Coagulatory Defects in Type-1 and Type-2 Diabetes



Abstract
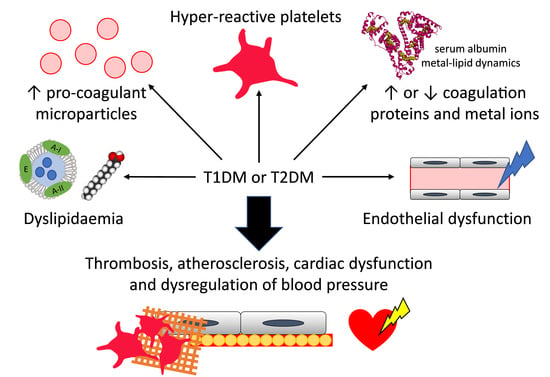
1. Introduction
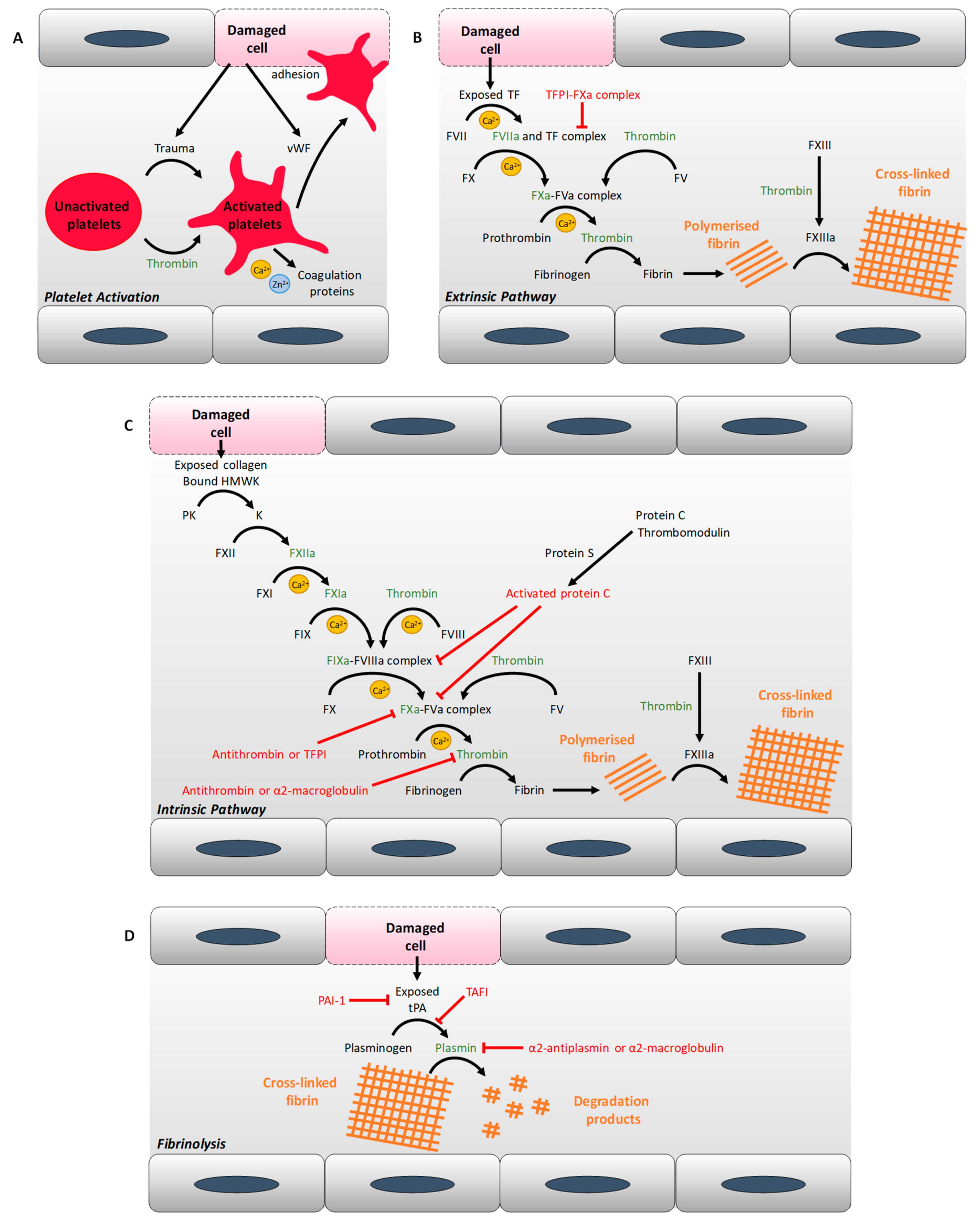
2. Diabetes and Thrombosis: Abnormal Coagulation Mechanisms
2.1. Alterations of Plasma Protein Concentrations
2.2. Changes in Metal Ion Homeostasis
2.3. Changes in Lipid Metabolism at the Origin of Atherosclerosis and Lipotoxicity
2.4. Endothelial Dysfunction
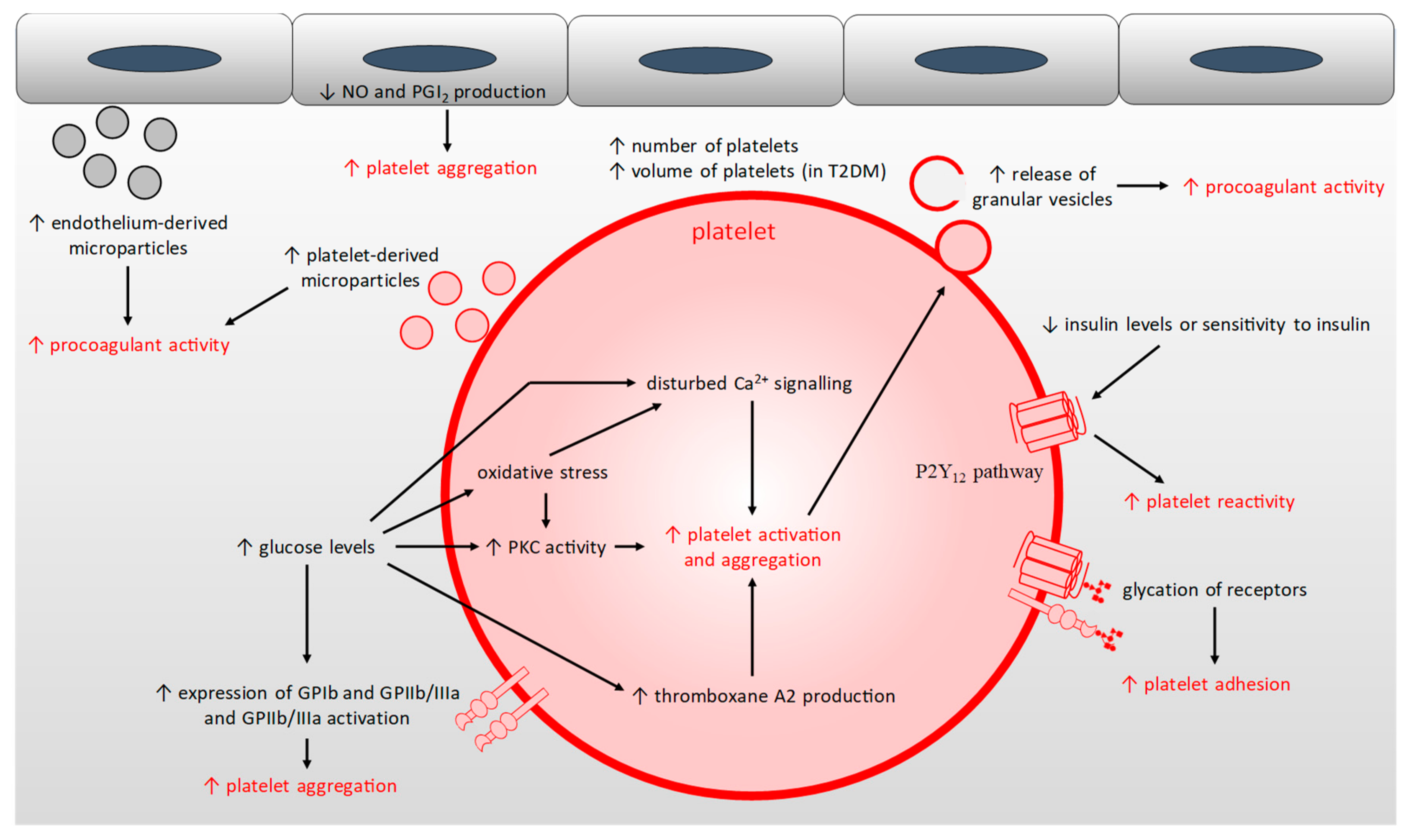
2.5. Platelet Hyper-Activation
2.6. Pro-Coagulatory Microparticles
3. Differences in Thrombotic Risks Between T1DM and T2DM
4. Current Treatment and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AGE | Advanced glycation end-products |
| CETP | Cholesterol ester transfer protein |
| DPP-4 | Dipeptidyl peptidase-4 |
| GLP-1 | Glucagon-like peptide-1 |
| HDL | High-density lipoprotein |
| LDL | Low-density lipoprotein |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PKC | Protein kinase C |
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 |
| RAGE | Advanced glycation end-product receptors |
| S1P | HDL-associated sphingosine-1-phosphate |
| SGLT2 | Sodium–glucose cotransporter 2 |
| T1DM | Type-1 diabetes mellitus |
| T2DM | Type-2 diabetes mellitus |
| vWF | von Willebrand factor |
References
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Al Alawi, A.M.; Majoni, S.W.; Falhammar, H. Magnesium and Human Health: Perspectives and Research Directions. Int. J. Endocrinol. 2018, 2018, 9041694. [Google Scholar] [CrossRef]
- Cerf, M.E. Beta cell dysfunction and insulin resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Alzahrani, S.H.; Ajjan, R.A. Coagulation and fibrinolysis in diabetes. Diab. Vasc. Dis. Res. 2010, 7, 260–273. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Blindauer, C.A.; Stewart, A.J. Changes in Plasma Free Fatty Acids Associated with Type-2 Diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Adjedj, J.; Varenne, O. Diabetes Mellitus, a prothrombotic disease. Ann. Cardiol. Angeiol. 2017, 66, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Haffner, S.M.; Lehto, S.; Ronnemaa, T.; Pyorala, K.; Laakso, M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N. Engl. J. Med. 1998, 339, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Rivas Rios, J.R.; Franchi, F.; Rollini, F.; Angiolillo, D.J. Diabetes and antiplatelet therapy: From bench to bedside. Cardiovasc. Diagn. Ther. 2018, 8, 594–609. [Google Scholar] [CrossRef]
- Verkleij, C.J.; Bruijn, R.E.; Meesters, E.W.; Gerdes, V.E.; Meijers, J.C.; Marx, P.F. The hemostatic system in patients with type 2 diabetes with and without cardiovascular disease. Clin. Appl. Thromb. Hemost. 2011, 17, E57–E63. [Google Scholar] [CrossRef]
- Galajda, P.; Martinka, E.; Mokan, M.; Kubisz, P. Endothelial markers in diabetes mellitus. Thromb. Res. 1997, 85, 63–65. [Google Scholar] [CrossRef]
- De Mattia, G.; Bravi, M.C.; Laurenti, O.; Moretti, A.; Cipriani, R.; Gatti, A.; Mandosi, E.; Morano, S. Endothelial dysfunction and oxidative stress in type 1 and type 2 diabetic patients without clinical macrovascular complications. Diabetes Res. Clin. Pract. 2008, 79, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Kedzierska, K.; Ciechanowski, K.; Golembiewska, E.; Safranow, K.; Ciechanowicz, A.; Domanski, L.; Myslak, M.; Rozanski, J. Plasma prekallikrein as a risk factor for diabetic retinopathy. Arch. Med. Res. 2005, 36, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Patrassi, G.M.; Vettor, R.; Padovan, D.; Girolami, A. Contact phase of blood coagulation in diabetes mellitus. Eur. J. Clin. Invest. 1982, 12, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, J.E.; Park, S.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S. High coagulation factor levels and low protein C levels contribute to enhanced thrombin generation in patients with diabetes who do not have macrovascular complications. J. Diabetes Complicat. 2014, 28, 365–369. [Google Scholar] [CrossRef] [PubMed]
- El-Hagracy, R.S.; Kamal, G.M.; Sabry, I.M.; Saad, A.A.; Abou El Ezz, N.F.; Nasr, H.A. Tissue Factor, Tissue Factor Pathway Inhibitor and Factor VII Activity in Cardiovascular Complicated Type 2 Diabetes Mellitus. Oman Med. J. 2010, 25, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Boden, G.; Homko, C.; Gunawardana, J.; Rao, A.K. Whole-blood tissue factor procoagulant activity is elevated in type 1 diabetes: Effects of hyperglycemia and hyperinsulinemia. Diabetes Care 2012, 35, 1322–1327. [Google Scholar] [CrossRef]
- Barillari, G.; Fabbro, E.; Pasca, S.; Bigotto, E. Coagulation and oxidative stress plasmatic levels in a type 2 diabetes population. Blood Coagul. Fibrinolysis 2009, 20, 290–296. [Google Scholar] [CrossRef]
- Fattah, M.A.; Shaheen, M.H.; Mahfouz, M.H. Disturbances of haemostasis in diabetes mellitus. Dis. Markers 2003, 19, 251–258. [Google Scholar] [CrossRef]
- Madan, R.; Gupt, B.; Saluja, S.; Kansra, U.C.; Tripathi, B.K.; Guliani, B.P. Coagulation profile in diabetes and its association with diabetic microvascular complications. J. Assoc. Physicians India 2010, 58, 481–484. [Google Scholar]
