Depression and Sleep


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Sleep EEG in Patients with Depression
- (i)
- Impaired sleep continuity (prolonged sleep latency, increased intermittent awakenings, early morning awakenings).
- (ii)
- Disinhibition of REM sleep: shortened REM latency, or sleep onset REM periods (SOREMs, REM latency 0–20 min), prolonged first REM period, enhanced REM density (measure of frequency of rapid eye movements) particularly during first REM period.
- (iii)
- Changes in non-REM sleep (decreased stage N2 and SWS, in younger patients shift of SWS from the first to the second sleep cycle).
3. Sleep EEG in High-Risk Probands for Affective Disorders
4. Sleep EEG and Risk Genes for Depression
5. Effects of Antidepressants on Sleep EEG
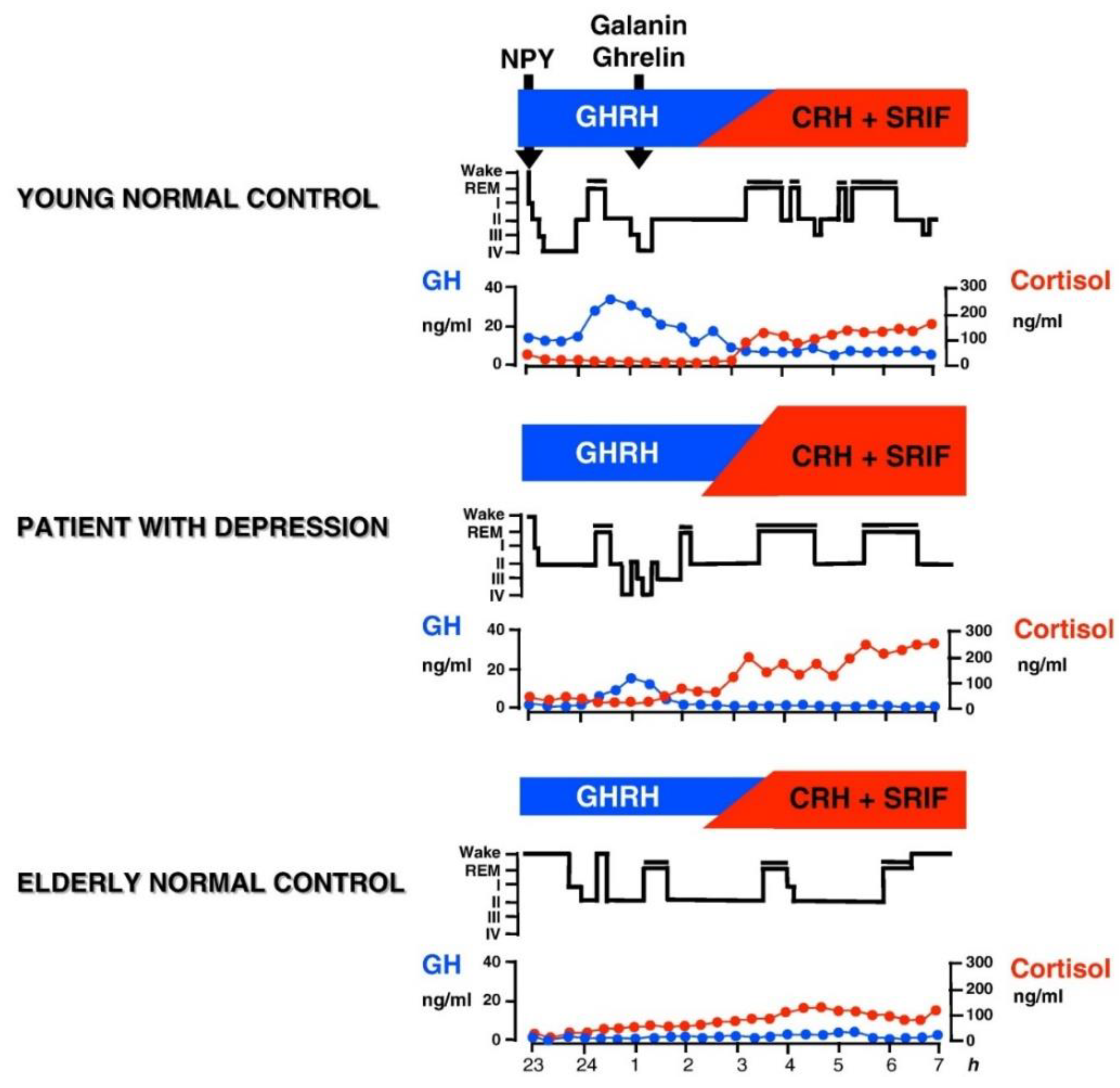
6. Contribution of the HPA System to Sleep-EEG Abnormalities in Depression
7. Amyloid-β and Sleep
8. State and Vulnerability Markers Related to Antidepressant Therapy
9. Cordance Derived from REM Sleep as a Predictor of Therapy Response
10. Heart Rate Variability Derived from REM Sleep in Depressed Patients
11. Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Baglioni, C.; Battagliese, G.; Feige, B.; Spiegelhalder, K.; Nissen, C.; Voderholzer, U.; Lombardo, C.; Riemann, D. Insomnia as a predictor of depression: A meta-analytic evaluation of longitudinal epidemiological studies. J. Affect. Disord. 2011, 135, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, D.; Foster, F.G. Interval between onset of sleep and rapid-eye-movement sleep as an indicator of depression. Lancet 1972, 300, 684–686. [Google Scholar] [CrossRef]
- Chen, C.-N. Sleep, depression and antidepressants. Br. J. Psychiatry 1979, 135, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Vogel, G.W.; Thurmond, A.; Gibbons, P.; Sloan, K.; Boyd, M.; Walker, M. REM sleep reduction effects on depression syndromes. Arch. Gen. Psychiatry 1975, 32, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Kales, A.; Rechtschaffen, A. A Manual of Standardized Terminology, Techniques and Scoring System for Sleep Stages of Human Subjects; US Department of Health, Education and Welfare, Public Health Service, National Institutes of Health, National Institute of Neurological Diseases and Blindness, Neurological Information Network: Bethesda, MD, USA, 1968.
