Partners in Crime: Towards New Ways of Targeting Calcium Channels




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
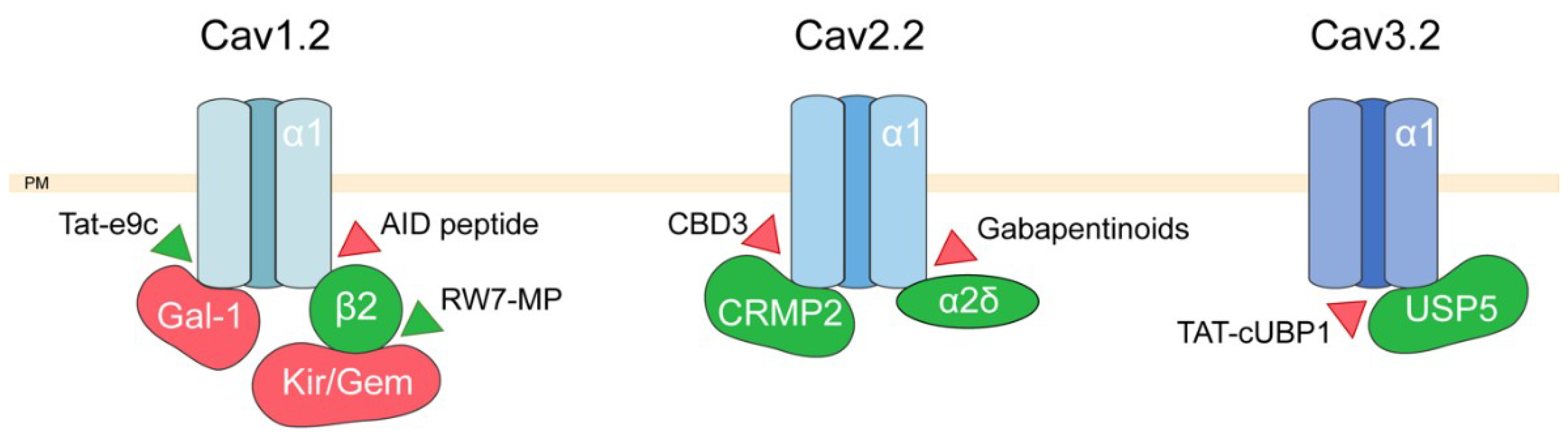
2. Targeting Voltage-Dependent Calcium Channel Interactions
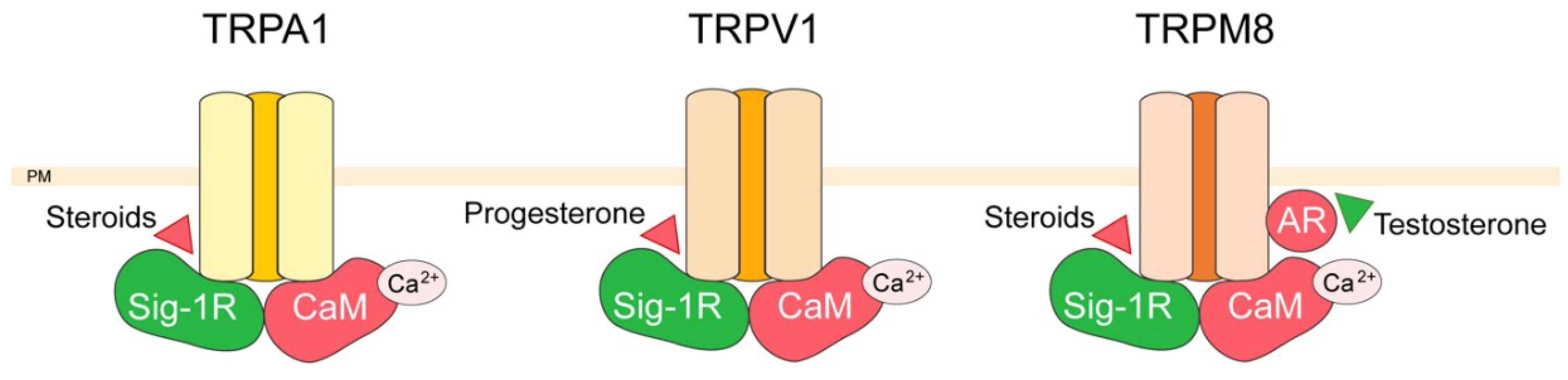
3. TRP Channel Interactors
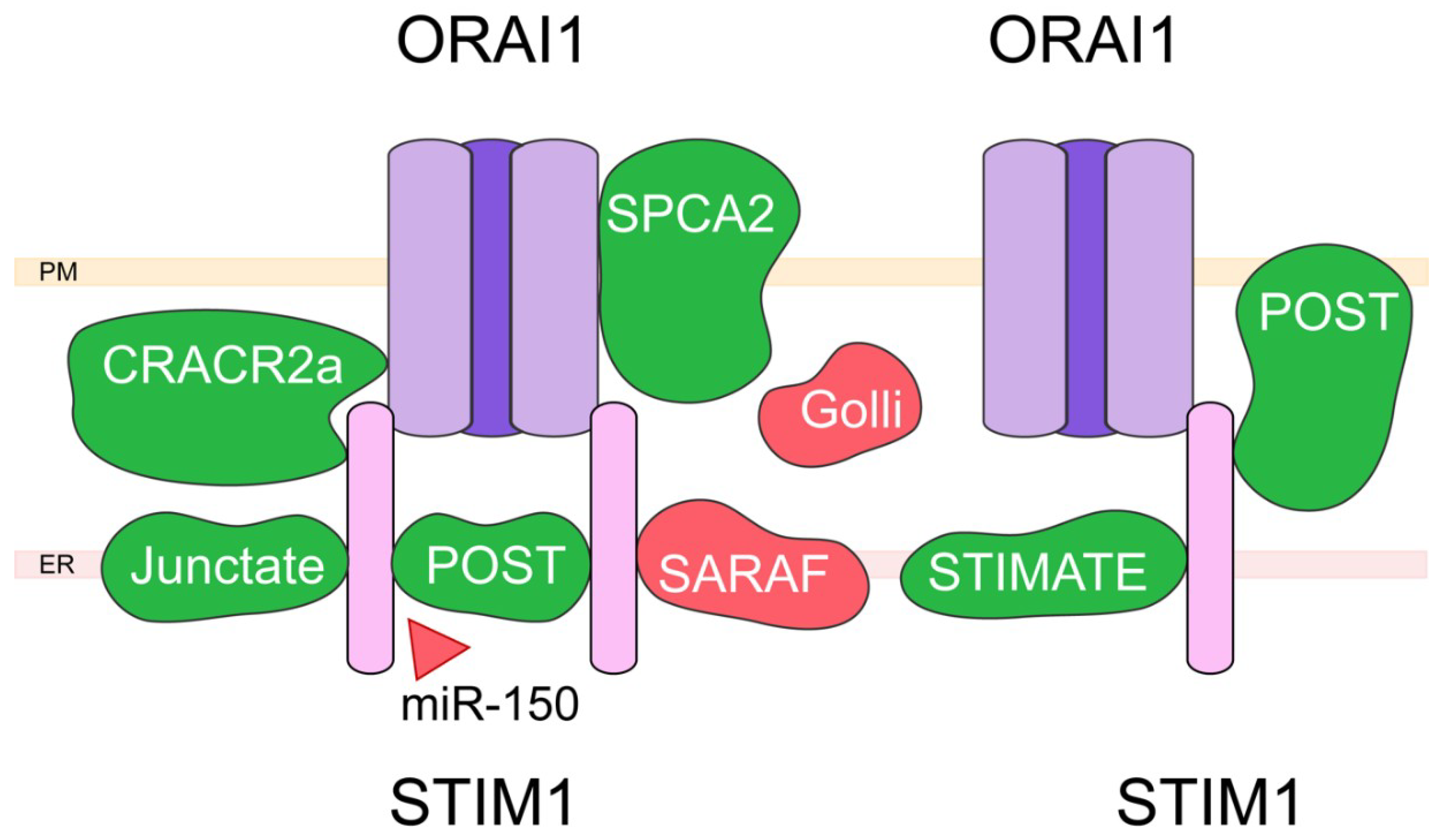
4. Capacitative Calcium Entry (CCE) and Associated Proteins
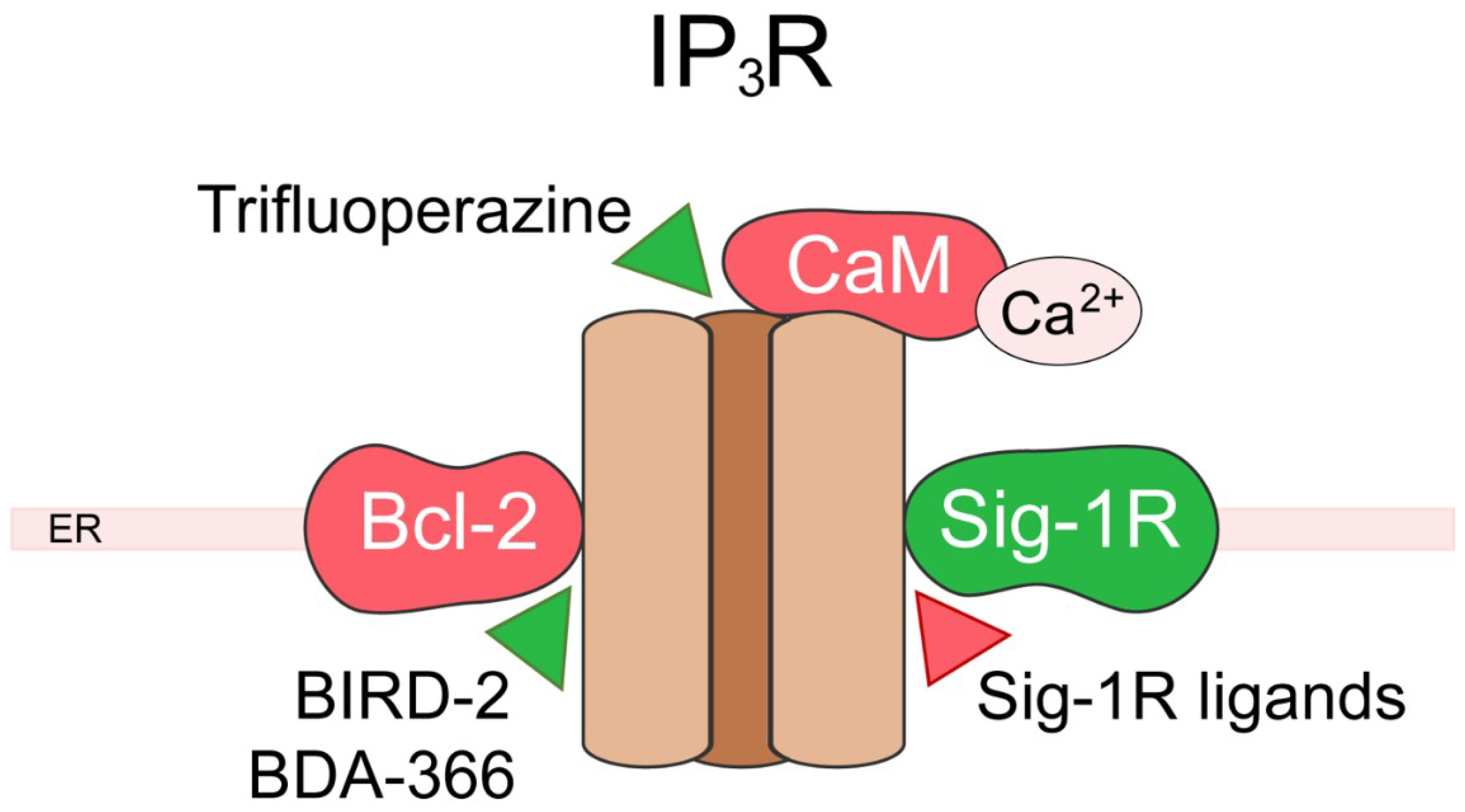
5. Inositol 1,4,5-Trisphosphate Receptor (IP3R)
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AAVis | Adeno-associated virus |
| ABP | AID-binding pocket |
| ALS | Amyotrophic lateral sclerosis |
| Bcl-2 | B-cell lymphoma-2 |
| BiP | Binding immunoglobulin protein |
| BIRD-2 | Bcl-2-IP3R disrupter-2 |
| CBD | Calcium channel binding domain |
| CC | Coiled-coil |
| CCE | Capacitative calcium entry |
| CRAC CRACR2a | Calcium release activated calcium channel Calcium release activated calcium channel regulator 2a |
| DHP | Dihydropyridines |
| ER | Endoplasmic reticulum |
| GK | Guanylate kinase |
| HVA | High-voltage activated channels |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| IP3R | Inositol 1,4,5-trisphosphate receptor |
| LVA | Low-voltage activated |
| MAM | Mitochondria-associated membranes |
| MBP | Myelin basic proteins |
| PEST | Proline/lysin rich sequence |
| PM | Plasma membrane |
| SARAF | SOCE-associated regulatory factor |
| SCDI | Slow calcium dependent inactivation |
| SCDI | Slow calcium dependent inactivation |
| Sig-1R | Sigma-1 receptor |
| SOC | Store-operated calcium |
| SOCE | Store-operated calcium entry |
| SPCA2 | Secretory pathway calcium ATPase 2 |
| Stim STIMATE | Stromal interaction molecule Stim-activating enhancer |
| TRP | Transient receptor potential |
| VDCC | Voltage-dependent calcium channels |
References
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A.; Becchetti, A. Novel perspectives in cancer therapy: Targeting ion channels. Drug Resist. Updat 2015, 21–22, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.M. TRP Channels as Potential Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2018, 58, 309–330. [Google Scholar] [CrossRef] [PubMed]
- Snutch, T.P. Targeting chronic and neuropathic pain: The N-type calcium channel comes of age. NeuroRx 2005, 2, 662–670. [Google Scholar] [CrossRef]
- Zamponi, G.W. Targeting voltage-gated calcium channels in neurological and psychiatric diseases. Nat. Rev. Drug Discov. 2016, 15, 19–34. [Google Scholar] [CrossRef]
- Poteser, M.; Groschner, K. Studying Subunit Interaction and Complex Assembly of TRP Channels. In TRP Channels; Zhu, M.X., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2011; ISBN 978-1-4398-1860-2. [Google Scholar]
- Dolphin, A.C. Calcium channel auxiliary alpha2delta and beta subunits: Trafficking and one step beyond. Nat. Rev. Neurosci. 2012, 13, 542–555. [Google Scholar] [CrossRef]
- Nowycky, M.C.; Fox, A.P.; Tsien, R.W. Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 1985, 316, 440–443. [Google Scholar] [CrossRef]
- Fox, A.P.; Nowycky, M.C.; Tsien, R.W. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J. Physiol. 1987, 394, 149–172. [Google Scholar] [CrossRef]
- Llinas, R.; Sugimori, M.; Lin, J.W.; Cherksey, B. Blocking and isolation of a calcium channel from neurons in mammals and cephalopods utilizing a toxin fraction (FTX) from funnel-web spider poison. Proc Natl. Acad. Sci. USA 1989, 86, 1689–1693. [Google Scholar] [CrossRef]
- Huc, S.; Monteil, A.; Bidaud, I.; Barbara, G.; Chemin, J.; Lory, P. Regulation of T-type calcium channels: Signalling pathways and functional implications. Biochim. Biophys. Acta 2009, 1793, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Lacinova, L. Voltage-dependent calcium channels. Gen. Physiol. Biophys. 2005, 24 (Suppl. 1), 1–78. [Google Scholar]
- Lee, J.; Ishihara, A.; Oxford, G.; Johnson, B.; Jacobson, K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 1999, 400, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [PubMed]
- Kuzmenkina, E.; Novikova, E.; Jangsangthong, W.; Matthes, J.; Herzig, S. Single-Channel Resolution of the Interaction between C-Terminal CaV1.3 Isoforms and Calmodulin. Biophys. J. 2019, 116, 836–846. [Google Scholar] [CrossRef]
- Fan, M.; Zhang, W.K.; Buraei, Z.; Yang, J. Molecular determinants of Gem protein inhibition of P/Q-type Ca2+ channels. J. Biol. Chem. 2012, 287, 22749–22758. [Google Scholar] [CrossRef]
- Jarvis, S.E.; Barr, W.; Feng, Z.-P.; Hamid, J.; Zamponi, G.W. Molecular determinants of syntaxin 1 modulation of N-type calcium channels. J. Biol. Chem. 2002, 277, 44399–44407. [Google Scholar] [CrossRef]
- Kim, D.K.; Catterall, W.A. Ca2+-dependent and -independent interactions of the isoforms of the alpha1A subunit of brain Ca2+ channels with presynaptic SNARE proteins. Proc. Natl. Acad. Sci. USA 1997, 94, 14782–14786. [Google Scholar] [CrossRef]