- Klein, R.L.; Hunter, S.J.; Jenkins, A.J.; Zheng, D.; Semler, A.J.; Clore, J.; Garvey, W.T.; Dcct/Ecic Study, G. Fibrinogen is a marker for nephropathy and peripheral vascular disease in type 1 diabetes: Studies of plasma fibrinogen and fibrinogen gene polymorphism in the DCCT/EDIC cohort. Diabetes Care 2003, 26, 1439–1448. [Google Scholar] [CrossRef]
- Agren, A.; Jorneskog, G.; Elgue, G.; Henriksson, P.; Wallen, H.; Wiman, B. Increased incorporation of antiplasmin into the fibrin network in patients with type 1 diabetes. Diabetes Care 2014, 37, 2007–2014. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, A.M.; Reis, M.L.; Melo, V.L.; Foss, M.C.; Gallo, L., Jr. Increased kininogen levels observed in plasma of diabetic patients are corrected by the administration of insulin. Horm. Metab. Res. 1999, 31, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Sommeijer, D.W.; Hansen, H.R.; van Oerle, R.; Hamulyak, K.; van Zanten, A.P.; Meesters, E.; Spronk, H.M.; ten Cate, H. Soluble tissue factor is a candidate marker for progression of microvascular disease in patients with Type 2 diabetes. J. Thromb. Haemost. 2006, 4, 574–580. [Google Scholar] [CrossRef] [PubMed]
- Song, D.Y.; Gu, J.Y.; Yoo, H.J.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Activation of Factor XII and Kallikrein-kinin System Combined with Neutrophil Extracellular Trap Formation in Diabetic Retinopathy. Exp. Clin. Endocrinol. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Mansfield, M.W.; Kohler, H.P.; Ariens, R.A.; McCormack, L.J.; Grant, P.J. Circulating levels of coagulation factor XIII in subjects with type 2 diabetes and in their first-degree relatives. Diabetes Care 2000, 23, 703–705. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Colhoun, H.M.; Zito, F.; Norman Chan, N.; Rubens, M.B.; Fuller, J.H.; Humphries, S.E. Activated factor XII levels and factor XII 46C>T genotype in relation to coronary artery calcification in patients with type 1 diabetes and healthy subjects. Atherosclerosis 2002, 163, 363–369. [Google Scholar] [CrossRef]
- Reverter, J.L.; Reverter, J.C.; Tassies, D.; Rius, F.; Monteagudo, J.; Rubies-Prat, J.; Escolar, G.; Ordinas, A.; Sanmarti, A. Thrombomodulin and induced tissue factor expression on monocytes as markers of diabetic microangiopathy: A prospective study on hemostasis and lipoproteins in insulin-dependent diabetes mellitus. Am. J. Hematol. 1997, 56, 93–99. [Google Scholar] [CrossRef]
- Aslan, B.; Eren, N.; Ciğerli, Ş.; Müldür, F.; Yücel, N. Evaluation of Plasma Protein C Antigen, Protein C Activity and Thrombomodulin Levels in Type 2 Diabetic Patients. Turk. J. Med. Sci. 2005, 35, 305–310. [Google Scholar]
- Yasuma, T.; Yano, Y.; D’Alessandro-Gabazza, C.N.; Toda, M.; Gil-Bernabe, P.; Kobayashi, T.; Nishihama, K.; Hinneh, J.A.; Mifuji-Moroka, R.; Roeen, Z.; et al. Amelioration of Diabetes by Protein S. Diabetes 2016, 65, 1940–1951. [Google Scholar] [CrossRef]
- Rigla, M.; Mateo, J.; Fontcuberta, J.; Souto, J.C.; de Leiva, A.; Perez, A. Normalisation of tissue factor pathway inhibitor activity after glycaemic control optimisation in type 1 diabetic patients. Thromb. Haemost. 2000, 84, 223–227. [Google Scholar] [CrossRef]
- Ceriello, A.; Giugliano, D.; Quatraro, A.; Stante, A.; Dello Russo, P.; Torella, R. Increased alpha 2-macroglobulin in diabetes: A hyperglycemia related phenomenon associated with reduced antithrombin III activity. Acta Diabetol. Lat. 1989, 26, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Borsey, D.Q.; Prowse, C.V.; Gray, R.S.; Dawes, J.; James, K.; Elton, R.A.; Clarke, B.F. Platelet and coagulation factors in proliferative diabetic retinopathy. J. Clin. Pathol. 1984, 37, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Glycosaminoglycan Neutralization in Coagulation Control. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Wasty, F.; Alavi, M.Z.; Moore, S. Distribution of glycosaminoglycans in the intima of human aortas: Changes in atherosclerosis and diabetes mellitus. Diabetologia 1993, 36, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Meigs, J.B.; Mittleman, M.A.; Nathan, D.M.; Tofler, G.H.; Singer, D.E.; Murphy-Sheehy, P.M.; Lipinska, I.; D’Agostino, R.B.; Wilson, P.W. Hyperinsulinemia, hyperglycemia, and impaired hemostasis: The Framingham Offspring Study. JAMA 2000, 283, 221–228. [Google Scholar] [CrossRef]
- Kubisz, P.; Stanciakova, L.; Stasko, J.; Galajda, P.; Mokan, M. Endothelial and platelet markers in diabetes mellitus type 2. World J. Diabetes 2015, 6, 423–431. [Google Scholar] [CrossRef]
- Juhan-Vague, I.; Roul, C.; Alessi, M.C.; Ardissone, J.P.; Heim, M.; Vague, P. Increased plasminogen activator inhibitor activity in non insulin dependent diabetic patients—Relationship with plasma insulin. Thromb. Haemost. 1989, 61, 370–373. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Phoenix, F.A.; Pitt, S.J.; Ajjan, R.A.; Stewart, A.J. Plasma magnesium levels associate with increased fibrin clot density and lysis time in individuals with type-1 diabetes and controls. Thromb. Haemost. (in press).