- Iber, C.; Ancoli-Israel, S.; Chesson, A.L.; Quan, S.F. The AASM Manual for the Scoring of Sleep and Associated Events: Rules, Terminology and Technical Specifications, 1st ed.; American Academy of Sleep Medicine: Westchester, IL, USA, 2007. [Google Scholar]
- Hawkins, D.R.; Taub, J.M.; Van de Castle, R.L. Extended sleep (hypersomnia) in young depressed patients. Am. J. Psychiatry 1985, 142, 905–910. [Google Scholar] [CrossRef] [PubMed]
- Armitage, R. Sleep and circadian rhythms in mood disorders. Acta Psychiatr. Scand. 2007, 115, 104–115. [Google Scholar] [CrossRef]
- Reynolds, C.F.; Kupfer, D.J. Sleep research in affective illness: State of the art circa 1987. Sleep 1987, 10, 199–215. [Google Scholar] [CrossRef]
- Benca, R.M.; Okawa, M.; Uchiyama, M.; Ozaki, S.; Nakajima, T.; Shibui, K.; Obermeyer, W.H. Sleep and mood disorders. Sleep Med. Rev. 1997, 1, 45–56. [Google Scholar] [CrossRef]
- Borbély, A.A.; Tobler, I.; Loepfe, M.; Kupfer, D.J.; Ulrich, R.F.; Grochocinski, V.; Doman, J.; Matthews, G. All-night spectral analysis of the sleep EEG in untreated depressives and normal controls. Psychiatry Res. 1984, 12, 27–33. [Google Scholar] [CrossRef]
- Kupfer, D.J.; Ulrich, R.F.; Coble, P.A.; Jarrett, D.B.; Grochocinski, V.; Doman, J.; Matthews, G.; Borbély, A.A. Application of automated REM and slow wave sleep analysis: II. Testing the assumptions of the two-process model of sleep regulation in normal and depressed subjects. Psychiatry Res. 1984, 13, 335–343. [Google Scholar] [CrossRef]
- Kupfer, D.J.; Reynolds, C.F.; Ulrich, R.F.; Grochocinski, V.J. Comparison of automated REM and slow-wave sleep analysis in young and middle-aged depressed subjects. Biol. Psychiatry 1986, 21, 189–200. [Google Scholar] [CrossRef]
- Kupfer, D.J.; Reynolds, C.F., III; Grochocinski, V.J.; Ulrich, R.F.; McEachran, A. Aspects of short REM latency in affective states: A revisit. Psychiatry Res. 1986, 17, 49–59. [Google Scholar] [CrossRef]
- Lauer, C.J.; Riemann, D.; Wiegand, M.; Berger, M. From early to late adulthood changes in EEG sleep of depressed patients and healthy volunteers. Biol. Psychiatry 1991, 29, 979–993. [Google Scholar] [CrossRef]
- Riemann, D.; Lauer, C.; Hohagen, F.; Berger, M. Longterm evolution of sleep in depression. In Sleep and Aging; Masson Press: Milano, Italy, 1991; pp. 195–204. [Google Scholar]
- Rush, A.J.; Erman, M.K.; Giles, D.E.; Schlesser, M.A.; Carpenter, G.; Vasavada, N.; Roffwarg, H.P. Polysomnographic findings in recently drug-free and clinically remitted depressed patients. Arch. Gen. Psychiatry 1986, 43, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Steiger, A.; von Bardeleben, U.; Herth, T.; Holsboer, F. Sleep EEG and nocturnal secretion of cortisol and growth hormone in male patients with endogenous depression before treatment and after recovery. J. Affect. Disord. 1989, 16, 189–195. [Google Scholar] [CrossRef]
- Rao, U.; Poland, R.E. Electroencephalographic sleep and hypothalamic–pituitary–adrenal changes from episode to recovery in depressed adolescents. J. Child Adolesc. Psychopharmacol. 2008, 18, 607–613. [Google Scholar] [CrossRef]
- Kupfer, D.J.; Ehlers, C.L.; Frank, E.; Grochocinski, V.J.; McEachran, A.B. EEG sleep profiles and recurrent depression. Biol. Psychiatry 1991, 30, 641–655. [Google Scholar] [CrossRef]
- Hudson, J.I.; Lipinski, J.F.; Frankenburg, F.R.; Grochocinski, V.J.; Kupfer, D.J. Electroencephalographic sleep in mania. Arch. Gen. Psychiatry 1988, 45, 267–273. [Google Scholar] [CrossRef]
- Zarcone, V.P.; Benson, K.L.; Berger, P.A. Abnormal rapid eye movement latencies in schizophrenia. Arch. Gen. Psychiatry 1987, 44, 45–48. [Google Scholar] [CrossRef]