- Berkefeld, H.; Fakler, B.; Schulte, U. Ca2+-activated K+ channels: From protein complexes to function. Physiol. Rev. 2010, 90, 1437–1459. [Google Scholar] [CrossRef]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2, 239821281879480. [Google Scholar] [CrossRef]
- Tibbs, G.R.; Posson, D.J.; Goldstein, P.A. Voltage-Gated Ion Channels in the PNS: Novel Therapies for Neuropathic Pain? Trends Pharmacol. Sci. 2016, 37, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Sueta, D.; Tabata, N.; Hokimoto, S. Clinical roles of calcium channel blockers in ischemic heart diseases. Hypertens Res. 2017, 40, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Clemons, T.D.; Viola, H.M.; House, M.J.; Iyer, K.S.; Hool, L.C. Examining efficacy of “TAT-less” delivery of a peptide against the L-type calcium channel in cardiac ischemia-reperfusion injury. ACS Nano 2013, 7, 2212–2220. [Google Scholar] [CrossRef] [PubMed]
- Buraei, Z.; Yang, J. Structure and function of the beta subunit of voltage-gated Ca(2)(+) channels. Biochim. Biophys. Acta 2013, 1828, 1530–1540. [Google Scholar] [CrossRef]
- Hohaus, A.; Poteser, M.; Romanin, C.; Klugbauer, N.; Hofmann, F.; Morano, I.; Haase, H.; Groschner, K. Modulation of the smooth-muscle L-type Ca2+ channel alpha1 subunit (alpha1C-b) by the beta2a subunit: A peptide which inhibits binding of beta to the I-II linker of alpha1 induces functional uncoupling. Biochem. J. 2000, 348 Pt 3, 657–665. [Google Scholar]
- Viola, H.M.; Jordan, M.C.; Roos, K.P.; Hool, L.C. Decreased myocardial injury and improved contractility after administration of a peptide derived against the alpha-interacting domain of the L-type calcium channel. J. Am. Heart Assoc. 2014, 3, e000961. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, F.; Campiglio, M.; Jo, H.; Abderemane-Ali, F.; Rumpf, C.H.; Pope, L.; Rossen, N.D.; Flucher, B.E.; DeGrado, W.F.; Minor, D.L., Jr. Stapled Voltage-Gated Calcium Channel (CaV) alpha-Interaction Domain (AID) Peptides Act As Selective Protein-Protein Interaction Inhibitors of CaV Function. ACS Chem. Neurosci. 2017, 8, 1313–1326. [Google Scholar] [CrossRef]
- Weiss N, K.A. Pathologies of Calcium Channels; Springer: Berlin, Germany, 2014; pp. 47–114. [Google Scholar]
- Rusconi, F.; Ceriotti, P.; Miragoli, M.; Carullo, P.; Salvarani, N.; Rocchetti, M.; Di Pasquale, E.; Rossi, S.; Tessari, M.; Caprari, S.; et al. Peptidomimetic Targeting of Cavbeta2 Overcomes Dysregulation of the L-Type Calcium Channel Density and Recovers Cardiac Function. Circulation 2016, 134, 534–546. [Google Scholar] [CrossRef]
- Beguin, P.; Nagashima, K.; Gonoi, T.; Shibasaki, T.; Takahashi, K.; Kashima, Y.; Ozaki, N.; Geering, K.; Iwanaga, T.; Seino, S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature 2001, 411, 701–706. [Google Scholar] [CrossRef]
- Beguin, P.; Ng, Y.J.; Krause, C.; Mahalakshmi, R.N.; Ng, M.Y.; Hunziker, W. RGK small GTP-binding proteins interact with the nucleotide kinase domain of Ca2+-channel beta-subunits via an uncommon effector binding domain. J. Biol. Chem. 2007, 282, 11509–11520. [Google Scholar] [CrossRef]
- Hofmann, F.; Flockerzi, V.; Kahl, S.; Wegener, J.W. L-type CaV1.2 calcium channels: From in vitro findings to in vivo function. Physiol. Rev. 2014, 94, 303–326. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Liang, M.C.; Soong, T.W. Alternative Splicing of L-type CaV1.2 Calcium Channels: Implications in Cardiovascular Diseases. Genes (Basel) 2017, 8, 344. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, G.; Wang, J.W.; Chong, S.Y.; Yu, D.; Wang, X.; Soon, J.L.; Liang, M.C.; Wong, Y.P.; Huang, N.; et al. Regulation of Blood Pressure by Targeting CaV1.2-Galectin-1 Protein Interaction. Circulation 2018, 138, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Doggrell, S.A. Intrathecal ziconotide for refractory pain. Expert Opin. Investig. Drugs 2004, 13, 875–877. [Google Scholar] [CrossRef] [PubMed]
- Alles, S.R.A.; Smith, P.A. Etiology and Pharmacology of Neuropathic Pain. Pharmacol. Rev. 2018, 70, 315–347. [Google Scholar] [CrossRef]
- Feldman, P.; Khanna, R. Challenging the catechism of therapeutics for chronic neuropathic pain: Targeting CaV2.2 interactions with CRMP2 peptides. Neurosci. Lett. 2013, 557 Pt A, 27–36. [Google Scholar] [CrossRef]
- Cai, S.; Shan, Z.; Zhang, Z.; Moutal, A.; Khanna, R. Activity of T-type calcium channels is independent of CRMP2 in sensory neurons. Channels (Austin) 2019, 13, 147–152. [Google Scholar] [CrossRef]
- Yu, H.; Shin, S.M.; Xiang, H.; Chao, D.; Cai, Y.; Xu, H.; Khanna, R.; Pan, B.; Hogan, Q.H. AAV-encoded CaV2.2 peptide aptamer CBD3A6K for primary sensory neuron-targeted treatment of established neuropathic pain. Gene Ther. 2019, 26, 308–323. [Google Scholar] [CrossRef]
- Garcia-Caballero, A.; Gadotti, V.M.; Stemkowski, P.; Weiss, N.; Souza, I.A.; Hodgkinson, V.; Bladen, C.; Chen, L.; Hamid, J.; Pizzoccaro, A.; et al. The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 2014, 83, 1144–1158. [Google Scholar] [CrossRef]