- Hori, Y.; Gabazza, E.C.; Yano, Y.; Katsuki, A.; Suzuki, K.; Adachi, Y.; Sumida, Y. Insulin resistance is associated with increased circulating level of thrombin-activatable fibrinolysis inhibitor in type 2 diabetic patients. J. Clin. Endocrinol. Metab 2002, 87, 660–665. [Google Scholar] [CrossRef]
- Sherif, E.M.; Elbarbary, N.S.; Abd Al Aziz, M.M.; Mohamed, S.F. Plasma thrombin-activatable fibrinolysis inhibitor levels in children and adolescents with type 1 diabetes mellitus: Possible relation to diabetic microvascular complications. Blood Coagul. Fibrinolysis 2014, 25, 451–457. [Google Scholar] [CrossRef]
- Yoshino, S.; Fujimoto, K.; Takada, T.; Kawamura, S.; Ogawa, J.; Kamata, Y.; Kodera, Y.; Shichiri, M. Molecular form and concentration of serum alpha2-macroglobulin in diabetes. Sci. Rep. 2019, 9, 12927. [Google Scholar] [CrossRef] [PubMed]
- Polat, S.B.; Ugurlu, N.; Yulek, F.; Simavli, H.; Ersoy, R.; Cakir, B.; Erel, O. Evaluation of serum fibrinogen, plasminogen, alpha2-anti-plasmin, and plasminogen activator inhibitor levels (PAI) and their correlation with presence of retinopathy in patients with type 1 DM. J. Diabetes Res. 2014, 2014, 317292. [Google Scholar] [CrossRef] [PubMed]
- Jax, T.W.; Peters, A.J.; Plehn, G.; Schoebel, F.C. Hemostatic risk factors in patients with coronary artery disease and type 2 diabetes—A two year follow-up of 243 patients. Cardiovasc. Diabetol. 2009, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Vazquez, B.Y.; Vazquez, M.A.; Jaquez, M.G.; Huemoeller, A.H.; Intaglietta, M.; Cabrales, P. Blood pressure directly correlates with blood viscosity in diabetes type 1 children but not in normals. Clin. Hemorheol. Microcirc. 2010, 44, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Singh, N. Blood viscosity, lipid profile, and lipid peroxidation in type-1 diabetic patients with good and poor glycemic control. N. Am. J. Med. Sci. 2013, 5, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Addai-Mensah, O.; Annani-Akollor, M.E.; Nsafoah, F.O.; Fondjo, L.A.; Owiredu, E.W.; Danquah, K.O.; Duneeh, R.V.; Amponsah, F.A. Effect of poor glycaemic control on plasma levels and activity of protein C, protein S, and antithrombin III in type 2 diabetes mellitus. PLoS ONE 2019, 14, e0223171. [Google Scholar] [CrossRef]
- Mansfield, M.W.; Heywood, D.M.; Grant, P.J. Circulating levels of factor VII, fibrinogen, and von Willebrand factor and features of insulin resistance in first-degree relatives of patients with NIDDM. Circulation 1996, 94, 2171–2176. [Google Scholar] [CrossRef]
- Leurs, P.B.; van Oerle, R.; Wolffenbuttel, B.H.; Hamulyak, K. Increased tissue factor pathway inhibitor (TFPI) and coagulation in patients with insulin-dependent diabetes mellitus. Thromb. Haemost. 1997, 77, 472–476. [Google Scholar] [CrossRef]
- Ceriello, A.; Quatraro, A.; Marchi, E.; Barbanti, M.; Dello Russo, P.; Lefebvre, P.; Giugliano, D. The role of hyperglycaemia-induced alterations of antithrombin III and factor X activation in the thrombin hyperactivity of diabetes mellitus. Diabet. Med. 1990, 7, 343–348. [Google Scholar] [CrossRef]
- Ajjan, R.A.; Gamlen, T.; Standeven, K.F.; Mughal, S.; Hess, K.; Smith, K.A.; Dunn, E.J.; Anwar, M.M.; Rabbani, N.; Thornalley, P.J.; et al. Diabetes is associated with posttranslational modifications in plasminogen resulting in reduced plasmin generation and enzyme-specific activity. Blood 2013, 122, 134–142. [Google Scholar] [CrossRef]
- Walmsley, D.; Hampton, K.K.; Grant, P.J. Contrasting fibrinolytic responses in type 1 (insulin-dependent) and type 2 (non-insulin-dependent) diabetes. Diabet. Med. 1991, 8, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Dunn, E.J.; Ariens, R.A.; Grant, P.J. The influence of type 2 diabetes on fibrin structure and function. Diabetologia 2005, 48, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Pieters, M.; van Zyl, D.G.; Rheeder, P.; Jerling, J.C.; Loots du, T.; van der Westhuizen, F.H.; Gottsche, L.T.; Weisel, J.W. Glycation of fibrinogen in uncontrolled diabetic patients and the effects of glycaemic control on fibrinogen glycation. Thromb. Res. 2007, 120, 439–446. [Google Scholar] [CrossRef]
- Pieters, M.; Covic, N.; van der Westhuizen, F.H.; Nagaswami, C.; Baras, Y.; Toit Loots, D.; Jerling, J.C.; Elgar, D.; Edmondson, K.S.; van Zyl, D.G.; et al. Glycaemic control improves fibrin network characteristics in type 2 diabetes - a purified fibrinogen model. Thromb. Haemost. 2008, 99, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, R.; Mignemi, N.; Rose, K.; O’Rear, L.; Sarilla, S.; Hamm, H.E.; Barnett, J.V.; Verhamme, I.M.; Schoenecker, J. The hyperglycemic byproduct methylglyoxal impairs anticoagulant activity through covalent adduction of antithrombin III. Thromb. Res. 2014, 134, 1350–1357. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.K.; Chouhan, V.; Chen, X.; Sun, L.; Boden, G. Activation of the tissue factor pathway of blood coagulation during prolonged hyperglycemia in young healthy men. Diabetes 1999, 48, 1156–1161. [Google Scholar] [CrossRef]
- Davi, G.; Gennaro, F.; Spatola, A.; Catalano, I.; Averna, M.; Montalto, G.; Amato, S.; Notarbartolo, A. Thrombin-antithrombin III complexes in type II diabetes mellitus. J. Diabetes Complicat. 1992, 6, 7–11. [Google Scholar] [CrossRef]
- Osende, J.I.; Badimon, J.J.; Fuster, V.; Herson, P.; Rabito, P.; Vidhun, R.; Zaman, A.; Rodriguez, O.J.; Lev, E.I.; Rauch, U.; et al. Blood thrombogenicity in type 2 diabetes mellitus patients is associated with glycemic control. J. Am. Coll. Cardiol. 2001, 38, 1307–1312. [Google Scholar] [CrossRef]
- Hess, K.; Alzahrani, S.H.; Mathai, M.; Schroeder, V.; Carter, A.M.; Howell, G.; Koko, T.; Strachan, M.W.; Price, J.F.; Smith, K.A.; et al. A novel mechanism for hypofibrinolysis in diabetes: The role of complement C3. Diabetologia 2012, 55, 1103–1113. [Google Scholar] [CrossRef]
- Hess, K.; Alzahrani, S.H.; Price, J.F.; Strachan, M.W.; Oxley, N.; King, R.; Gamlen, T.; Schroeder, V.; Baxter, P.D.; Ajjan, R.A. Hypofibrinolysis in type 2 diabetes: The role of the inflammatory pathway and complement C3. Diabetologia 2014, 57, 1737–1741. [Google Scholar] [CrossRef]
- Dunn, E.J.; Philippou, H.; Ariens, R.A.; Grant, P.J. Molecular mechanisms involved in the resistance of fibrin to clot lysis by plasmin in subjects with type 2 diabetes mellitus. Diabetologia 2006, 49, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Kearney, K.; Tomlinson, D.; Smith, K.; Ajjan, R. Hypofibrinolysis in diabetes: A therapeutic target for the reduction of cardiovascular risk. Cardiovasc. Diabetol. 2017, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Konieczynska, M.; Fil, K.; Bazanek, M.; Undas, A. Prolonged duration of type 2 diabetes is associated with increased thrombin generation, prothrombotic fibrin clot phenotype and impaired fibrinolysis. Thromb. Haemost. 2014, 111, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.H.; Hess, K.; Price, J.F.; Strachan, M.; Baxter, P.D.; Cubbon, R.; Phoenix, F.; Gamlen, T.; Ariens, R.A.; Grant, P.J.; et al. Gender-specific alterations in fibrin structure function in type 2 diabetes: Associations with cardiometabolic and vascular markers. J. Clin. Endocrinol. Metab. 2012, 97, E2282–E2287. [Google Scholar] [CrossRef] [PubMed]
- Bryk, A.H.; Siudut, J.; Broniatowska, E.; Bagoly, Z.; Barath, B.; Katona, E.; Undas, A. Sex-specific alteration to alpha2-antiplasmin incorporation in patients with type 2 diabetes. Thromb. Res. 2019, 185, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, S.; Jorneskog, G.; Agren, A.; Lins, P.E.; Wallen, H.; Antovic, A. Fibrin clot properties and haemostatic function in men and women with type 1 diabetes. Thromb. Haemost. 2015, 113, 312–318. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Stefanowicz, F.; Pitt, S.J.; Ajjan, R.A.; Stewart, A.J. Total plasma magnesium, zinc, copper and selenium concentrations in type-I and type-II diabetes. Biometals 2019. [Google Scholar] [CrossRef]