- Reich, L.; Weiss, B.L.; Coble, P.; McPartland, R.; Kupfer, D.J. Sleep disturbance in schizophrenia: A revisit. Arch. Gen. Psychiatry 1975, 32, 51–55. [Google Scholar] [CrossRef]
- Insel, T.R.; Gillin, J.C.; Moore, A.; Mendelson, W.B.; Loewenstein, R.J.; Murphy, D.L. The sleep of patients with obsessive-compulsive disorder. Arch. Gen. Psychiatry 1982, 39, 1372–1377. [Google Scholar] [CrossRef] [PubMed]
- Uhde, T.W.; Roy-Byrne, P.; Gillin, J.C.; Mendelson, W.B.; Boulenger, J.-P.; Vittone, B.J.; Post, R.M. The sleep of patients with panic disorder: A preliminary report. Psychiatry Res. 1984, 12, 251–259. [Google Scholar] [CrossRef]
- Katz, J.L.; Kuperberg, A.; Pollack, C.P.; Walsh, B.T.; Zumoff, B.; Weiner, H. Is there a relationship between eating disorder and affective disorder? New evidence from sleep recordings. Am. J. Psychiatry 1984. [Google Scholar] [CrossRef]
- Schmidt, H.S.; Nofzinger, E.A. Short REM latency in impotence without depression. Biol. Psychiatry 1988, 24, 25–32. [Google Scholar] [CrossRef]
- Lauer, C.J.; Krieg, J.-C.; Riemann, D.; Zulley, J.; Berger, M. A polysomnographic study in young psychiatric inpatients: Major depression, anorexia nervosa, bulimia nervosa. J. Affect. Disord. 1990, 18, 235–245. [Google Scholar] [CrossRef]
- Lauer, C.J.; Krieg, J.-C.; Garcia-Borreguero, D.; Özdaglar, A.; Holsboer, F. Panic disorder and major depression: A comparative electroencephalographic sleep study. Psychiatry Res. 1992, 44, 41–54. [Google Scholar] [CrossRef]
- Lauer, C.J.; Schreiber, W.; Holsboer, F.; Krieg, J.-C. In quest of identifying vulnerability markers for psychiatric disorders by all-night polysomnography. Arch. Gen. Psychiatry 1995, 52, 145–153. [Google Scholar] [CrossRef]
- Modell, S.; Ising, M.; Holsboer, F.; Lauer, C.J. The Munich Vulnerability Study on Affective Disorders: Stability of polysomnographic findings over time. Biol. Psychiatry 2002, 52, 430–437. [Google Scholar] [CrossRef]
- Schreiber, W.; Lauer, C.J.; Krumrey, K.; Holsboer, F.; Krieg, J.-C. Cholinergic REM sleep induction test in subjects at high risk for psychiatric disorders. Biol. Psychiatry 1992, 32, 79–90. [Google Scholar] [CrossRef]
- Lauer, C.J.; Modell, S.; Schreiber, W.; Krieg, J.-C.; Holsboer, F. Prediction of the development of a first major depressive episode with a rapid eye movement sleep induction test using the cholinergic agonist RS 86. J. Clin. Psychopharmacol. 2004, 24, 356–357. [Google Scholar] [CrossRef] [PubMed]
- Modell, S.; Ising, M.; Holsboer, F.; Lauer, C.J. The Munich vulnerability study on affective disorders: Premorbid polysomnographic profile of affected high-risk probands. Biol. Psychiatry 2005, 58, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Abkevich, V.; Camp, N.J.; Hensel, C.H.; Neff, C.D.; Russell, D.L.; Hughes, D.C.; Plenk, A.M.; Lowry, M.R.; Richards, R.L.; Carter, C. Predisposition locus for major depression at chromosome 12q22-12q23.2. Am. J. Hum. Genet. 2003, 73, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Degn, B.; Lundorf, M.; Wang, A.; Vang, M.; Mors, O.; Kruse, T.; Ewald, H. Further evidence for a bipolar risk gene on chromosome 12q24 suggested by investigation of haplotype sharing and allelic association in patients from the Faroe Islands. Mol. Psychiatry 2001, 6, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.; Sluyter, R.; Gu, B.; Stokes, L.; Fuller, S. The human P2X7 receptor and its role in innate immunity. Tissue Antigens 2011, 78, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Barden, N.; Harvey, M.; Gagné, B.; Shink, E.; Tremblay, M.; Raymond, C.; Labbé, M.; Villeneuve, A.; Rochette, D.; Bordeleau, L.; et al. Analysis of single nucleotide polymorphisms in genes in the chromosome 12Q24. 31 region points to P2RX7 as a susceptibility gene to bipolar affective disorder. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2006, 141, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Lucae, S.; Salyakina, D.; Barden, N.; Harvey, M.; Gagné, B.; Labbé, M.; Binder, E.B.; Uhr, M.; Paez-Pereda, M.; Sillaber, I.; et al. P2RX7, a gene coding for a purinergic ligand-gated ion channel, is associated with major depressive disorder. Hum. Mol. Genet. 2006, 15, 2438–2445. [Google Scholar] [CrossRef] [PubMed]
