Complement and Complement Targeting Therapies in Glomerular Diseases




Abstract
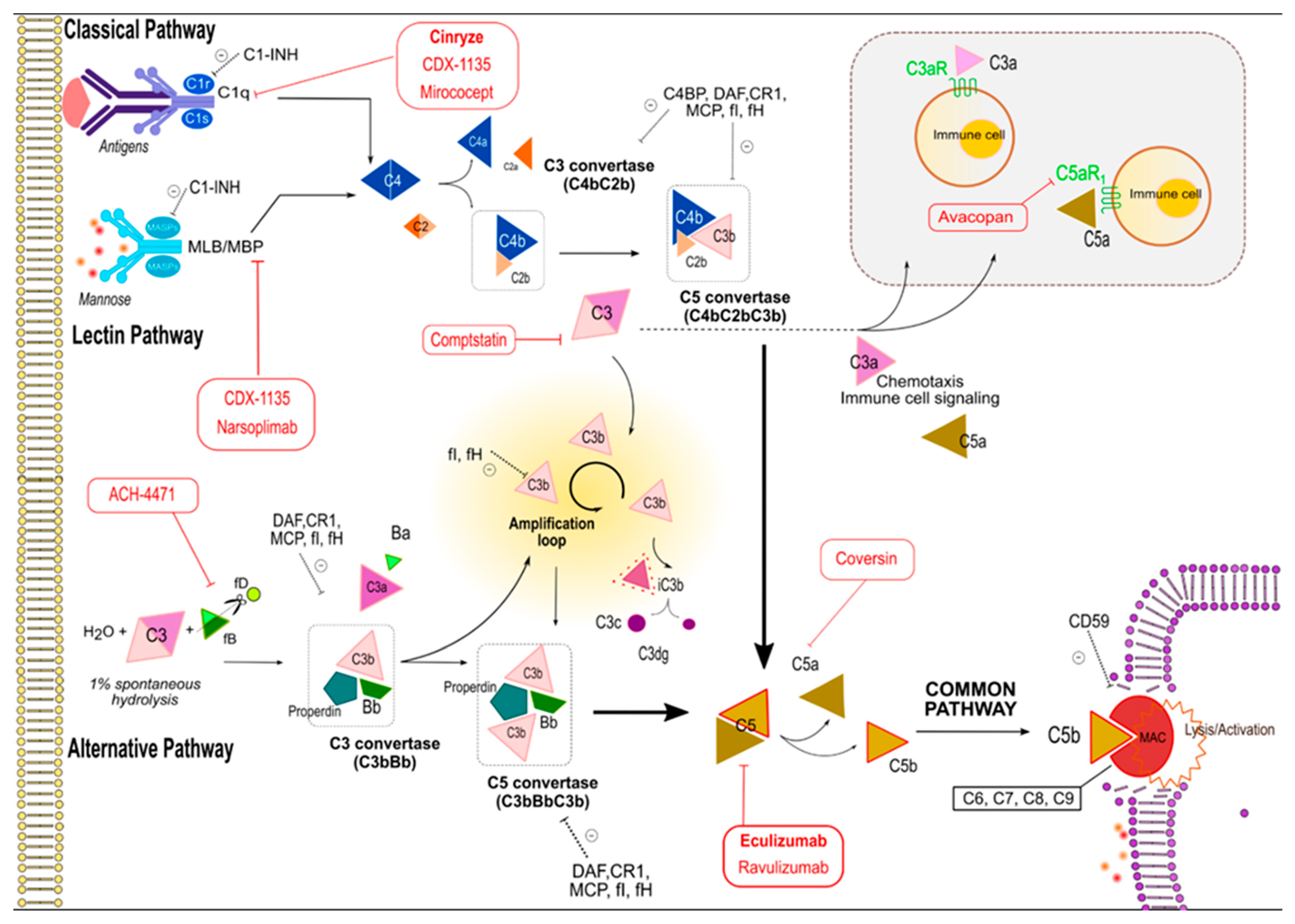
1. Complement Cascade
2. Complement Cascade Activation and Regulation
3. Effector Functions
4. Complement in Glomerular Diseases
4.1. Diseases with Antibody-Mediated Complement Activation
4.2. IgA Nephropathy
4.3. Membranous Nephropathy
4.4. Post Infectious Glomerulonephritis
4.5. Immune Complex-Mediated Membranoproliferative Glomerulonephritis (MPGN)
4.6. Anti-GBM Glomerulonephritis
4.7. ANCA Induced Renal Vasculitis
4.8. Lupus Nephritis
5. Disease with Complement Activation in the Absence of Detectable Serum Antibodies
5.1. Atypical Hemolytic Uremic Syndrome
5.2. C3 Nephropathy
6. Other Glomerular Diseases
Focal Segmental Glomerulosclerosis
7. Complement Inhibitory Drugs in Kidney Diseases
7.1. Eculizumab
7.2. C1 Inhibitor
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Walport, M.J. Complement−First of Two Parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Walport, M.J. Complement: Second of two parts: Complement at the interface between innate and adaptive immunity. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, M.; Suzuki, Y.; Ito, Y. Complement regulation and kidney diseases: Recent knowledge of the double-edged roles of complement activation in nephrology. Clin. Exp. Nephrol. 2018, 22, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Hourcade, D.E.; Spitzer, D.; Mitchell, L.M.; Atkinson, J.P. Properdin Can Initiate Complement Activation by Binding Specific Target Surfaces and Providing a Platform for De Novo. J. Immunol. Ref. 2007, 179, 2600–2608. [Google Scholar]
- Holmskov, U.; Thiel, S.; Jensenius, J.C. Collectin and Ficolins: Humoral Lectins of the Innate Immune Defense. Annu. Rev. Immunol. 2003, 21, 547–578. [Google Scholar] [CrossRef]
- Endo, Y.; Matsushita, M.; Fujita, T. The role of ficolins in the lectin pathway of innate immunity. Int. J. Biochem. Cell Biol. 2011, 43, 705–712. [Google Scholar] [CrossRef]