- Vu, T.T.; Fredenburgh, J.C.; Weitz, J.I. Zinc, an important cofactor in haemostasis and thrombosis. Thomb. Haemost. 2013, 109, 421–430. [Google Scholar] [CrossRef]
- Ripoll, L.; Mazoyer, E.; Baudin, B.; Lacam, S.; Varsat, B.; Drouet, L.O. Can high plasma ceruloplasmin levels cause acquired activated protein C resistance? Thromb. Haemost. 1998, 79, 449–450. [Google Scholar]
- Vasilyev, V.B. Looking for a partner: Ceruloplasmin in protein-protein interactions. Biometals 2019, 32, 195–210. [Google Scholar] [CrossRef]
- Mann, K.G.; Lawler, C.M.; Vehar, G.A.; Church, W.R. Coagulation Factor V contains copper ion. J. Biol. Chem. 1984, 259, 12949–12951. [Google Scholar] [PubMed]
- Tagliavacca, L.; Moon, N.; Dunham, W.R.; Kaufman, R.J. Identification and functional requirement of Cu(I) and its ligands within coagulation factor VIII. J. Biol. Chem. 1997, 272, 27428–27434. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Tomas, N.; Estruch, R.; Bullo, M.; Casas, R.; Diaz-Lopez, A.; Basora, J.; Fito, M.; Serra-Majem, L.; Salas-Salvado, J. Increased serum calcium levels and risk of type 2 diabetes in individuals at high cardiovascular risk. Diabetes Care 2014, 37, 3084–3091. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.R.; Pankow, J.S.; Sibley, S.D.; Selvin, E.; Reis, J.P.; Michos, E.D.; Lutsey, P.L. Serum calcium and incident type 2 diabetes: The Atherosclerosis Risk in Communities (ARIC) study. Am. J. Clin. Nutr. 2016, 104, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, C.; Hanley, A.J.; Rewers, M.J.; Haffner, S.M. Calcium and phosphate concentrations and future development of type 2 diabetes: The Insulin Resistance Atherosclerosis Study. Diabetologia 2014, 57, 1366–1374. [Google Scholar] [CrossRef]
- Jorde, R.; Schirmer, H.; Njolstad, I.; Lochen, M.L.; Bogeberg Mathiesen, E.; Kamycheva, E.; Figenschau, Y.; Grimnes, G. Serum calcium and the calcium-sensing receptor polymorphism rs17251221 in relation to coronary heart disease, type 2 diabetes, cancer and mortality: The Tromso Study. Eur. J. Epidemiol. 2013, 28, 569–578. [Google Scholar] [CrossRef]
- Levy, J.; Stern, Z.; Gutman, A.; Naparstek, Y.; Gavin, J.R., 3rd; Avioli, L.V. Plasma calcium and phosphate levels in an adult noninsulin-dependent diabetic population. Calcif. Tissue Int. 1986, 39, 316–318. [Google Scholar] [CrossRef]
- Singh, D.K.; Winocour, P.; Summerhayes, B.; Viljoen, A.; Sivakumar, G.; Farrington, K. Low serum osteoprotegerin levels in normoalbuminuric type 1 diabetes mellitus. Acta Diabetol. 2010, 47 (Suppl. 1), 105–110. [Google Scholar] [CrossRef]
- Wierzbicka, E.; Szalecki, M.; Pludowski, P.; Jaworski, M.; Brzozowska, A. Vitamin D status, body composition and glycemic control in Polish adolescents with type 1 diabetes. Minerva Endocrinol. 2016, 41, 445–455. [Google Scholar]
- Tankeu, A.T.; Ndip Agbor, V.; Noubiap, J.J. Calcium supplementation and cardiovascular risk: A rising concern. J. Clin. Hypertens. 2017, 19, 640–646. [Google Scholar] [CrossRef]
- Sobczak, A.I.S.; Pitt, S.J.; Stewart, A.J. Influence of zinc on glycosaminoglycan neutralisation during coagulation. Metallomics 2018, 10, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Sanjeevi, N.; Freeland-Graves, J.; Beretvas, S.N.; Sachdev, P.K. Trace element status in type 2 diabetes: A meta-analysis. J. Clin. Diagn. Res. 2018, 12, OE01–OE08. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, C.; Li, L.; Shao, M.; Wang, L.; Lv, X.; Gao, C.; Niu, H.; Li, B. The Study on the Correlation Between Six Kinds of Mineral Elements and Diabetes. Biol. Trace Elem. Res. 2018, 183, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, A.I.S.; Katundu, K.G.H.; Phoenix, F.A.; Khazaipoul, S.; Yu, R.; Lampiao, F.; Stefanowicz, F.; Blindauer, C.A.; Pitt, S.J.; Smith, T.K.; et al. Plasma non-esterified fatty acids contribute to increased coagulability in type-2 diabetes through altered plasma zinc speciation (preprint). bioRxiv 2019. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Barnett, J.P.; Adamu, A.H.; Griffiths, E.J.; Stewart, A.J.; Blindauer, C.A. A metalloproteomic analysis of interactions between plasma proteins and zinc: Elevated fatty acid levels affect zinc distribution. Metallomics 2019. [Google Scholar] [CrossRef]
- Sekiya, F.; Yoshida, M.; Yamashita, T.; Morita, T. Magnesium(II) is a crucial constituent of the blood coagulation cascade. Potentiation of coagulant activities of factor IX by Mg2+ ions. J. Biol. Chem. 1996, 271, 8541–8544. [Google Scholar] [CrossRef]
- Vadivel, K.; Agah, S.; Messer, A.S.; Cascio, D.; Bajaj, M.S.; Krishnaswamy, S.; Esmon, C.T.; Padmanabhan, K.; Bajaj, S.P. Structural and functional studies of gamma-carboxyglutamic acid domains of factor VIIa and activated Protein C: Role of magnesium at physiological calcium. J. Mol. Biol. 2013, 425, 1961–1981. [Google Scholar] [CrossRef][Green Version]
- Tokutake, T.; Baba, H.; Shimada, Y.; Takeda, W.; Sato, K.; Hiroshima, Y.; Kirihara, T.; Shimizu, I.; Nakazawa, H.; Kobayashi, H.; et al. Exogenous Magnesium Chloride Reduces the Activated Partial Thromboplastin Times of Lupus Anticoagulant-Positive Patients. PLoS ONE 2016, 11, e0157835. [Google Scholar] [CrossRef]
- Van den Besselaar, A.M.; Witteveen, E.; Meeuwisse-Braun, J.; van der Meer, F.J. The influence of exogenous magnesium chloride on the apparent INR determined with human, rabbit, and bovine thromboplastin reagents. Thromb. Haemost. 2003, 89, 43–47. [Google Scholar]
- Jankun, J.; Skrzypczak-Jankun, E.; Lipinski, B. Complex function of magnesium in blood clot formation and lysis. Cent. Eur. J. Immunol. 2013, 38, 149–153. [Google Scholar] [CrossRef]
- Anstall, H.B.; Hayward, G.H.; Huntsman, R.G.; Weitzman, D.; Lehmann, H. The effect of magnesium on blood coagulation in human subjects. Lancet 1959, 1, 814–815. [Google Scholar] [CrossRef]
- Efstratiadis, G.; Sarigianni, M.; Gougourelas, I. Hypomagnesemia and cardiovascular system. Hippokratia 2006, 10, 147–152. [Google Scholar] [PubMed]
- Barbagallo, M.; Dominguez, L.J. Magnesium and type 2 diabetes. World J. Diabetes 2015, 6, 1152–1157. [Google Scholar] [CrossRef]
- Squitti, R.; Negrouk, V.; Perera, M.; Llabre, M.M.; Ricordi, C.; Rongioletti, M.C.A.; Mendez, A.J. Serum copper profile in patients with type 1 diabetes in comparison to other metals. J. Trace Elem. Med. Biol. 2019, 56, 156–161. [Google Scholar] [CrossRef]
- Keen, C.L.; Feldman, B.F.; Knight, J.; O’Neill, S.; Ferrell, F.; Hurley, L.S. The influence of high concentrations of dietary copper on vitamin K-dependent coagulation factors. Proc. Soc. Exp. Biol. Med. 1982, 170, 471–475. [Google Scholar] [CrossRef]
- Memisogullari, R.; Bakan, E. Levels of ceruloplasmin, transferrin, and lipid peroxidation in the serum of patients with Type 2 diabetes mellitus. J. Diabetes Complicat. 2004, 18, 193–197. [Google Scholar] [CrossRef]
- Bergis, D.; Tessmer, L.; Badenhoop, K. Iron deficiency in long standing type 1 diabetes mellitus and its association with depression and impaired quality of life. Diabetes Res. Clin. Pract. 2019, 151, 74–81. [Google Scholar] [CrossRef]
- Akkermans, M.D.; Mieke Houdijk, E.C.A.; Bakker, B.; Boers, A.C.; van der Kaay, D.C.M.; de Vries, M.C.; Claire Woltering, M.; Mul, D.; van Goudoever, J.B.; Brus, F. Iron status and its association with HbA1c levels in Dutch children with diabetes mellitus type 1. Eur. J. Pediatr. 2018, 177, 603–610. [Google Scholar] [CrossRef]