- Soronen, P.; Mantere, O.; Melartin, T.; Suominen, K.; Vuorilehto, M.; Rytsälä, H.; Arvilommi, P.; Holma, I.; Holma, M.; Jylhä, P.; et al. P2RX7 gene is associated consistently with mood disorders and predicts clinical outcome in three clinical cohorts. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2011, 156, 435–447. [Google Scholar] [CrossRef]
- Metzger, M.W.; Walser, S.M.; Dedic, N.; Aprile-Garcia, F.; Jakubcakova, V.; Adamczyk, M.; Webb, K.J.; Uhr, M.; Refojo, D.; Schmidt, M.V. Heterozygosity for the mood disorder-associated variant Gln460Arg alters P2X7 receptor function and sleep quality. J. Neurosci. 2017, 37, 11688–11700. [Google Scholar] [CrossRef]
- Steiger, A.; Von Bardeleben, U.; Guldner, J.; Lauer, C.; Rothe, B.; Holsboer, F. The sleep EEG and nocturnal hormonal secretion studies on changes during the course of depression and on effects of CNS-active drugs. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 1993, 17, 125–137. [Google Scholar] [CrossRef]
- Dunleavy, D.; Brezinova, V.; Oswald, I.; Maclean, A.; Tinker, M. Changes during weeks in effects of tricyclic drugs on the human sleeping brain. Br. J. Psychiatry 1972, 120, 663–672. [Google Scholar] [CrossRef]
- Passouant, P.; Cadilhac, J.; Billiard, M.; Besset, A. La suppression du sommeil paradoxal par la clomipramine. Thérapie 1973, 28, 379–392. [Google Scholar] [PubMed]
- Shipley, J.E.; Kupfer, D.J.; Dealy, R.S.; Griffin, S.J.; Coble, P.A.; McEachran, A.B.; Grochocinski, V.J. Differential effects of amitriptyline and of zimelidine on the sleep electroencephalogram of depressed patients. Clin. Pharmacol. Ther. 1984, 36, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Von Bardeleben, U.; Steiger, A.; Gerken, A.; Holsboer, F. Effects of fluoxetine upon pharmacoendocrine and sleep-EEG parameters in normal controls. Int. Clin. Psychopharmacol. 1989, 4, 1–5. [Google Scholar]
- Künzel, H.; Murck, H.; Held, K.; Ziegenbein, M.; Steiger, A. Reboxetine induces similar sleep-EEG changes like SSRI’s in patients with depression. Pharmacopsychiatry 2004, 37, 193–195. [Google Scholar] [CrossRef]
- Kluge, M.; Schüssler, P.; Steiger, A. Duloxetine increases stage 3 sleep and suppresses rapid eye movement (REM) sleep in patients with major depression. Eur. Neuropsychopharmacol. 2007, 17, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Cramer, H.; Ohlmeier, D. Ein Fall von Tranylcypromin-und Trifluoperazin-(Jatrosom®)-Sucht: Psychopathologische, schlafphysiologische und biochemische Untersuchungen. Archiv für Psychiatrie und Nervenkrankheiten 1967, 210, 182–197. [Google Scholar] [CrossRef]
- Wyatt, R.J.; Fram, D.H.; Kupfer, D.J.; Snyder, F. Total prolonged drug-induced REM sleep suppression in anxious-depressed patients. Arch. Gen. Psychiatry 1971, 24, 145–155. [Google Scholar] [CrossRef]
- Landolt, H.-P.; Raimo, E.B.; Schnierow, B.J.; Kelsoe, J.R.; Rapaport, M.H.; Gillin, J.C. Sleep and sleep electroencephalogram in depressed patients treated with phenelzine. Arch. Gen. Psychiatry 2001, 58, 268–276. [Google Scholar] [CrossRef]
- Steiger, A.; Benkert, O.; Holsboer, F. Effects of long-term treatment with the MAO-A inhibitor moclobemide on sleep EEG and nocturnal hormonal secretion in normal men. Neuropsychobiology 1994, 30, 101–105. [Google Scholar] [CrossRef]