- Khanna, R.; Yu, J.; Yang, X.; Moutal, A.; Chefdeville, A.; Gokhale, V.; Shuja, Z.; Chew, L.A.; Bellampalli, S.S.; Luo, S.; et al. Targeting the CaValpha-CaVbeta interaction yields an antagonist of the N-type CaV2.2 channel with broad antinociceptive efficacy. Pain 2019, 160, 1644–1661. [Google Scholar] [CrossRef]
- Chen, X.; Liu, D.; Zhou, D.; Si, Y.; Xu, D.; Stamatkin, C.W.; Ghozayel, M.K.; Ripsch, M.S.; Obukhov, A.G.; White, F.A.; et al. Small-molecule CaValpha1CaVbeta antagonist suppresses neuronal voltage-gated calcium-channel trafficking. Proc Natl. Acad Sci. USA 2018, 115, E10566–E10575. [Google Scholar] [CrossRef] [PubMed]
- Montell, C.; Rubin, G.M. Molecular characterization of the Drosophila trp locus: A putative integral membrane protein required for phototransduction. Neuron 1989, 2, 1313–1323. [Google Scholar] [CrossRef]
- Montell, C.; Birnbaumer, L.; Flockerzi, V.; Bindels, R.J.; Bruford, E.A.; Caterina, M.J.; Clapham, D.E.; Harteneck, C.; Heller, S.; Julius, D.; et al. A Unified Nomenclature for the Superfamily of TRP Cation Channels. Mol. Cell. 2002, 9, 229–231. [Google Scholar] [CrossRef]
- Owsianik, G.; Talavera, K.; Voets, T.; Nilius, B. Permeation and selectivity of TRP channels. Annu. Rev. Physiol. 2006, 68, 685–717. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Shin, Y.-C.; Shin, S.-Y.; Chun, J.N.; Cho, H.S.; Lim, J.M.; Kim, H.-G.; So, I.; Kwon, D.; Jeon, J.-H. TRIP Database 2.0: A Manually Curated Information Hub for Accessing TRP Channel Interaction Network. PLoS ONE 2012, 7, e47165. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Chaperones at the ER- Mitochondrion Interface Regulate Ca2+ Signaling and Cell Survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef]
- Kourrich, S.; Hayashi, T.; Chuang, J.-Y.; Tsai, S.-Y.; Su, T.-P.; Bonci, A. Dynamic Interaction between Sigma-1 Receptor and Kv1.2 Shapes Neuronal and Behavioral Responses to Cocaine. Cell 2013, 152, 236–247. [Google Scholar] [CrossRef]
- Kinoshita, M.; Matsuoka, Y.; Suzuki, T.; Mirrielees, J.; Yang, J. Sigma-1 receptor alters the kinetics of Kv1.3 voltage gated potassium channels but not the sensitivity to receptor ligands. Brain Res. 2012, 1452, 1–9. [Google Scholar] [CrossRef]
- Balasuriya, D.; Stewart, A.P.; Crottès, D.; Borgese, F.; Soriani, O.; Edwardson, J.M. The Sigma-1 Receptor Binds to the Nav1.5 Voltage-gated Na + Channel with 4-Fold Symmetry. J. Biol. Chem. 2012, 287, 37021–37029. [Google Scholar] [CrossRef]
- Balasuriya, D.; D’Sa, L.; Talker, R.; Dupuis, E.; Maurin, F.; Martin, P.; Borgese, F.; Soriani, O.; Edwardson, J.M. A Direct Interaction between the Sigma-1 Receptor and the hERG Voltage-gated K + Channel Revealed by Atomic Force Microscopy and Homogeneous Time-resolved Fluorescence (HTRF®). J. Biol. Chem. 2014, 289, 32353–32363. [Google Scholar] [CrossRef] [PubMed]
- Morales-Lázaro, S.L.; González-Ramírez, R.; Rosenbaum, T. Molecular Interplay Between the Sigma-1 Receptor, Steroids, and Ion Channels. Front. Pharmacol. 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Montero, E.; Sánchez-Blázquez, P.; Onetti, Y.; Merlos, M.; Garzón, J. Ligands Exert Biased Activity to Regulate Sigma 1 Receptor Interactions With Cationic TRPA1, TRPV1, and TRPM8 Channels. Front. Pharmacol. 2019, 10, 634. [Google Scholar] [CrossRef] [PubMed]
- Ortíz-Rentería, M.; Juárez-Contreras, R.; González-Ramírez, R.; Islas, L.D.; Sierra-Ramírez, F.; Llorente, I.; Simon, S.A.; Hiriart, M.; Rosenbaum, T.; Morales-Lázaro, S.L. TRPV1 channels and the progesterone receptor Sig-1R interact to regulate pain. Proc. Natl. Acad. Sci. USA 2018, 115, E1657–E1666. [Google Scholar] [CrossRef]
- Hasan, R.; Leeson-Payne, A.T.S.; Jaggar, J.H.; Zhang, X. Calmodulin is responsible for Ca2+-dependent regulation of TRPA1 Channels. Sci. Rep. 2017, 7, 45098. [Google Scholar] [CrossRef]
- Numazaki, M.; Tominaga, T.; Takeuchi, K.; Murayama, N.; Toyooka, H.; Tominaga, M. Structural determinant of TRPV1 desensitization interacts with calmodulin. Proc. Natl. Acad. Sci. USA 2003, 100, 8002–8006. [Google Scholar] [CrossRef]
- Sarria, I.; Ling, J.; Zhu, M.X.; Gu, J.G. TRPM8 acute desensitization is mediated by calmodulin and requires PIP 2: Distinction from tachyphylaxis. J. Neurophysiol. 2011, 106, 3056–3066. [Google Scholar] [CrossRef]
- Rosenbaum, T.; Gordon-Shaag, A.; Munari, M.; Gordon, S.E. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J. Gen. Physiol. 2004, 123, 53–62. [Google Scholar] [CrossRef]
- Caterina, M.J.; Rosen, T.A.; Tominaga, M.; Brake, A.J.; Julius, D. A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 1999, 398, 436–441. [Google Scholar] [CrossRef]
- Moran, M.M.; McAlexander, M.A.; Biro, T.; Szallasi, A. Transient receptor potential channels as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 601–620. [Google Scholar] [CrossRef]