- Jankun, J.; Landeta, P.; Pretorius, E.; Skrzypczak-Jankun, E.; Lipinski, B. Unusual clotting dynamics of plasma supplemented with iron(III). Int. J. Mol. Med. 2014, 33, 367–372. [Google Scholar] [CrossRef]
- Lipinski, B.; Pretorius, E. Iron-induced fibrin in cardiovascular disease. Curr. Neurovasc. Res. 2013, 10, 269–274. [Google Scholar] [CrossRef] [PubMed]
- De Ferranti, S.D.; de Boer, I.H.; Fonseca, V.; Fox, C.S.; Golden, S.H.; Lavie, C.J.; Magge, S.N.; Marx, N.; McGuire, D.K.; Orchard, T.J.; et al. Type 1 diabetes mellitus and cardiovascular disease: A scientific statement from the American Heart Association and American Diabetes Association. Circulation 2014, 130, 1110–1130. [Google Scholar] [CrossRef] [PubMed]
- Lazarte, J.; Hegele, R.A. Dyslipidemia Management in Adults With Diabetes. Can. J. Diabetes 2019. [Google Scholar] [CrossRef] [PubMed]
- Schofield, J.; Ho, J.; Soran, H. Cardiovascular Risk in Type 1 Diabetes Mellitus. Diabetes Ther. 2019, 10, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Costacou, T.; Evans, R.W.; Orchard, T.J. High-density lipoprotein cholesterol in diabetes: Is higher always better? J. Clin. Lipidol. 2011, 5, 387–394. [Google Scholar] [CrossRef] [PubMed]
- Ravnskov, U.; de Lorgeril, M.; Diamond, D.M.; Hama, R.; Hamazaki, T.; Hammarskjold, B.; Hynes, N.; Kendrick, M.; Langsjoen, P.H.; Mascitelli, L.; et al. LDL-C does not cause cardiovascular disease: A comprehensive review of the current literature. Expert Rev. Clin. Pharmacol. 2018, 11, 959–970. [Google Scholar] [CrossRef]
- Hero, C.; Svensson, A.M.; Gidlund, P.; Gudbjornsdottir, S.; Eliasson, B.; Eeg-Olofsson, K. LDL cholesterol is not a good marker of cardiovascular risk in Type 1 diabetes. Diabet. Med. 2016, 33, 316–323. [Google Scholar] [CrossRef]
- Soedamah-Muthu, S.S.; Chaturvedi, N.; Toeller, M.; Ferriss, B.; Reboldi, P.; Michel, G.; Manes, C.; Fuller, J.H.; Group, E.P.C.S. Risk factors for coronary heart disease in type 1 diabetic patients in Europe: The EURODIAB Prospective Complications Study. Diabetes Care 2004, 27, 530–537. [Google Scholar] [CrossRef]
- Berneis, K.K.; Krauss, R.M. Metabolic origins and clinical significance of LDL heterogeneity. J. Lipid Res. 2002, 43, 1363–1379. [Google Scholar] [CrossRef]
- Jenkins, A.J.; Best, J.D.; Klein, R.L.; Lyons, T.J. ‘Lipoproteins, glycoxidation and diabetic angiopathy’. Diabetes Metab Res. Rev. 2004, 20, 349–368. [Google Scholar] [CrossRef]
- Taskinen, M.R. LDL-cholesterol, HDL-cholesterol or triglycerides--which is the culprit? Diabetes Res. Clin. Pract 2003, 61 (Suppl. 1), S19–S26. [Google Scholar] [CrossRef]
- Soran, H.; Durrington, P.N. Susceptibility of LDL and its subfractions to glycation. Curr. Opin. Lipidol. 2011, 22, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Deckert, V.; Brunet, A.; Lantoine, F.; Lizard, G.; Millanvoye-van Brussel, E.; Monier, S.; Lagrost, L.; David-Dufilho, M.; Gambert, P.; Devynck, M.A. Inhibition by cholesterol oxides of NO release from human vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1054–1060. [Google Scholar] [CrossRef] [PubMed]
- Steinbrecher, U.P.; Zhang, H.F.; Lougheed, M. Role of oxidatively modified LDL in atherosclerosis. Free Radic. Biol. Med. 1990, 9, 155–168. [Google Scholar] [CrossRef]
- Li, X.; Guan, B.; Wang, Y.; Tse, G.; Zou, F.; Khalid, B.W.; Xia, Y.; Wu, S.; Sun, J. Association between high-density lipoprotein cholesterol and all-cause mortality in the general population of northern China. Sci. Rep. 2019, 9, 14426. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Trigatti, B.L.; Mineo, C.; Knaack, D.; Wilkins, J.T.; Sahoo, D.; Asztalos, B.F.; Mora, S.; Cuchel, M.; Pownall, H.J.; et al. Proceedings of the Ninth HDL (High-Density Lipoprotein) Workshop: Focus on Cardiovascular Disease. Arterioscler. Thromb. Vasc. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Kothari, V.; Bornfeldt, K.E. High-Density Lipoprotein Function in Cardiovascular Disease and Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e10–e16. [Google Scholar] [CrossRef]
- Shea, S.; Stein, J.H.; Jorgensen, N.W.; McClelland, R.L.; Tascau, L.; Shrager, S.; Heinecke, J.W.; Yvan-Charvet, L.; Tall, A.R. Cholesterol Mass Efflux Capacity, Incident Cardiovascular Disease, and Progression of Carotid Plaque. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 89–96. [Google Scholar] [CrossRef]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef]
- Ogura, M.; Hori, M.; Harada-Shiba, M. Association Between Cholesterol Efflux Capacity and Atherosclerotic Cardiovascular Disease in Patients With Familial Hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 181–188. [Google Scholar] [CrossRef]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef]
- Khera, A.V.; Demler, O.V.; Adelman, S.J.; Collins, H.L.; Glynn, R.J.; Ridker, P.M.; Rader, D.J.; Mora, S. Cholesterol Efflux Capacity, High-Density Lipoprotein Particle Number, and Incident Cardiovascular Events: An Analysis From the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2017, 135, 2494–2504. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L.E.; Sacks, F.M.; Rimm, E.B.; Jensen, M.K. Cholesterol efflux capacity, HDL cholesterol, and risk of coronary heart disease: A nested case-control study in men. J. Lipid Res. 2019, 60, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- El Khoudary, S.R.; Ceponiene, I.; Samargandy, S.; Stein, J.H.; Li, D.; Tattersall, M.C.; Budoff, M.J. HDL (High-Density Lipoprotein) Metrics and Atherosclerotic Risk in Women. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2236–2244. [Google Scholar] [CrossRef] [PubMed]
- Zimetti, F.; Freitas, W.M.; Campos, A.M.; Daher, M.; Adorni, M.P.; Bernini, F.; Sposito, A.C.; Zanotti, I.; Brazilian Study on Healthy, A. Cholesterol efflux capacity does not associate with coronary calcium, plaque vulnerability, and telomere length in healthy octogenarians. J. Lipid Res. 2018, 59, 714–721. [Google Scholar] [CrossRef]
- Saleheen, D.; Scott, R.; Javad, S.; Zhao, W.; Rodrigues, A.; Picataggi, A.; Lukmanova, D.; Mucksavage, M.L.; Luben, R.; Billheimer, J.; et al. Association of HDL cholesterol efflux capacity with incident coronary heart disease events: A prospective case-control study. Lancet Diabetes Endocrinol. 2015, 3, 507–513. [Google Scholar] [CrossRef]
- Apro, J.; Tietge, U.J.; Dikkers, A.; Parini, P.; Angelin, B.; Rudling, M. Impaired Cholesterol Efflux Capacity of High-Density Lipoprotein Isolated From Interstitial Fluid in Type 2 Diabetes Mellitus-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 787–791. [Google Scholar] [CrossRef]
- Gourgari, E.; Playford, M.P.; Campia, U.; Dey, A.K.; Cogen, F.; Gubb-Weiser, S.; Mete, M.; Desale, S.; Sampson, M.; Taylor, A.; et al. Low cholesterol efflux capacity and abnormal lipoprotein particles in youth with type 1 diabetes: A case control study. Cardiovasc. Diabetol. 2018, 17, 158. [Google Scholar] [CrossRef]
- Mehta, N.U.; Grijalva, V.; Hama, S.; Wagner, A.; Navab, M.; Fogelman, A.M.; Reddy, S.T. Apolipoprotein E-/- Mice Lacking Hemopexin Develop Increased Atherosclerosis via Mechanisms That Include Oxidative Stress and Altered Macrophage Function. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1152–1163. [Google Scholar] [CrossRef]
- Sorrentino, S.A.; Besler, C.; Rohrer, L.; Meyer, M.; Heinrich, K.; Bahlmann, F.H.; Mueller, M.; Horvath, T.; Doerries, C.; Heinemann, M.; et al. Endothelial-vasoprotective effects of high-density lipoprotein are impaired in patients with type 2 diabetes mellitus but are improved after extended-release niacin therapy. Circulation 2010, 121, 110–122. [Google Scholar] [CrossRef]