- Sonntag, A.; Rothe, B.; Guldner, J.; Yassouridis, A.; Holsboer, F.; Steiger, A. Trimipramine and imipramine exert different effects on the sleep EEG and on nocturnal hormone secretion during treatment of major depression. Depression 1996, 4, 1–13. [Google Scholar] [CrossRef]
- Nofzinger, E.A.; Reynolds, C.F., III; Thase, M.E.; Frank, E. REM sleep enhancement by bupropion in depressed men. Am. J. Psychiatry 1995, 152, 274–276. [Google Scholar] [PubMed]
- Murck, H.; Nickel, T.; Künzel, H.; Antonijevic, I.; Schill, J.; Zobel, A.; Steiger, A.; Sonntag, A.; Holsboer, F. State markers of depression in sleep EEG: Dependency on drug and gender in patients treated with tianeptine or paroxetine. Neuropsychopharmacology 2003, 28, 348–358. [Google Scholar] [CrossRef] [PubMed]
- Ruigt, G.; Kemp, B.; Groenhout, C.; Kamphuisen, H. Effect of the antidepressant Org 3770 on human sleep. Eur. J. clin. Pharmacol. 1990, 38, 551–554. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.A.; Wichniak, A.; Uhr, M.; Ising, M.; Brunner, H.; Held, K.; Weikel, J.C.; Sonntag, A.; Steiger, A. Changes of sleep architecture, spectral composition of sleep EEG, the nocturnal secretion of cortisol, ACTH, GH, prolactin, melatonin, ghrelin, and leptin, and the DEX-CRH test in depressed patients during treatment with mirtazapine. Neuropsychopharmacology 2006, 31, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Steiger, A. Effects of clomipramine on sleep EEG and nocturnal penile tumescence: A long-term study in a healthy man. J. Clin. Psychopharmacol. 1988, 8, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Akindele, M.; Evans, J.; Oswald, I. Mono-amine oxidase inhibitors, sleep and mood. Electroencephalogr. Clin. Neurophysiol. 1970, 29, 47–56. [Google Scholar] [CrossRef]
- Murck, H.; Frieboes, R.; Antonijevic, I.; Steiger, A. Distinct temporal pattern of the effects of the combined serotonin-reuptake inhibitor and 5-HT 1A agonist EMD 68843 on the sleep EEG in healthy men. Psychopharmacology 2001, 155, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Grözinger, M.; Kögel, P.; Röschke, J. Effects of REM sleep awakenings and related wakening paradigms on the ultradian sleep cycle and the symptoms in depression. J. Psychiatr. Res. 2002, 36, 299–308. [Google Scholar] [CrossRef]
- Steiger, A.; Gerken, A.; Benkert, O.; Holsboer, F. Differential effects of the enantiomers R (−) and S (+) oxaprotiline on major endogenous depression, the sleep EEG and neuroendocrine secretion: Studies on depressed patients and normal controls. Eur. Neuropsychopharmacol. 1993, 3, 117–126. [Google Scholar] [CrossRef]
- Saletu, B.; Frey, R.; Krupka, M.; Anderer, P.; Grfulberger, J.; See, W.R. Sleep laboratory studies on the single-dose effects of serotonin reuptake inhibitors paroxetine and fluoxetine on human sleep and awakening qualities. Sleep 1991, 14, 439–447. [Google Scholar] [CrossRef]
- Sharpley, A.; Williamson, D.; Attenburrow, M.; Pearson, G.; Sargent, P.; Cowen, P. The effects of paroxetine and nefazodone on sleep: A placebo controlled trial. Psychopharmacology 1996, 126, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Kupfer, D. REM latency: A psychobiologic marker for primary depressive disease. Biol. Psychiatry 1976, 11, 159–174. [Google Scholar] [PubMed]
- Riemann, D.; Berger, M. The effects of total sleep deprivation and subsequent treatment with clomipramine on depressive symptoms and sleep electroencephalography in patients with a major depressive disorder. Acta Psychiatr. Scand. 1990, 81, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Holsboer, F.; Ising, M. Stress hormone regulation: Biological role and translation into therapy. Annu. Rev. Psychol. 2010, 61, 81–109. [Google Scholar] [CrossRef] [PubMed]