- Su, T.; London, E.; Jaffe, J. Steroid binding at sigma receptors suggests a link between endocrine, nervous, and immune systems. Science 1988, 240, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Cendán, C.M.; Pujalte, J.M.; Portillo-Salido, E.; Montoliu, L.; Baeyens, J.M. Formalin-induced pain is reduced in σ1 receptor knockout mice. Eur. J. Pharmacol. 2005, 511, 73–74. [Google Scholar] [CrossRef] [PubMed]
- Entrena, J.M.; Cobos, E.J.; Nieto, F.R.; Cendán, C.M.; Gris, G.; Del Pozo, E.; Zamanillo, D.; Baeyens, J.M. Sigma-1 receptors are essential for capsaicin-induced mechanical hypersensitivity: Studies with selective sigma-1 ligands and sigma-1 knockout mice. Pain 2009, 143, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Grolez, G.; Gkika, D. TRPM8 Puts the Chill on Prostate Cancer. Pharmaceuticals 2016, 9, 44. [Google Scholar] [CrossRef] [PubMed]
- Tsavaler, L.; Shapero, M.H.; Morkowski, S.; Laus, R. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001, 61, 3760–3769. [Google Scholar]
- Henshall, S.M.; Afar, D.E.H.; Hiller, J.; Horvath, L.G.; Quinn, D.I.; Rasiah, K.K.; Gish, K.; Willhite, D.; Kench, J.G.; Gardiner-Garden, M.; et al. Survival analysis of genome-wide gene expression profiles of prostate cancers identifies new prognostic targets of disease relapse. Cancer Res. 2003, 63, 4196–4203. [Google Scholar]
- Zhu, G.; Wang, X.; Yang, Z.; Cao, H.; Meng, Z.; Wang, Y.; Chen, D. Effects of TRPM8 on the proliferation and angiogenesis of prostate cancer PC-3 cells in vivo. Oncol. Lett. 2011, 2, 1213–1217. [Google Scholar] [CrossRef]
- Gkika, D.; Flourakis, M.; Lemonnier, L.; Prevarskaya, N. PSA reduces prostate cancer cell motility by stimulating TRPM8 activity and plasma membrane expression. Oncogene 2010, 29, 4611–4616. [Google Scholar] [CrossRef]
- Genova, T.; Grolez, G.P.; Camillo, C.; Bernardini, M.; Bokhobza, A.; Richard, E.; Scianna, M.; Lemonnier, L.; Valdembri, D.; Munaron, L.; et al. TRPM8 inhibits endothelial cell migration via a non-channel function by trapping the small GTPase Rap1. J. Cell Biol. 2017, 216, 2107–2130. [Google Scholar] [CrossRef]
- Grolez, G.P.; Gordiendko, D.V.; Clarisse, M.; Hammadi, M.; Desruelles, E.; Fromont, G.; Prevarskaya, N.; Slomianny, C.; Gkika, D. TRPM8-androgen receptor association within lipid rafts promotes prostate cancer cell migration. Cell Death Dis. 2019, 10, 652. [Google Scholar] [CrossRef]
- Tu, H.; Gu, J.; Meng, Q.H.; Kim, J.; Strom, S.; Davis, J.W.; He, Y.; Wagar, E.A.; Thompson, T.C.; Logothetis, C.J.; et al. Low serum testosterone is associated with tumor aggressiveness and poor prognosis in prostate cancer. Oncol. Lett. 2017, 13, 1949–1957. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, M.; Schindl, R.; Romanin, C. Studies of Structure-Function and Subunit Composition of Orai/STIM Channel. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; ISBN 978-1-4987-5272-5. [Google Scholar]
- Putney, J.W.J. A model for receptor-regulated calcium entry. Cell Calcium 1986, 7, 1–12. [Google Scholar] [CrossRef]
- Feske, S.; Gwack, Y.; Prakriya, M.; Srikanth, S.; Puppel, S.-H.; Tanasa, B.; Hogan, P.G.; Lewis, R.S.; Daly, M.; Rao, A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 2006, 441, 179–185. [Google Scholar] [CrossRef]
- Roos, J.; DiGregorio, P.J.; Yeromin, A.V.; Ohlsen, K.; Lioudyno, M.; Zhang, S.; Safrina, O.; Kozak, J.A.; Wagner, S.L.; Cahalan, M.D.; et al. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 2005, 169, 435–445. [Google Scholar] [CrossRef]
- Lunz, V.; Romanin, C.; Frischauf, I. STIM1 activation of Orai1. Cell Calcium 2019, 77, 29–38. [Google Scholar] [CrossRef]
- Sharma, S.; Quintana, A.; Findlay, G.M.; Mettlen, M.; Baust, B.; Jain, M.; Nilsson, R.; Rao, A.; Hogan, P.G. An siRNA screen for NFAT activation identifies septins as coordinators of store-operated Ca2+ entry. Nature 2013, 499, 238–242. [Google Scholar] [CrossRef]
- Ong, H.L.; Ambudkar, I.S. STIM-TRP Pathways and Microdomain Organization: Contribution of TRPC1 in Store-Operated Ca2+ Entry: Impact on Ca2+ Signaling and Cell Function. Adv. Exp. Med. Biol. 2017, 993, 159–188. [Google Scholar] [PubMed]
- Groschner, K.; Shrestha, N.; Fameli, N. Non-Orai Partners of STIM Proteins: Role in ER-PM Communication and Ca2+ Signaling. In Calcium Entry Channels in Non-Excitable Cells; Kozak, J.A., Putney, J.W., Eds.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2018; ISBN 978-1-4987-5272-5. [Google Scholar]
- Putney, J.W. Forms and functions of store-operated calcium entry mediators, STIM and Orai. Adv. Biol. Regul. 2018, 68, 88–96. [Google Scholar] [CrossRef]
- Srikanth, S.; Jung, H.-J.; Kim, K.-D.; Souda, P.; Whitelegge, J.; Gwack, Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat. Cell Biol. 2010, 12, 436–446. [Google Scholar] [CrossRef]