- Brinck, J.W.; Thomas, A.; Lauer, E.; Jornayvaz, F.R.; Brulhart-Meynet, M.C.; Prost, J.C.; Pataky, Z.; Lofgren, P.; Hoffstedt, J.; Eriksson, M.; et al. Diabetes Mellitus Is Associated With Reduced High-Density Lipoprotein Sphingosine-1-Phosphate Content and Impaired High-Density Lipoprotein Cardiac Cell Protection. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Frej, C.; Mendez, A.J.; Ruiz, M.; Castillo, M.; Hughes, T.A.; Dahlback, B.; Goldberg, R.B. A Shift in ApoM/S1P Between HDL-Particles in Women With Type 1 Diabetes Mellitus Is Associated With Impaired Anti-Inflammatory Effects of the ApoM/S1P Complex. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Hartz, J.C.; de Ferranti, S.; Gidding, S. Hypertriglyceridemia in Diabetes Mellitus: Implications for Pediatric Care. J. Endocr. Soc. 2018, 2, 497–512. [Google Scholar] [CrossRef] [PubMed]
- Selim, S. Frequency and pattern of chronic complications of diabetes and their association with glycemic control. Diabetes Metab. Syndr. 2017, 11 (Suppl. 1), S311–S314. [Google Scholar] [CrossRef]
- Kushner, P.A.; Cobble, M.E. Hypertriglyceridemia: The importance of identifying patients at risk. Postgrad. Med. 2016, 128, 848–858. [Google Scholar] [CrossRef]
- Girona, J.; Amigo, N.; Ibarretxe, D.; Plana, N.; Rodriguez-Borjabad, C.; Heras, M.; Ferre, R.; Gil, M.; Correig, X.; Masana, L. HDL Triglycerides: A New Marker of Metabolic and Cardiovascular Risk. Int. J. Mol. Sci. 2019, 20, 3151. [Google Scholar] [CrossRef]
- Amigo, N.; Mallol, R.; Heras, M.; Martinez-Hervas, S.; Blanco Vaca, F.; Escola-Gil, J.C.; Plana, N.; Yanes, O.; Masana, L.; Correig, X. Lipoprotein hydrophobic core lipids are partially extruded to surface in smaller HDL: “Herniated” HDL, a common feature in diabetes. Sci. Rep. 2016, 6, 19249. [Google Scholar] [CrossRef]
- Verges, B. Lipid modification in type 2 diabetes: The role of LDL and HDL. Fundam. Clin. Pharmacol. 2009, 23, 681–685. [Google Scholar] [CrossRef]
- Holmes, M.V.; Millwood, I.Y.; Kartsonaki, C.; Hill, M.R.; Bennett, D.A.; Boxall, R.; Guo, Y.; Xu, X.; Bian, Z.; Hu, R.; et al. Lipids, Lipoproteins, and Metabolites and Risk of Myocardial Infarction and Stroke. J. Am. Coll. Cardiol. 2018, 71, 620–632. [Google Scholar] [CrossRef]
- Dutta, T.; Kudva, Y.C.; Persson, X.M.; Schenck, L.A.; Ford, G.C.; Singh, R.J.; Carter, R.; Nair, K.S. Impact of Long-Term Poor and Good Glycemic Control on Metabolomics Alterations in Type 1 Diabetic People. J. Clin. Endocrinol. Metab. 2016, 101, 1023–1033. [Google Scholar] [CrossRef]
- Ghosh, A.; Gao, L.; Thakur, A.; Siu, P.M.; Lai, C.W.K. Role of free fatty acids in endothelial dysfunction. J. Biomed. Sci. 2017, 24, 50. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, R.L.; Li, W.; Park, Y.M.; Rahaman, S.O. Mechanisms of cell signaling by the scavenger receptor CD36: Implications in atherosclerosis and thrombosis. Trans. Am. Clin. Climatol. Assoc. 2010, 121, 206–220. [Google Scholar] [PubMed]
- Tanka-Salamon, A.; Komorowicz, E.; Szabo, L.; Tenekedjiev, K.; Kolev, K. Free Fatty Acids Modulate Thrombin Mediated Fibrin Generation Resulting in Less Stable Clots. PLoS ONE 2016, 11, e0167806. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.C. Abnormal myocardial dietary fatty acid metabolism and diabetic cardiomyopathy. Can. J. Cardiol. 2018, 34, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Palomer, X.; Salvado, L.; Barroso, E.; Vazquez-Carrera, M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int. J. Cardiol. 2013, 168, 3160–3172. [Google Scholar] [CrossRef]
- Ali, A.; Boutjdir, M.; Aromolaran, A.S. Cardiolipotoxicity, inflammation, and arrhythmias: Role for Interleukin-6 molecular mechanisms. Front. Physiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Westein, E.; Hoefer, T.; Calkin, A.C. Thrombosis in diabetes: A shear flow effect? Clin. Sci. 2017, 131, 1245–1260. [Google Scholar] [CrossRef]
- Muniyappa, R.; Montagnani, M.; Koh, K.K.; Quon, M.J. Cardiovascular actions of insulin. Endocr. Rev. 2007, 28, 463–491. [Google Scholar] [CrossRef]
- Chen, Q.; Jin, M.; Yang, F.; Zhu, J.; Xiao, Q.; Zhang, L. Matrix metalloproteinases: Inflammatory regulators of cell behaviors in vascular formation and remodeling. Mediators Inflamm. 2013, 2013, 928315. [Google Scholar] [CrossRef]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Schalkwijk, C.G.; Stehouwer, C.D.A. Plasma matrix metalloproteinases are associated with incident cardiovascular disease and all-cause mortality in patients with type 1 diabetes: A 12-year follow-up study. Cardiovasc. Diabetol. 2017, 16, 55. [Google Scholar] [CrossRef]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Chaturvedi, N.; Fuller, J.H.; Schalkwijk, C.G.; Stehouwer, C.D.; Group, E.P.C.S. Plasma levels of matrix metalloproteinase-2, -3, -10, and tissue inhibitor of metalloproteinase-1 are associated with vascular complications in patients with type 1 diabetes: The EURODIAB Prospective Complications Study. Cardiovasc. Diabetol. 2015, 14, 31. [Google Scholar] [CrossRef]
- Peeters, S.A.; Engelen, L.; Buijs, J.; Chaturvedi, N.; Fuller, J.H.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Theilade, S.; Rossing, P.; et al. Circulating matrix metalloproteinases are associated with arterial stiffness in patients with type 1 diabetes: Pooled analysis of three cohort studies. Cardiovasc. Diabetol. 2017, 16, 139. [Google Scholar] [CrossRef]
- Kozakova, M.; Morizzo, C.; Goncalves, I.; Natali, A.; Nilsson, J.; Palombo, C. Cardiovascular organ damage in type 2 diabetes mellitus: The role of lipids and inflammation. Cardiovasc. Diabetol. 2019, 18, 61. [Google Scholar] [CrossRef]
- Chase, A.J.; Newby, A.C. Regulation of matrix metalloproteinase (matrixin) genes in blood vessels: A multi-step recruitment model for pathological remodelling. J. Vasc. Res. 2003, 40, 329–343. [Google Scholar] [CrossRef]
- Giebel, S.J.; Menicucci, G.; McGuire, P.G.; Das, A. Matrix metalloproteinases in early diabetic retinopathy and their role in alteration of the blood-retinal barrier. Lab. Invest. 2005, 85, 597–607. [Google Scholar] [CrossRef]
- Shah, P.K. Plaque disruption and thrombosis: Potential role of inflammation and infection. Cardiol. Rev. 2000, 8, 31–39. [Google Scholar] [CrossRef]
- Kadoglou, N.P.; Daskalopoulou, S.S.; Perrea, D.; Liapis, C.D. Matrix metalloproteinases and diabetic vascular complications. Angiology 2005, 56, 173–189. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Abbas, M.; Morel, N. Prothrombotic changes in diabetes mellitus. Semin. Thromb. Hemost. 2013, 39, 477–488. [Google Scholar] [CrossRef]
- Trovati, M.; Anfossi, G.; Massucco, P.; Mattiello, L.; Costamagna, C.; Piretto, V.; Mularoni, E.; Cavalot, F.; Bosia, A.; Ghigo, D. Insulin stimulates nitric oxide synthesis in human platelets and, through nitric oxide, increases platelet concentrations of both guanosine-3′, 5′-cyclic monophosphate and adenosine-3′, 5′-cyclic monophosphate. Diabetes 1997, 46, 742–749. [Google Scholar] [CrossRef]
- Westerbacka, J.; Yki-Jarvinen, H.; Turpeinen, A.; Rissanen, A.; Vehkavaara, S.; Syrjala, M.; Lassila, R. Inhibition of platelet-collagen interaction: An in vivo action of insulin abolished by insulin resistance in obesity. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 167–172. [Google Scholar] [CrossRef]
- Winocour, P.D.; Watala, C.; Perry, D.W.; Kinlough-Rathbone, R.L. Decreased platelet membrane fluidity due to glycation or acetylation of membrane proteins. Thromb. Haemost. 1992, 68, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Assert, R.; Scherk, G.; Bumbure, A.; Pirags, V.; Schatz, H.; Pfeiffer, A.F. Regulation of protein kinase C by short term hyperglycaemia in human platelets in vivo and in vitro. Diabetologia 2001, 44, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, G.; Wascher, T.C.; Kostner, G.M.; Graier, W.F. Alterations in platelet Ca2+ signalling in diabetic patients is due to increased formation of superoxide anions and reduced nitric oxide production. Diabetologia 1999, 42, 167–176. [Google Scholar] [CrossRef]