- Linkowski, P.; Mendlewicz, J.; Kerkhofs, M.; Leclercq, R.; Golstein, J.; Brasseur, M.; Copinschi, G.; Cauter, E.V. 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: Effect of antidepressant treatment. J. Clin. Endocrinol. MeTable 1987, 65, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, C.L.; Reed, T.K.; Henriksen, S.J. Effects of corticotropin-releasing factor and growth hormone-releasing factor on sleep and activity in rats. Neuroendocrinology 1986, 42, 467–474. [Google Scholar] [CrossRef]
- Opp, M.; Obal, F., Jr.; Krueger, J. Corticotropin-releasing factor attenuates interleukin 1-induced sleep and fever in rabbits. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1989, 257, R528–R535. [Google Scholar] [CrossRef] [PubMed]
- Romanowski, C.; Fenzl, T.; Flachskamm, C.; Deussing, J.; Kimura, M. CRH-R1 is involved in effects of CRH on NREM, but not REM, sleep suppression. Sleep Biol. Rhythms 2007, 5, A53. [Google Scholar]
- Sanford, L.; Yang, L.; Wellman, L.; Dong, E.; Tang, X. Mouse strain differences in the effects of corticotropin releasing hormone (CRH) on sleep and wakefulness. Brain Res. 2008, 1190, 94–104. [Google Scholar] [CrossRef]
- Holsboer, F.; Von Bardeleben, U.; Steiger, A. Effects of intravenous corticotropin-releasing hormone upon sleep-related growth hormone surge and sleep EEG in man. Neuroendocrinology 1988, 48, 32–38. [Google Scholar] [CrossRef]
- Kimura, M.; Müller-Preuss, P.; Lu, A.; Wiesner, E.; Flachskamm, C.; Wurst, W.; Holsboer, F.; Deussing, J. Conditional corticotropin-releasing hormone overexpression in the mouse forebrain enhances rapid eye movement sleep. Mol. Psychiatry 2010, 15, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Schüssler, P.; Kluge, M.; Gamringer, W.; Wetter, T.; Yassouridis, A.; Uhr, M.; Rupprecht, R.; Steiger, A. Corticotropin-releasing hormone induces depression-like changes of sleep electroencephalogram in healthy women. Psychoneuroendocrinology 2016, 74, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Hatzinger, M.; Brand, S.; Perren, S.; Stadelmann, S.; von Wyl, A.; von Klitzing, K.; Holsboer-Trachsler, E. Electroencephalographic sleep profiles and hypothalamic–pituitary–adrenocortical (HPA)-activity in kindergarten children: Early indication of poor sleep quality associated with increased cortisol secretion. J. Psychiatr. Res. 2008, 42, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Held, K.; Künzel, H.; Ising, M.; Schmid, D.; Zobel, A.; Murck, H.; Holsboer, F.; Steiger, A. Treatment with the CRH1-receptor-antagonist R121919 improves sleep-EEG in patients with depression. J. Psychiatr. Res. 2004, 38, 129–136. [Google Scholar] [CrossRef]
- Antonijevic, I.A.; Steiger, A. Depression-like changes of the sleep-EEG during high dose corticosteroid treatment in patients with multiple sclerosis. Psychoneuroendocrinology 2003, 28, 780–795. [Google Scholar] [CrossRef]
- Obal, F., Jr.; Alfoldi, P.; Cady, A.; Johannsen, L.; Sáry, G.; Krueger, J. Growth hormone-releasing factor enhances sleep in rats and rabbits. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 1988, 255, R310–R316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Obál, F.; Zheng, T.; Fang, J.; Taishi, P.; Krueger, J.M. Intrapreoptic microinjection of GHRH or its antagonist alters sleep in rats. J. Neurosci. 1999, 19, 2187–2194. [Google Scholar] [CrossRef]
- Obál, F., Jr.; Floyd, R.; Kapas, L.E.; Bodosi, B.; Krueger, J. Effects of systemic GHRH on sleep in intact and hypophysectomized rats. Am. J. Physiol.-Endocrinol. MeTable 1996, 270, E230–E237. [Google Scholar] [CrossRef]