- Srikanth, S.; Jew, M.; Kim, K.-D.; Yee, M.-K.; Abramson, J.; Gwack, Y. Junctate is a Ca2+-sensing structural component of Orai1 and stromal interaction molecule 1 (STIM1). Proc. Natl. Acad. Sci. USA 2012, 109, 8682–8687. [Google Scholar] [CrossRef]
- Krapivinsky, G.; Krapivinsky, L.; Stotz, S.C.; Manasian, Y.; Clapham, D.E. POST, partner of stromal interaction molecule 1 (STIM1), targets STIM1 to multiple transporters. Proc. Natl. Acad. Sci. USA 2011, 108, 19234–19239. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Grice, D.M.; Faddy, H.M.; Nguyen, N.; Leitch, S.; Wang, Y.; Muend, S.; Kenny, P.A.; Sukumar, S.; Roberts-Thomson, S.J.; et al. Store-Independent Activation of Orai1 by SPCA2 in Mammary Tumors. Cell 2010, 143, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Smaardijk, S.; Chen, J.; Wuytack, F.; Vangheluwe, P. SPCA2 couples Ca2+ influx via Orai1 to Ca2+ uptake into the Golgi/secretory pathway. Tissue Cell 2017, 49, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Jing, J.; He, L.; Sun, A.; Quintana, A.; Ding, Y.; Ma, G.; Tan, P.; Liang, X.; Zheng, X.; Chen, L.; et al. Proteomic mapping of ER-PM junctions identifies STIMATE as regulator of Ca2+ influx. Nat. Cell Biol. 2015, 17, 1339–1347. [Google Scholar] [CrossRef]
- Quintana, A.; Rajanikanth, V.; Farber-Katz, S.; Gudlur, A.; Zhang, C.; Jing, J.; Zhou, Y.; Rao, A.; Hogan, P.G. TMEM110 regulates the maintenance and remodeling of mammalian ER–plasma membrane junctions competent for STIM–ORAI signaling. Proc. Natl. Acad. Sci. USA 2015, 112, E7083–E7092. [Google Scholar] [CrossRef]
- Feng, J.-M.; Hu, Y.K.; Xie, L.-H.; Colwell, C.S.; Shao, X.M.; Sun, X.-P.; Chen, B.; Tang, H.; Campagnoni, A.T. Golli Protein Negatively Regulates Store Depletion-Induced Calcium Influx in T Cells. Immunity 2006, 24, 717–727. [Google Scholar] [CrossRef]
- Walsh, C.M.; Doherty, M.K.; Tepikin, A.V.; Burgoyne, R.D. Evidence for an interaction between Golli and STIM1 in store-operated calcium entry. Biochem. J. 2010, 430, 453–460. [Google Scholar] [CrossRef][Green Version]
- Palty, R.; Raveh, A.; Kaminsky, I.; Meller, R.; Reuveny, E. SARAF Inactivates the Store Operated Calcium Entry Machinery to Prevent Excess Calcium Refilling. Cell 2012, 149, 425–438. [Google Scholar] [CrossRef]
- Jha, A.; Ahuja, M.; Maléth, J.; Moreno, C.M.; Yuan, J.P.; Kim, M.S.; Muallem, S. The STIM1 CTID domain determines access of SARAF to SOAR to regulate Orai1 channel function. J. Cell Biol. 2013, 202, 71–79. [Google Scholar] [CrossRef]
- Albarran, L.; Lopez, J.J.; Amor, N.B.; Martin-Cano, F.E.; Berna-Erro, A.; Smani, T.; Salido, G.M.; Rosado, J.A. Dynamic interaction of SARAF with STIM1 and Orai1 to modulate store-operated calcium entry. Sci. Rep. 2016, 6, 24452. [Google Scholar] [CrossRef]
- Jardín, I.; Albarran, L.; Salido, G.M.; López, J.J.; Sage, S.O.; Rosado, J.A. Fine-tuning of store-operated calcium entry by fast and slow Ca2+-dependent inactivation: Involvement of SARAF. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.-D.; Jung, H.-R.; Seo, S.-H.; Oh, S.-C.; Ban, Y.; Tan, X.; Min Kim, J.; Hyun Lee, S.; Koh, D.-S.; Jung, H.; et al. MicroRNA-150 modulates intracellular Ca2+ levels in naïve CD8+ T cells by targeting TMEM20. Sci. Rep. 2017, 7. [Google Scholar]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Ion Channels in Cancer: Are Cancer Hallmarks Oncochannelopathies? Physiol Rev. 2018, 98, 559–621. [Google Scholar] [CrossRef] [PubMed]
- Kappel, S.; Borgström, A.; Stokłosa, P.; Dörr, K.; Peinelt, C. Store-operated calcium entry in disease: Beyond STIM/Orai expression levels. Semin. Cell Dev. Biol. 2019, 94, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Supattapone, S.; Worley, P.F.; Baraban, J.M.; Snyder, S.H. Solubilization, purification, and characterization of an inositol trisphosphate receptor. J. Biol. Chem. 1988, 263, 1530–1534. [Google Scholar] [PubMed]
- Ferris, C.D.; Huganir, R.L.; Supattapone, S.; Snyder, S.H. Purified inositol 1,4,5-trisphosphate receptor mediates calcium flux in reconstituted lipid vesicles. Nature 1989, 342, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Meldolesi, J.; Milner, T.A.; Satoh, T.; Supattapone, S.; Snyder, S.H. Inositol 1,4,5-trisphosphate receptor localized to endoplasmic reticulum in cerebellar Purkinje neurons. Nature 1989, 339, 468–470. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.-H.; Mak, D.-O.D. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, C.; Mikoshiba, K. IP3 receptor mutations and brain diseases in human and rodents. J. Neurochem. 2017, 141, 790–807. [Google Scholar] [CrossRef]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a Well-Known Antipsychotic, Inhibits Glioblastoma Invasion by Binding to Calmodulin and Disinhibiting Calcium Release Channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef]