- Thomas, G.; Skrinska, V.; Lucas, F.V.; Schumacher, O.P. Platelet glutathione and thromboxane synthesis in diabetes. Diabetes 1985, 34, 951–954. [Google Scholar] [CrossRef]
- Randriamboavonjy, V.; Pistrosch, F.; Bolck, B.; Schwinger, R.H.; Dixit, M.; Badenhoop, K.; Cohen, R.A.; Busse, R.; Fleming, I. Platelet sarcoplasmic endoplasmic reticulum Ca2+-ATPase and mu-calpain activity are altered in type 2 diabetes mellitus and restored by rosiglitazone. Circulation 2008, 117, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Keating, F.K.; Sobel, B.E.; Schneider, D.J. Effects of increased concentrations of glucose on platelet reactivity in healthy subjects and in patients with and without diabetes mellitus. Am. J. Cardiol. 2003, 92, 1362–1365. [Google Scholar] [CrossRef]
- Tschoepe, D.; Roesen, P.; Esser, J.; Schwippert, B.; Nieuwenhuis, H.K.; Kehrel, B.; Gries, F.A. Large platelets circulate in an activated state in diabetes mellitus. Semin. Thromb. Hemost. 1991, 17, 433–438. [Google Scholar] [CrossRef]
- Zaccardi, F.; Rocca, B.; Rizzi, A.; Ciminello, A.; Teofili, L.; Ghirlanda, G.; De Stefano, V.; Pitocco, D. Platelet indices and glucose control in type 1 and type 2 diabetes mellitus: A case-control study. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 902–909. [Google Scholar] [CrossRef]
- Santilli, F.; Marchisio, M.; Lanuti, P.; Boccatonda, A.; Miscia, S.; Davi, G. Microparticles as new markers of cardiovascular risk in diabetes and beyond. Thromb. Haemost. 2016, 116, 220–234. [Google Scholar] [CrossRef]
- Zara, M.; Guidetti, G.F.; Camera, M.; Canobbio, I.; Amadio, P.; Torti, M.; Tremoli, E.; Barbieri, S.S. Biology and Role of Extracellular Vesicles (EVs) in the Pathogenesis of Thrombosis. Int. J. Mol. Sci. 2019, 20, 2840. [Google Scholar] [CrossRef]
- Tramontano, A.F.; Lyubarova, R.; Tsiakos, J.; Palaia, T.; Deleon, J.R.; Ragolia, L. Circulating endothelial microparticles in diabetes mellitus. Mediators Inflamm. 2010, 2010, 250476. [Google Scholar] [CrossRef] [PubMed]
- Stec, J.J.; Silbershatz, H.; Tofler, G.H.; Matheney, T.H.; Sutherland, P.; Lipinska, I.; Massaro, J.M.; Wilson, P.F.; Muller, J.E.; D’Agostino, R.B., Sr. Association of fibrinogen with cardiovascular risk factors and cardiovascular disease in the Framingham Offspring Population. Circulation 2000, 102, 1634–1638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; McGeoch, S.C.; Johnstone, A.M.; Holtrop, G.; Sneddon, A.A.; MacRury, S.M.; Megson, I.L.; Pearson, D.W.; Abraham, P.; De Roos, B.; et al. Platelet-derived microparticle count and surface molecule expression differ between subjects with and without type 2 diabetes, independently of obesity status. J. Thromb. Thrombolysis 2014, 37, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.D.; Wu, X.Z.; Zhou, Y.; Xue, Y.; Zhang, K.Q. Proteomic characteristics of circulating microparticles in patients with newly-diagnosed type 2 diabetes. Am. J. Transl. Res. 2016, 8, 209–220. [Google Scholar] [PubMed]
- Zahran, A.M.; Mohamed, I.L.; El Asheer, O.M.; Tamer, D.M.; Abo, E.M.G.M.; Abdel-Rahim, M.H.; El-Badawy, O.H.B.; Elsayh, K.I. Circulating Endothelial Cells, Circulating Endothelial Progenitor Cells, and Circulating Microparticles in Type 1 Diabetes Mellitus. Clin. Appl. Thromb. Hemost. 2019, 25, 1076029618825311. [Google Scholar] [CrossRef] [PubMed]
- Sabatier, F.; Darmon, P.; Hugel, B.; Combes, V.; Sanmarco, M.; Velut, J.G.; Arnoux, D.; Charpiot, P.; Freyssinet, J.M.; Oliver, C.; et al. Type 1 and type 2 diabetic patients display different patterns of cellular microparticles. Diabetes 2002, 51, 2840–2845. [Google Scholar] [CrossRef]
- Salem, M.A.; Adly, A.A.; Ismail, E.A.; Darwish, Y.W.; Kamel, H.A. Platelets microparticles as a link between micro- and macro-angiopathy in young patients with type 1 diabetes. Platelets 2015, 26, 682–688. [Google Scholar] [CrossRef]
- Van Beers, E.J.; Schaap, M.C.; Berckmans, R.J.; Nieuwland, R.; Sturk, A.; van Doormaal, F.F.; Meijers, J.C.; Biemond, B.J.; group, C.s. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 2009, 94, 1513–1519. [Google Scholar] [CrossRef]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Invest. 1997, 99, 2118–2127. [Google Scholar] [CrossRef]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renne, T.; ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef]
- Donga, E.; Dekkers, O.M.; Corssmit, E.P.; Romijn, J.A. Insulin resistance in patients with type 1 diabetes assessed by glucose clamp studies: Systematic review and meta-analysis. Eur. J. Endocrinol. 2015, 173, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Cucuianu, M.; Coca, M. Thrombotic tendency in diabetes mellitus. Revisiting and revising a study initiated 30 years ago. Rom. J. Intern. Med. 2012, 50, 107–115. [Google Scholar] [PubMed]
- Zheng, Q.; Jiang, J.; Huo, Y.; Chen, D. Genetic predisposition to type 2 diabetes is associated with severity of coronary artery disease in patients with acute coronary syndromes. Cardiovasc. Diabetol. 2019, 18, 131. [Google Scholar] [CrossRef] [PubMed]
- Pfister, R.; Barnes, D.; Luben, R.N.; Khaw, K.T.; Wareham, N.J.; Langenberg, C. Individual and cumulative effect of type 2 diabetes genetic susceptibility variants on risk of coronary heart disease. Diabetologia 2011, 54, 2283–2287. [Google Scholar] [CrossRef] [PubMed]
- Qi, Q.; Meigs, J.B.; Rexrode, K.M.; Hu, F.B.; Qi, L. Diabetes genetic predisposition score and cardiovascular complications among patients with type 2 diabetes. Diabetes Care 2013, 36, 737–739. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.; Arcidiacono, B.; Chiefari, E.; Brunetti, A.; Indolfi, C.; Foti, D.P. Type 2 Diabetes Mellitus and Cardiovascular Disease: Genetic and Epigenetic Links. Front. Endocrinol. 2018, 9, 2. [Google Scholar] [CrossRef]
- Takaike, H.; Uchigata, Y.; Nakagami, T.; Iwamoto, Y. Incidence and development of diabetic microangiopathy of fulminant type 1 diabetes—Comparison with non-fulminant type 1 diabetes. Intern. Med. 2010, 49, 1079–1083. [Google Scholar] [CrossRef]
- Murase, Y.; Imagawa, A.; Hanafusa, T.; Iwahashi, H.; Uchigata, Y.; Kanatsuka, A.; Kawasaki, E.; Kobayashi, T.; Shimada, A.; Shimizu, I.; et al. Fulminant type 1 diabetes as a high risk group for diabetic microangiopathy—A nationwide 5-year-study in Japan. Diabetologia 2007, 50, 531–537. [Google Scholar] [CrossRef]
- Guarnotta, V.; Vigneri, E.; Pillitteri, G.; Ciresi, A.; Pizzolanti, G.; Giordano, C. Higher cardiometabolic risk in idiopathic versus autoimmune type 1 diabetes: A retrospective analysis. Diabetol. Metab. Syndr. 2018, 10, 40. [Google Scholar] [CrossRef]
- National Institute for Health and Clinical Excellence. Type 1 Diabetes in Adults: Diagnosis and Management, NICE Guideline (NG17). Available online: https://www.nice.org.uk/guidance/ng17 (accessed on 4 September 2019).
- National Institute for Health and Clinical Excellence. Diabetes (Type 1 and Type 2) in Children and Young People: Diagnosis and Management, NICE Guideline (NG18). Available online: https://www.nice.org.uk/guidance/ng18 (accessed on 4 September 2019).
- National Institute for Health and Clinical Excellence. National Institute for Health and Clinical Excellence. Type 2 Diabetes in Adults: Management, NICE Guideline (NG28). Available online: https://www.nice.org.uk/guidance/ng28 (accessed on 31 October 2019).