- Steiger, A.; Guldner, J.; Hemmeter, U.; Rothe, B.; Wiedemann, K.; Holsboer, F. Effects of growth hormone-releasing hormone and somatostatin on sleep EEG and nocturnal hormone secretion in male controls. Neuroendocrinology 1992, 56, 566–573. [Google Scholar] [CrossRef]
- Antonijevic, I.A.; Murck, H.; Frieboes, R.-M.; Barthelmes, J.; Steiger, A. Sexually dimorphic effects of GHRH on sleep-endocrine activity in patients with depression and normal controls—p#art I: The sleep EEG. Sleep Res. Online 2000, 3, 5–13. [Google Scholar]
- Antonijevic, I.A.; Murck, H.; Frieboes, R.-M.; Steiger, A. Sexually dimorphic effects of GHRH on sleep-endocrine activity in patients with depression and normal controls—p#art II: Hormone secretion. Sleep Res. Online 2000, 3, 15–21. [Google Scholar] [PubMed]
- Mayeux, R.; Stern, Y. Epidemiology of Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Shokri-Kojori, E.; Wang, G.-J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Huang, J.; Yang, L.; Zeng, X.-A.; Zhang, Y.; Wang, X.; Chen, M.; Li, X.; Zhang, Y.; Zhang, M. Sleep deprivation accelerates the progression of Alzheimer’s disease by influencing Aβ-related metabolism. Neurosci. Lett. 2017, 650, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Lucey, B.P.; Hicks, T.J.; McLeland, J.S.; Toedebusch, C.D.; Boyd, J.; Elbert, D.L.; Patterson, B.W.; Baty, J.; Morris, J.C.; Ovod, V.; et al. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann. Neurol. 2018, 83, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kincheski, G.C.; Valentim, I.S.; Clarke, J.R.; Cozachenco, D.; Castelo-Branco, M.T.; Ramos-Lobo, A.M.; Rumjanek, V.M.; Donato, J., Jr.; De Felice, F.G.; Ferreira, S.T. Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-β oligomers in mice. Brain. Behav. Immun. 2017, 64, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, E.N.; Miyazaki, K.; Maruo, K.; Yagihara, H.; Fujita, H.; Wada, K.; Nagai, Y. Chronic sleep fragmentation exacerbates amyloid β deposition in Alzheimer’s disease model mice. Neurosci. Lett. 2017, 653, 362–369. [Google Scholar] [CrossRef]
- Ju, Y.-E.S.; Ooms, S.J.; Sutphen, C.; Macauley, S.L.; Zangrilli, M.A.; Jerome, G.; Fagan, A.M.; Mignot, E.; Zempel, J.M.; Claassen, J.A. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain 2017, 140, 2104–2111. [Google Scholar] [CrossRef]
- Chen, D.-W.; Wang, J.; Zhang, L.-L.; Wang, Y.-J.; Gao, C.-Y. Cerebrospinal fluid amyloid-β levels are increased in patients with insomnia. J. Alzheimers Dis. 2018, 61, 645–651. [Google Scholar] [CrossRef]
- Park, H.J.; Ran, Y.; Jung, J.I.; Holmes, O.; Price, A.R.; Smithson, L.; Ceballos-Diaz, C.; Han, C.; Wolfe, M.S.; Daaka, Y.; et al. The stress response neuropeptide CRF increases amyloid-β production by regulating γ-secretase activity. EMBO J. 2015, 34, 1674–1686. [Google Scholar] [CrossRef]
- Morgese, M.G.; Schiavone, S.; Trabace, L. Emerging role of amyloid beta in stress response: Implication for depression and diabetes. Eur. J. Pharmacol. 2017, 817, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Hatzinger, M.; Hemmeter, U.M.; Brand, S.; Ising, M.; Holsboer-Trachsler, E. Electroencephalographic sleep profiles in treatment course and long-term outcome of major depression: Association with DEX/CRH-test response. J. Psychiatr. Res. 2004, 38, 453–465. [Google Scholar] [CrossRef] [PubMed]
- Leuchter, A.F.; Uijtdehaage, S.H.; Cook, I.A.; O’Hara, R.; Mandelkern, M. Relationship between brain electrical activity and cortical perfusion in normal subjects. Psychiatry Res. Neuroimaging 1999, 90, 125–140. [Google Scholar] [CrossRef]