- Sakakura, C.; Hagiwara, A.; Fukuda, K.; Shimomura, K.; Takagi, T.; Kin, S.; Nakase, Y.; Fujiyama, J.; Mikoshiba, K.; Okazaki, Y.; et al. Possible involvement of inositol 1,4,5-trisphosphate receptor type 3 (IP3R3) in the peritoneal dissemination of gastric cancers. Anticancer Res. 2003, 23, 3691–3697. [Google Scholar] [PubMed]
- Shibao, K.; Fiedler, M.J.; Nagata, J.; Minagawa, N.; Hirata, K.; Nakayama, Y.; Iwakiri, Y.; Nathanson, M.H.; Yamaguchi, K. The type III inositol 1,4,5-trisphosphate receptor is associated with aggressiveness of colorectal carcinoma. Cell Calcium 2010, 48, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Hedberg, M.L.; Goh, G.; Chiosea, S.I.; Bauman, J.E.; Freilino, M.L.; Zeng, Y.; Wang, L.; Diergaarde, B.B.; Gooding, W.E.; Lui, V.W.Y.; et al. Genetic landscape of metastatic and recurrent head and neck squamous cell carcinoma. J. Clin. Invest. 2016, 126, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgó, J.; Distelhorst, C.W.; Missiaen, L.; Mikoshiba, K.; et al. IP3R2 levels dictate the apoptotic sensitivity of diffuse large B-cell lymphoma cells to an IP3R-derived peptide targeting the BH4 domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta 2014, 1843, 2240–2252. [Google Scholar] [CrossRef]
- Rong, Y.; Distelhorst, C.W. Bcl-2 Protein Family Members: Versatile Regulators of Calcium Signaling in Cell Survival and Apoptosis. Annu. Rev. Physiol. 2008, 70, 73–91. [Google Scholar] [CrossRef]
- Chen, R.; Valencia, I.; Zhong, F.; McColl, K.S.; Roderick, H.L.; Bootman, M.D.; Berridge, M.J.; Conway, S.J.; Holmes, A.B.; Mignery, G.A.; et al. Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J. Cell Biol. 2004, 166, 193–203. [Google Scholar] [CrossRef]
- Rong, Y.-P.; Bultynck, G.; Aromolaran, A.S.; Zhong, F.; Parys, J.B.; Smedt, H.D.; Mignery, G.A.; Roderick, H.L.; Bootman, M.D.; Distelhorst, C.W. The BH4 domain of Bcl-2 inhibits ER calcium release and apoptosis by binding the regulatory and coupling domain of the IP3 receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 14397–14402. [Google Scholar] [CrossRef]
- Ivanova, H.; Ritaine, A.; Wagner, L.; Luyten, T.; Shapovalov, G.; Welkenhuyzen, K.; Seitaj, B.; Monaco, G.; De Smedt, H.; Prevarskaya, N.; et al. The trans-membrane domain of Bcl-2α, but not its hydrophobic cleft, is a critical determinant for efficient IP3 receptor inhibition. Oncotarget 2016, 7, 55704–55720. [Google Scholar] [CrossRef]
- Ivanova, H.; Wagner, L.E.; Tanimura, A.; Vandermarliere, E.; Luyten, T.; Welkenhuyzen, K.; Alzayady, K.J.; Wang, L.; Hamada, K.; Mikoshiba, K.; et al. Bcl-2 and IP3 compete for the ligand-binding domain of IP3Rs modulating Ca2+ signaling output. Cell. Mol. Life Sci. 2019, 76, 3843–3859. [Google Scholar] [CrossRef]
- Vervliet, T.; Parys, J.B.; Bultynck, G. Bcl-2 proteins and calcium signaling: Complexity beneath the surface. Oncogene 2016, 35, 5079–5092. [Google Scholar] [CrossRef]
- Foyouzi-Youssefi, R.; Arnaudeau, S.; Borner, C.; Kelley, W.L.; Tschopp, J.; Lew, D.P.; Demaurex, N.; Krause, K.-H. Bcl-2 decreases the free Ca2+ concentration within the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2000, 97, 5723–5728. [Google Scholar] [CrossRef]
- Eckenrode, E.F.; Yang, J.; Velmurugan, G.V.; Foskett, J.K.; White, C. Apoptosis Protection by Mcl-1 and Bcl-2 Modulation of Inositol 1,4,5-Trisphosphate Receptor-dependent Ca2+ Signaling. J. Biol. Chem. 2010, 285, 13678–13684. [Google Scholar] [CrossRef]
- Chang, M.-J.; Zhong, F.; Lavik, A.R.; Parys, J.B.; Berridge, M.J.; Distelhorst, C.W. Feedback regulation mediated by Bcl-2 and DARPP-32 regulates inositol 1,4,5-trisphosphate receptor phosphorylation and promotes cell survival. Proc. Natl. Acad. Sci. USA 2014, 111, 1186–1191. [Google Scholar] [CrossRef]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef]
- Vervloessem, T.; Kerkhofs, M.; La Rovere, R.M.; Sneyers, F.; Parys, J.B.; Bultynck, G. Bcl-2 inhibitors as anti-cancer therapeutics: The impact of and on calcium signaling. Cell Calcium 2018, 70, 102–116. [Google Scholar] [CrossRef]
- Akl, H.; La Rovere, R.M.L.; Janssens, A.; Vandenberghe, P.; Parys, J.B.; Bultynck, G. HA14-1 potentiates apoptosis in B-cell cancer cells sensitive to a peptide disrupting IP 3 receptor / Bcl-2 complexes. Int. J. Dev. Biol. 2015, 59, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Lavik, A.R.; Zhong, F.; Chang, M.-J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A synthetic peptide targeting the BH4 domain of Bcl-2 induces apoptosis in multiple myeloma and follicular lymphoma cells alone or in combination with agents targeting the BH3-binding pocket of Bcl-2. Oncotarget 2015, 6, 27388–27402. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, M.; La Rovere, R.M.; Schuermans, M.; Luyten, T.; Mikoshiba, K.; Vangheluwe, P.; Parys, J.B.; Bultynck, G. Extracellular and ER-stored Ca2+ contribute to BIRD-2-induced cell death in diffuse large B-cell lymphoma cells. Cell Death Discov. 2018, 4, 101. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, M.; La Rovere, R.M.; Akl, H.; Martines, C.; Welkenhuyzen, K.; Dubron, K.; Baes, M.; Janssens, A.; Vandenberghe, P.; Laurenti, L.; et al. Constitutive IP3 signaling underlies the sensitivity of B-cell cancers to the Bcl-2/IP3 receptor disruptor BIRD-2. Cell Death Differ. 2019, 26, 531–547. [Google Scholar] [CrossRef] [PubMed]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.-P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-driven apoptosis in chronic lymphocytic leukemia cells by peptide-mediated disruption of Bcl-2-IP3 receptor interaction. Blood 2011, 117, 2924–2934. [Google Scholar] [CrossRef]