- Cefalu, W.T.; Schneider, D.J.; Carlson, H.E.; Migdal, P.; Gan Lim, L.; Izon, M.P.; Kapoor, A.; Bell-Farrow, A.; Terry, J.G.; Sobel, B.E. Effect of combination glipizide GITS/metformin on fibrinolytic and metabolic parameters in poorly controlled type 2 diabetic subjects. Diabetes Care 2002, 25, 2123–2128. [Google Scholar] [CrossRef]
- Zilov, A.V.; Abdelaziz, S.I.; AlShammary, A.; Al Zahrani, A.; Amir, A.; Assaad Khalil, S.H.; Brand, K.; Elkafrawy, N.; Hassoun, A.A.K.; Jahed, A.; et al. Mechanisms of action of metformin with special reference to cardiovascular protection. Diabetes Metab. Res. Rev. 2019, 35, e3173. [Google Scholar] [CrossRef] [PubMed]
- Lamanna, C.; Monami, M.; Marchionni, N.; Mannucci, E. Effect of metformin on cardiovascular events and mortality: A meta-analysis of randomized clinical trials. Diabetes Obes. Metab. 2011, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.J.; Leaver, J.K.; Irving, G.J. Impact of metformin on cardiovascular disease: A meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia 2017, 60, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Bellman, S.M.; Stephenson, M.D.; Lisy, K. Metformin reduces all-cause mortality and diseases of ageing independent of its effect on diabetes control: A systematic review and meta-analysis. Ageing Res. Rev. 2017, 40, 31–44. [Google Scholar] [CrossRef]
- UK Prospective Diabetes Study (UKPDS) Group. Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998, 352, 854–865. [Google Scholar] [CrossRef]
- Davies, M.J.; D‘Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of Hyperglycemia in Type 2 Diabetes, 2018. A Consensus Report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2018, 41, 2669–2701. [Google Scholar] [CrossRef]
- Fei, Y.; Tsoi, M.F.; Cheung, B.M.Y. Cardiovascular outcomes in trials of new antidiabetic drug classes: A network meta-analysis. Cardiovasc. Diabetol. 2019, 18, 112. [Google Scholar] [CrossRef]
- Khunti, K.; Chatterjee, S.; Gerstein, H.C.; Zoungas, S.; Davies, M.J. Do sulphonylureas still have a place in clinical practice? Lancet Diabetes Endocrinol. 2018, 6, 821–832. [Google Scholar] [CrossRef]
- Gupta, A.K.; Verma, A.K.; Kailashiya, J.; Singh, S.K.; Kumar, N. Sitagliptin: Anti-platelet effect in diabetes and healthy volunteers. Platelets 2012, 23, 565–570. [Google Scholar] [CrossRef]
- Aroor, A.R.; Manrique-Acevedo, C.; DeMarco, V.G. The role of dipeptidylpeptidase-4 inhibitors in management of cardiovascular disease in diabetes; focus on linagliptin. Cardiovasc. Diabetol. 2018, 17, 59. [Google Scholar] [CrossRef]
- Aroor, A.R.; Sowers, J.R.; Jia, G.; DeMarco, V.G. Pleiotropic effects of the dipeptidylpeptidase-4 inhibitors on the cardiovascular system. Am. J. Physiol. Heart. Circ. Physiol. 2014, 307, H477–H492. [Google Scholar] [CrossRef] [PubMed]
- Cameron-Vendrig, A.; Reheman, A.; Siraj, M.A.; Xu, X.R.; Wang, Y.; Lei, X.; Afroze, T.; Shikatani, E.; El-Mounayri, O.; Noyan, H.; et al. Glucagon-Like Peptide 1 Receptor Activation Attenuates Platelet Aggregation and Thrombosis. Diabetes 2016, 65, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Sposito, A.C.; Berwanger, O.; de Carvalho, L.S.F.; Saraiva, J.F.K. GLP-1RAs in type 2 diabetes: Mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc. Diabetol. 2018, 17, 157. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; McMurray, J.J.V. SGLT2 inhibitors and mechanisms of cardiovascular benefit: A state-of-the-art review. Diabetologia 2018, 61, 2108–2117. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.K.; Strong, J. The Pleiotropic Effects of Sodium-Glucose Cotransporter-2 Inhibitors: Beyond the Glycemic Benefit. Diabetes. Ther. 2019, 10, 1771–1792. [Google Scholar] [CrossRef]
- Mobarrez, F.; He, S.; Broijersen, A.; Wiklund, B.; Antovic, A.; Antovic, J.; Egberg, N.; Jorneskog, G.; Wallen, H. Atorvastatin reduces thrombin generation and expression of tissue factor, P-selectin and GPIIIa on platelet-derived microparticles in patients with peripheral arterial occlusive disease. Thromb. Haemost. 2011, 106, 344–352. [Google Scholar] [CrossRef]
- Park, H.S.; Gu, J.Y.; Yoo, H.J.; Han, S.E.; Park, C.H.; Kim, Y.I.; Nam-Goong, I.S.; Kim, E.S.; Kim, H.K. Thrombin Generation Assay Detects Moderate-Intensity Statin-Induced Reduction of Hypercoagulability in Diabetes. Clin. Appl. Thromb. Hemost. 2018, 24, 1095–1101. [Google Scholar] [CrossRef]
- American Diabetes, A. 9. Cardiovascular Disease and Risk Management: Standards of Medical Care in Diabetes-2018. Diabetes Care 2018, 41, S86–S104. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| T1DM | T2DM | |
|---|---|---|
| Pro-coagulant proteins | ↑ vWF [12] | ↑ vWF [10,11,12] |
| ↑ prekallikrein [13] | ↑ kininogen [23] | |
| ↑ tissue factor procoagulant activity [17] | ↑ kallikrein [14] | |
| ↑ factor V [15] | ↑ soluble tissue factor [16,24] | |
| ↑ (activated) factor VII [15,17] | ↑ factor V [15] | |
| ↑ factor VIII [15] | ↑ (activated) factor VII [15,16] | |
| ↑ factor X [15] | ↑ factor VIII [15,18] | |
| ↑ factor XI [14] | ↑ factor IX [18] | |
| ↓ activated factor XII [27] | ↑ factor X [15] | |
| ↑ prothrombin [15] | ↑ factor XI [14] | |
| ↓ fibrinogen [22], ↑ fibrinogen in diabetic complications [21] | ↑ (activated) factor XII [18,25] | |
| ↑ factor XIII [26] | ||
| ↑ prothrombin [15] | ||
| ↑ fibrinogen [19,20] | ||
| Anticoagulant proteins | ↓ antithrombin activity [49,50] | ↑ antithrombin [18,33], ↓ antithrombin [32], ↓ antithrombin activity with bad glycaemic control [47] |
| ↓ protein C [15,19,28] | ↓ protein C [15,19,29], ↓ protein C activity with bad glycaemic control [47] | |
| ↓ protein S [30] | ↓ protein S [30], ↓ protein S activity with bad glycaemic control [47] | |
| ↑ tissue factor pathway inhibitor activity [49] | ↑ tissue factor pathway inhibitor [16] | |
| ↑ thrombomodulin [31] | ↑ thrombomodulin [29] | |
| Pro-fibrinolytic proteins | ↑ tissue plasminogen activator in diabetic complications [37] | ↑ tissue plasminogen activator [24] |
| ↓ plasmin activity [51] | ||
| Anti-fibrinolytic proteins | ↓ PAI-1 [39], ↓ PAI-1 activity [22,52] | ↑ PAI-1 [10,19,20,24,38] |
| ↑ α2-antiplasmin [19,43], ↓ α2-antiplasmin [22] | ↑ α2-antiplasmin [19] | |
| ↑ thrombin-activatable fibrinolysis inhibitor [41] | ↑ thrombin-activatable fibrinolysis inhibitor [40] | |
| ↑ α2-maroglobulin [42] | ↑ α2-maroglobulin [32,42] |
| In T1DM | In T2DM | In both T1DM and T2DM | |
|---|---|---|---|
| Coagulation | Reduced PAI-1 levels | Increased levels of anti-fibrinolysis proteins, including PAI-1 | Increased levels of pro-coagulatory proteins |
| Reduced anticoagulant activity | |||
| Denser fibrin fibres, less porous fibrin clot, fibrin clot more resistant to fibrinolysis | |||
| Metal ions | Dysregulation of coagulation by Mg2+ deficiency | Possible dysregulation of coagulation by elevated Ca2+ levels | Possible dysregulation of coagulation by elevated ceruloplasmin levels |
| Possible dysregulation of coagulation by Fe3+ supplements | Dysregulation of coagulation by altered zinc speciation | ||
| Possible dysregulation of coagulation by elevated iron levels | |||
| Lipids | Unchanged or reduced HDL levels | Reduced HDL levels | Elevated levels of small dense LDL that favoured atherosclerotic plaque formation and endothelial dysfunction |
| Reduced plasma FFA levels | Elevated plasma FFA levels causing the destabilisation of fibrin clot, metabolism dysregulation, atherosclerotic plaques and cardiac lipotoxicity | HDL dysfunction causing reduced HDL efflux, reduced anti-inflammatory effects and endothelial dysfunction | |
| Hypertriglyceridemia causing HDL and endothelial dysfunction | |||
| Endothelial dysfunction | Excess FFA levels causing endothelial dysfunction | Endothelial dysfunction causes reduced nitric oxide production, dysregulation of vasodilators and vasoconstrictors | |
| Formation of AGEs dysregulating nitric oxide synthase and protein synthesis by the endothelium, causing endothelial dysfunction | |||
| Matrix metalloproteinases upregulation, causing inflammation, endothelial dysfunction, vascular remodelling, thrombus formation and atherosclerotic plaque formation and destabilisation | |||
| Platelets | Platelets have unchanged volume | Larger platelets | Higher platelet count |
| Hyper-activation, adherence and aggregation of platelets | |||
| Microparticles | Elevated levels of endothelial- and platelet-derived microparticles correlated with HbA1c and associated with pro-coagulatory activity | Elevated levels of endothelial- and platelet-derived microparticles enriched in coagulation proteins and associated with atherosclerosis and thrombosis | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sobczak, A.I.S.; Stewart, A.J. Coagulatory Defects in Type-1 and Type-2 Diabetes. Int. J. Mol. Sci. 2019, 20, 6345. https://doi.org/10.3390/ijms20246345
Sobczak AIS, Stewart AJ. Coagulatory Defects in Type-1 and Type-2 Diabetes. International Journal of Molecular Sciences. 2019; 20(24):6345. https://doi.org/10.3390/ijms20246345
Chicago/Turabian StyleSobczak, Amélie I. S., and Alan J. Stewart. 2019. "Coagulatory Defects in Type-1 and Type-2 Diabetes" International Journal of Molecular Sciences 20, no. 24: 6345. https://doi.org/10.3390/ijms20246345
APA StyleSobczak, A. I. S., & Stewart, A. J. (2019). Coagulatory Defects in Type-1 and Type-2 Diabetes. International Journal of Molecular Sciences, 20(24), 6345. https://doi.org/10.3390/ijms20246345