmann, J.; Zannas, A.S.; Kretzschmar, A.; Zschocke, J.; Maccarrone, G.; Hafner, K.; Zellner, A.; Kollmannsberger, L.K.; Wagner, K.V.; et al. FKBP51 inhibits GSK3beta and augments the effects of distinct psychotropic medications. Mol. Psychiatry 2016, 21, 277–289. [Google Scholar] [CrossRef]
- Gassen, N.C.; Hartmann, J.; Zschocke, J.; Stepan, J.; Hafner, K.; Zellner, A.; Kirmeier, T.; Kollmannsberger, L.; Wagner, K.V.; Dedic, N.; et al. Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: Evidence in cells, mice, and humans. PLoS. Med. 2014, 11, e1001755. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Fries, G.R.; Zannas, A.S.; Hartmann, J.; Zschocke, J.; Hafner, K.; Carrillo-Roa, T.; Steinbacher, J.; Preissinger, S.N.; Hoeijmakers, L.; et al. Chaperoning epigenetics: FKBP51 decreases the activity of DNMT1 and mediates epigenetic effects of the antidepressant paroxetine. Sci. Signal. 2015, 8, ra119. [Google Scholar] [CrossRef] [PubMed]
- Rein, T. FK506 binding protein 51 integrates pathways of adaptation: FKBP51 shapes the reactivity to environmental change. Bioessays 2016, 38, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, J.; Wagner, K.V.; Gaali, S.; Kirschner, A.; Kozany, C.; Ruhter, G.; Dedic, N.; Hausl, A.S.; Hoeijmakers, L.; Westerholz, S.; et al. Pharmacological Inhibition of the Psychiatric Risk Factor FKBP51 Has Anxiolytic Properties. J. Neurosci. 2015, 35, 9007–9016. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, J.J.; Cordova, R.A.; Zheng, D.; Criado-Marrero, M.; Lemus, A.; Li, P.; Baker, J.D.; Nordhues, B.A.; Darling, A.L.; Martinez-Licha, C.; et al. Targeting the FKBP51/GR/Hsp90 Complex to Identify Functionally Relevant Treatments for Depression and PTSD. ACS Chem. Biol. 2018, 13, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Ewald, E.R.; Wand, G.S.; Seifuddin, F.; Yang, X.; Tamashiro, K.L.; Potash, J.B.; Zandi, P.; Lee, R.S. Alterations in DNA methylation of Fkbp5 as a determinant of blood-brain correlation of glucocorticoid exposure. Psychoneuroendocrinology 2014, 44, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Hennings, J.M.; Owashi, T.; Binder, E.B.; Horstmann, S.; Menke, A.; Kloiber, S.; Dose, T.; Wollweber, B.; Spieler, D.; Messer, T.; et al. Clinical characteristics and treatment outcome in a representative sample of depressed inpatients—Findings from the Munich Antidepressant Response Signature (MARS) project. J. Psychiatr. Res. 2009, 43, 215–229. [Google Scholar] [CrossRef]
- Endicott, J.; Cohen, J.; Nee, J.; Fleiss, J.; Sarantakos, S. Hamilton Depression Rating Scale. Extracted from Regular and Change Versions of the Schedule for Affective Disorders and Schizophrenia. Arch. Gen. Psychiatry 1981, 38, 98–103. [Google Scholar] [CrossRef]
- Kearns, N.P.; Cruickshank, C.A.; McGuigan, K.J.; Riley, S.A.; Shaw, S.P.; Snaith, R.P. A comparison of depression rating scales. Br. J. Psychiatry 1982, 141, 45–49. [Google Scholar] [CrossRef]
- World Health Organization. ICD-10: International Statistical Classification of Diseases and Related Health Problems, Tenth Revision, 2nd ed.; World Health Organization: Geneva, Switzerland, 2004. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Pawlak, M.; Schick, E.; Bopp, M.A.; Schneider, M.J.; Oroszlan, P.; Ehrat, M. Zeptosens’ protein microarrays: A novel high performance microarray platform for low abundance protein analysis. Proteomics 2002, 2, 383–393. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Characteristics | Responders N = 173 | Nonresponders N = 124 | p |
|---|---|---|---|
| Female sex (%) | 77 (44.5%) | 63 (50.8%) | 0.284 |
| Mean age (SD) | 48.8 (14.0) | 47.0 (13.4) | 0.270 |
| Diagnosis 1 (%) | 0.121 | ||
| F31 | 22 (12.7%) | 7 (5.6%) | |
| F32 | 36 (20.8%) | 30 (24.2 %) | |
| F33 | 115 (66.5%) | 87 (70.2%) | |
| HAMD-21 2 (SD) | 26.5 (6.63) | 25.5 (5.07) | 0.156 |
| FKBP5 RNA 2 (SD) | 1.83 (0.88) | 1.75 (0.65) | 0.404 |
| FKBP51 2,3 (SD) | 0.035 (0.07) | 0.031 (0.06) | 0.080 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ising, M.; Maccarrone, G.; Brückl, T.; Scheuer, S.; Hennings, J.; Holsboer, F.; Turck, C.W.; Uhr, M.; Lucae, S. FKBP5 Gene Expression Predicts Antidepressant Treatment Outcome in Depression. Int. J. Mol. Sci. 2019, 20, 485. https://doi.org/10.3390/ijms20030485
Ising M, Maccarrone G, Brückl T, Scheuer S, Hennings J, Holsboer F, Turck CW, Uhr M, Lucae S. FKBP5 Gene Expression Predicts Antidepressant Treatment Outcome in Depression. International Journal of Molecular Sciences. 2019; 20(3):485. https://doi.org/10.3390/ijms20030485
Chicago/Turabian StyleIsing, Marcus, Giuseppina Maccarrone, Tanja Brückl, Sandra Scheuer, Johannes Hennings, Florian Holsboer, Christoph W. Turck, Manfred Uhr, and Susanne Lucae. 2019. "FKBP5 Gene Expression Predicts Antidepressant Treatment Outcome in Depression" International Journal of Molecular Sciences 20, no. 3: 485. https://doi.org/10.3390/ijms20030485
APA StyleIsing, M., Maccarrone, G., Brückl, T., Scheuer, S., Hennings, J., Holsboer, F., Turck, C. W., Uhr, M., & Lucae, S. (2019). FKBP5 Gene Expression Predicts Antidepressant Treatment Outcome in Depression. International Journal of Molecular Sciences, 20(3), 485. https://doi.org/10.3390/ijms20030485