- Greenberg, E.F.; McColl, K.S.; Zhong, F.; Wildey, G.; Dowlati, A.; Distelhorst, C.W. Synergistic killing of human small cell lung cancer cells by the Bcl-2-inositol 1,4,5-trisphosphate receptor disruptor BIRD-2 and the BH3-mimetic ABT-263. Cell Death Dis. 2015, 6, e2034. [Google Scholar] [CrossRef] [PubMed]
- Kerkhofs, M.; Vervloessem, T.; Bittremieux, M.; Bultynck, G. Recent advances in uncovering the mechanisms contributing to BIRD-2-induced cell death in B-cell cancer cells. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Xu, Y.; Gao, W.; Zhang, Y.; Su, J.; Liu, Y.; Guo, Y.; Dou, M.; Hu, K.; Sun, L. TAT-fused IP3R-derived peptide enhances cisplatin sensitivity of ovarian cancer cells by increasing ER Ca2+ release. Int. J. Mol. Med. 2018, 41, 809–817. [Google Scholar] [CrossRef]
- Han, B.; Park, D.; Li, R.; Xie, M.; Owonikoko, T.K.; Zhang, G.; Sica, G.L.; Ding, C.; Zhou, J.; Magis, A.T.; et al. Small-Molecule Bcl2 BH4 Antagonist for Lung Cancer Therapy. Cancer Cell 2015, 27, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Park, D.; Wang, M.; Nooka, A.; Deng, Q.; Matulis, S.; Kaufman, J.; Lonial, S.; Boise, L.H.; Galipeau, J.; et al. BCL2-BH4 antagonist BDA-366 suppresses human myeloma growth. Oncotarget 2016, 7, 27753–27763. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. A BH3 Mimetic for Killing Cancer Cells. Cell 2016, 165, 1560. [Google Scholar] [CrossRef]
- Hirota, J.; Michikawa, T.; Natsume, T.; Furuichi, T.; Mikoshiba, K. Calmodulin inhibits inositol 1,4,5-trisphosphate-induced calcium release through the purified and reconstituted inositol 1,4,5-trisphosphate receptor type 1. FEBS Lett. 1999, 456, 322–326. [Google Scholar] [CrossRef]
- Vandonselaar, M.; Hickie, R.A.; Quail, J.W.; Delbaere, L.T. Trifluoperazine-induced conformational change in Ca(2+)-calmodulin. Nat. Struct. Biol. 1994, 1, 795–801. [Google Scholar] [CrossRef]
- Urani, A.; Romieu, P.; Portales-Casamar, E.; Roman, F.J.; Maurice, T. The antidepressant-like effect induced by the sigma(1) (sigma(1)) receptor agonist igmesine involves modulation of intracellular calcium mobilization. Psychopharmacology (Berl.) 2002, 163, 26–35. [Google Scholar] [CrossRef]
- Ishima, T.; Nishimura, T.; Iyo, M.; Hashimoto, K. Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by donepezil: Role of sigma-1 receptors and IP3 receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1656–1659. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Ishima, T.; Iyo, M.; Hashimoto, K. Potentiation of nerve growth factor-induced neurite outgrowth by fluvoxamine: Role of sigma-1 receptors, IP3 receptors and cellular signaling pathways. PLoS ONE 2008, 3, e2558. [Google Scholar] [CrossRef] [PubMed]
- Natsvlishvili, N.; Goguadze, N.; Zhuravliova, E.; Mikeladze, D. Sigma-1 receptor directly interacts with Rac1-GTPase in the brain mitochondria. BMC Biochem. 2015, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Chakraborty, S.; Brailoiu, G.C.; Zhao, P.; Barr, J.L.; Ilies, M.A.; Unterwald, E.M.; Abood, M.E.; Taylor, C.W. Choline Is an Intracellular Messenger Linking Extracellular Stimuli to IP3-Evoked Ca2+ Signals through Sigma-1 Receptors. Cell Rep. 2019, 26, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Tagashira, H.; Bhuiyan, M.S.; Fukunaga, K. Diverse regulation of IP3 and ryanodine receptors by pentazocine through σ1-receptor in cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1201–H1212. [Google Scholar] [CrossRef] [PubMed]
- Tagashira, H.; Bhuiyan, M.S.; Shioda, N.; Fukunaga, K. Fluvoxamine rescues mitochondrial Ca2+ transport and ATP production through σ(1)-receptor in hypertrophic cardiomyocytes. Life Sci. 2014, 95, 89–100. [Google Scholar] [CrossRef]
- Ryskamp, D.; Wu, J.; Geva, M.; Kusko, R.; Grossman, I.; Hayden, M.; Bezprozvanny, I. The sigma-1 receptor mediates the beneficial effects of pridopidine in a mouse model of Huntington disease. Neurobiol. Dis. 2017, 97, 46–59. [Google Scholar] [CrossRef]
- Tagashira, H.; Shinoda, Y.; Shioda, N.; Fukunaga, K. Methyl pyruvate rescues mitochondrial damage caused by SIGMAR1 mutation related to amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1840, 3320–3334. [Google Scholar] [CrossRef]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Vilner, B.J.; Bowen, W.D. Modulation of cellular calcium by sigma-2 receptors: Release from intracellular stores in human SK-N-SH neuroblastoma cells. J. Pharmacol. Exp. Ther. 2000, 292, 900–911. [Google Scholar]