- Bares, M.; Brunovsky, M.; Kopecek, M.; Stopkova, P.; Novak, T.; Kozeny, J.; Höschl, C. Changes in QEEG prefrontal cordance as a predictor of response to antidepressants in patients with treatment resistant depressive disorder: A pilot study. J. Psychiatr. Res. 2007, 41, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Bares, M.; Brunovsky, M.; Novak, T.; Kopecek, M.; Stopkova, P.; Sos, P.; Krajca, V.; Höschl, C. The change of prefrontal QEEG theta cordance as a predictor of response to bupropion treatment in patients who had failed to respond to previous antidepressant treatments. Eur. Neuropsychopharmacol. 2010, 20, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Cook, I.A.; Leuchter, A.F.; Morgan, M.; Witte, E.; Stubbeman, W.F.; Abrams, M.; Rosenberg, S.; Uijtdehaage, S.H. Early changes in prefrontal activity characterize clinical responders to antidepressants. Neuropsychopharmacology 2002, 27, 120–131. [Google Scholar] [CrossRef]
- Cook, I.A.; Leuchter, A.F.; Morgan, M.L.; Stubbeman, W.; Siegman, B.; Abrams, M. Changes in prefrontal activity characterize clinical response in SSRI nonresponders: A pilot study. J. Psychiatr. Res. 2005, 39, 461–466. [Google Scholar] [CrossRef]
- Asada, H.; Fukuda, Y.; Tsunoda, S.; Yamaguchi, M.; Tonoike, M. Frontal midline theta rhythms reflect alternative activation of prefrontal cortex and anterior cingulate cortex in humans. Neurosci. Lett. 1999, 274, 29–32. [Google Scholar] [CrossRef]
- Drevets, W.C. Neuroimaging studies of mood disorders. Biol. Psychiatry 2000, 48, 813–829. [Google Scholar] [CrossRef]
- Braun, A.R.; Balkin, T.; Wesenten, N.; Carson, R.; Varga, M.; Baldwin, P.; Selbie, S.; Belenky, G.; Herscovitch, P. Regional cerebral blood flow throughout the sleep-wake cycle. An H2(15)O PET study. Brain 1997, 120, 1173–1197. [Google Scholar] [CrossRef]
- Hobson, J.A.; Pace-Schott, E.F. The cognitive neuroscience of sleep: Neuronal systems, consciousness and learning. Nat. Rev. Neurosci. 2002, 3, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Hirai, N.; Miwakeichi, F.; Maehara, T.; Kawai, K.; Shimizu, H.; Uchida, S. Theta oscillation in the human anterior cingulate cortex during all-night sleep: An electrocorticographic study. Neurosci. Res. 2004, 50, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, M.; Gazea, M.; Wollweber, B.; Holsboer, F.; Dresler, M.; Steiger, A.; Pawlowski, M. Cordance derived from REM sleep EEG as a biomarker for treatment response in depression—A naturalistic study after antidepressant medication. J. Psychiatr. Res. 2015, 63, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Kemp, A.H.; Quintana, D.S.; Gray, M.A.; Felmingham, K.L.; Brown, K.; Gatt, J.M. Impact of depression and antidepressant treatment on heart rate variability: A review and meta-analysis. Biol. Psychiatry 2010, 67, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
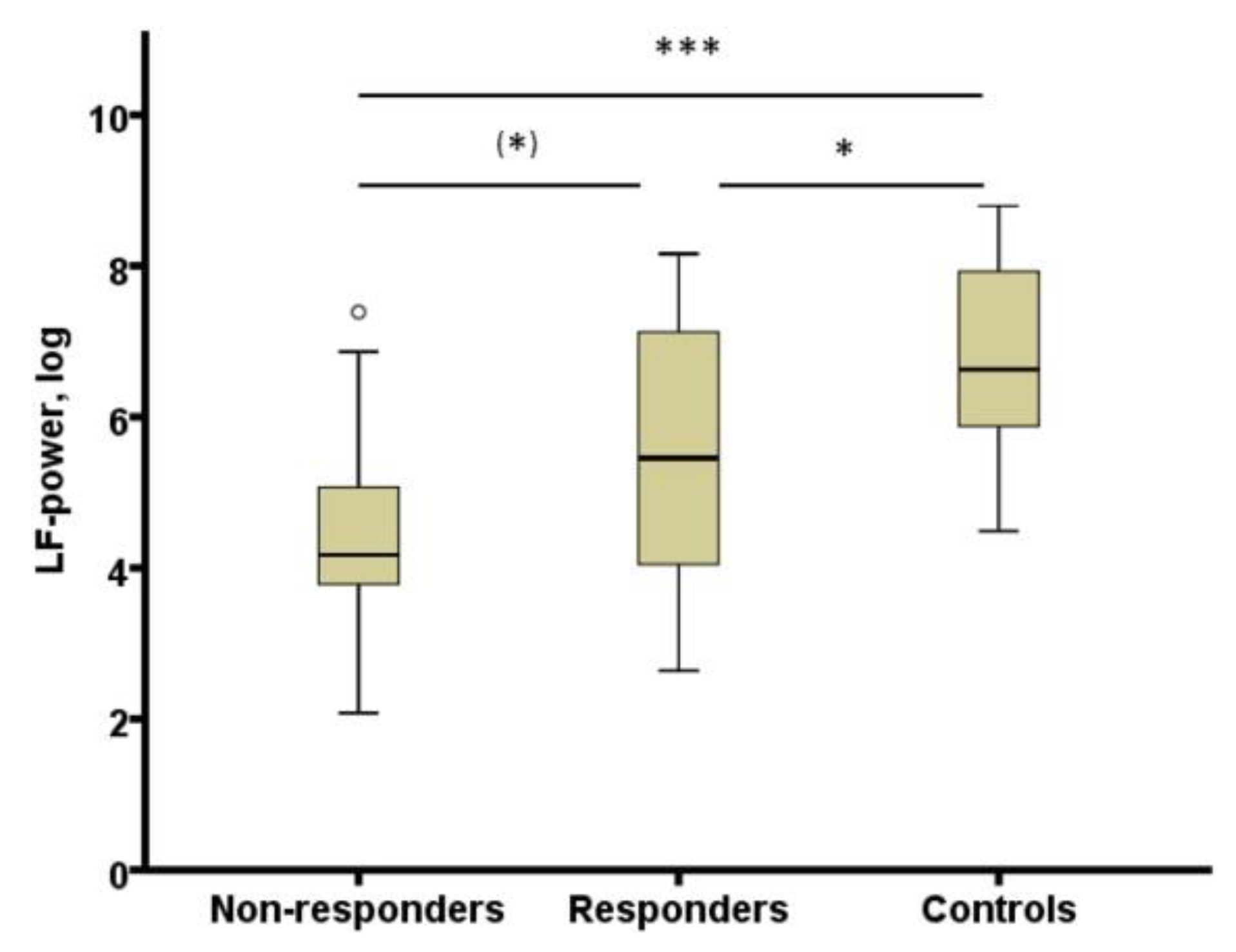
- Pawlowski, M.A.; Gazea, M.; Wollweber, B.; Dresler, M.; Holsboer, F.; Keck, M.E.; Steiger, A.; Adamczyk, M.; Mikoteit, T. Heart rate variability and cordance in rapid eye movement sleep as biomarkers of depression and treatment response. J. Psychiatr. Res. 2017, 92, 64–73. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steiger, A.; Pawlowski, M. Depression and Sleep. Int. J. Mol. Sci. 2019, 20, 607. https://doi.org/10.3390/ijms20030607
Steiger A, Pawlowski M. Depression and Sleep. International Journal of Molecular Sciences. 2019; 20(3):607. https://doi.org/10.3390/ijms20030607
Chicago/Turabian StyleSteiger, Axel, and Marcel Pawlowski. 2019. "Depression and Sleep" International Journal of Molecular Sciences 20, no. 3: 607. https://doi.org/10.3390/ijms20030607
APA StyleSteiger, A., & Pawlowski, M. (2019). Depression and Sleep. International Journal of Molecular Sciences, 20(3), 607. https://doi.org/10.3390/ijms20030607