- Wu, Z.; Bowen, W.D. Role of sigma-1 receptor C-terminal segment in inositol 1,4,5-trisphosphate receptor activation: Constitutive enhancement of calcium signaling in MCF-7 tumor cells. J. Biol. Chem. 2008, 283, 28198–28215. [Google Scholar] [CrossRef] [PubMed]
- Plácido, A.I.; Pereira, C.M.F.; Correira, S.C.; Carvalho, C.; Oliveira, C.R.; Moreira, P.I. Phosphatase 2A Inhibition Affects Endoplasmic Reticulum and Mitochondria Homeostasis Via Cytoskeletal Alterations in Brain Endothelial Cells. Mol. Neurobiol. 2017, 54, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Shioda, N.; Ishikawa, K.; Tagashira, H.; Ishizuka, T.; Yawo, H.; Fukunaga, K. Expression of a truncated form of the endoplasmic reticulum chaperone protein, σ1 receptor, promotes mitochondrial energy depletion and apoptosis. J. Biol. Chem. 2012, 287, 23318–23331. [Google Scholar] [CrossRef]
- Novakova, M.; Ela, C.; Bowen, W.D.; Hasin, Y.; Eilam, Y. Highly selective sigma receptor ligands elevate inositol 1,4,5-trisphosphate production in rat cardiac myocytes. Eur. J. Pharmacol. 1998, 353, 315–327. [Google Scholar] [CrossRef]
- Novakova, M.; Sedlakova, B.; Sirova, M.; Fialova, K.; Krizanova, O. Haloperidol increases expression of the inositol 1,4,5-trisphosphate receptors in rat cardiac atria, but not in ventricles. Gen. Physiol. Biophys. 2010, 29, 381–389. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Abou-Lovergne, A.; Collado-Hilly, M.; Monnet, F.P.; Koukoui, O.; Prigent, S.; Coquil, J.F.; Dupont, G.; Combettes, L. Investigation of the role of sigma1-receptors in inositol 1,4,5-trisphosphate dependent calcium signaling in hepatocytes. Cell Calcium 2011, 50, 62–72. [Google Scholar] [CrossRef]
- Kubickova, J.; Lencesova, L.; Csaderova, L.; Stracina, T.; Hudecova, S.; Babula, P.; Rozborilova, E.; Novakova, M.; Krizanova, O. Haloperidol Affects Plasticity of Differentiated NG-108 Cells Through σ1R/IP3R1 Complex. Cell. Mol. Neurobiol. 2018, 38, 181–194. [Google Scholar] [CrossRef]
- Meunier, J.; Hayashi, T. Sigma-1 receptors regulate Bcl-2 expression by reactive oxygen species-dependent transcriptional regulation of nuclear factor kappaB. J. Pharmacol. Exp. Ther. 2010, 332, 388–397. [Google Scholar] [CrossRef]
- Zhang, Y.; Shi, Y.; Qiao, L.; Sun, Y.; Ding, W.; Zhang, H.; Li, N.; Chen, D. Sigma-1 receptor agonists provide neuroprotection against gp120 via a change in bcl-2 expression in mouse neuronal cultures. Brain Res. 2012, 1431, 13–22. [Google Scholar] [CrossRef]
- Ha, Y.; Shanmugam, A.K.; Markand, S.; Zorrilla, E.; Ganapathy, V.; Smith, S.B. Sigma receptor 1 modulates ER stress and Bcl2 in murine retina. Cell Tissue Res. 2014, 356, 15–27. [Google Scholar] [CrossRef]
- Su, T.-P.; Su, T.-C.; Nakamura, Y.; Tsai, S.-Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Wu, M.; Zubcevic, L.; Borschel, W.F.; Lander, G.C.; Lee, S.-Y. Structure of the cold- and menthol-sensing ion channel TRPM8. Science 2018, 359, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Liao, M.; Cao, E.; Julius, D.; Cheng, Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature 2013, 504, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal structure of the human σ1 receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef]
- Murray, S.C.; Flanagan, J.; Popova, O.B.; Chiu, W.; Ludtke, S.J.; Serysheva, I.I. Validation of Cryo-EM Structure of IP3R1 Channel. Structure 2013, 21, 900–909. [Google Scholar] [CrossRef]
- Hou, X.; Pedi, L.; Diver, M.M.; Long, S.B. Crystal Structure of the Calcium Release-Activated Calcium Channel Orai. Science 2012, 338, 1308–1313. [Google Scholar] [CrossRef]
- Van Petegem, F.; Clark, K.A.; Chatelain, F.C.; Minor, D.L. Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature 2004, 429, 671–675. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noyer, L.; Lemonnier, L.; Mariot, P.; Gkika, D. Partners in Crime: Towards New Ways of Targeting Calcium Channels. Int. J. Mol. Sci. 2019, 20, 6344. https://doi.org/10.3390/ijms20246344
Noyer L, Lemonnier L, Mariot P, Gkika D. Partners in Crime: Towards New Ways of Targeting Calcium Channels. International Journal of Molecular Sciences. 2019; 20(24):6344. https://doi.org/10.3390/ijms20246344
Chicago/Turabian StyleNoyer, Lucile, Loic Lemonnier, Pascal Mariot, and Dimitra Gkika. 2019. "Partners in Crime: Towards New Ways of Targeting Calcium Channels" International Journal of Molecular Sciences 20, no. 24: 6344. https://doi.org/10.3390/ijms20246344
APA StyleNoyer, L., Lemonnier, L., Mariot, P., & Gkika, D. (2019). Partners in Crime: Towards New Ways of Targeting Calcium Channels. International Journal of Molecular Sciences, 20(24), 6344. https://doi.org/10.3390/ijms20246344