- Forneris, F.; Ricklin, D.; Wu, J.; Tzekou, A.; Wallace, R.S.; Lambris, J.D.; Gros, P. Structures of C3b in Complex with Factors B and D Give Insight into Complement Convertase Formation. Science 2010, 330, 1816–1820. [Google Scholar] [CrossRef]
- Mathern, D.R.; Heeger, P.S. Molecules Great and Small: The Complement System. Clin. J. Am. Soc. Nephrol. 2015, 10, 1636–1650. [Google Scholar] [CrossRef]
- Ricklin, D.; Hajishengallis, G.; Yang, K.; Lambris, J.D. Complement-a key system for immune surveillance and homeostasis. Nat. Immunol. 2010, 11, 785. [Google Scholar] [CrossRef]
- Guo, R.-F.; Ward, P.A. Role of C5a in inflamatory response. Annu. Rev. Immunol. 2005, 23, 821–852. [Google Scholar] [CrossRef]
- Kwan, W.H.; van der Touw, W.; Paz-Artal, E.; Li, M.O.; Heeger, P.S. Signaling through C5a receptor and C3a receptor diminishes function of murine natural regulatory T cells. J. Exp. Med. 2013, 210, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Van Der Touw, W.; Cravedi, P.; Kwan, W.-H.; Paz-Artal, E.; Merad, M.; Heeger, P.S. Receptors for C3a and C5a modulate stability of alloantigen-reactive induced regulatory T cells. J. Immunol. 2013, 190, 5921–5925. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Tenner, A.J.; Johswich, K.-O.; Ager, R.R.; Reis, E.S.; Köhl, J. The Role of the Anaphylatoxins in Health and Disease. Mol. Immunol. 2009, 46, 2753–2766. [Google Scholar] [CrossRef] [PubMed]
- Jane-wit, D.; Manes, T.D.; Yi, T.; Qin, L.; Clark, P.; Kirkiles-Smith, N.C.; Abrahimi, P.; Devalliere, J.; Moeckel, G.; Kulkarni, S.; et al. Alloantibody and Complement Promote T Cell-Mediated Cardiac Allograft Vasculopathy through Non-Canonical NF-κB Signaling in Endothelial Cells. Circulation 2013, 128, 2504–2516. [Google Scholar] [CrossRef]
- Adler, S.; Baker, P.J.; Johnson, R.J.; Ochi, R.F.; Pritzl, P.; Couser, W.G. Complement Membrane Attack Complex. Stimulates Production of Reactive Oxygen Metabolites by Cultured Rat Mesangial Cells. J. Clin. Investig. 1996, 77, 762–767. [Google Scholar] [CrossRef]
- Peake, P.W.; O’Grady, S.; Pussell, B.A.; Charlesworth, J.A. C3a is made by proximal tubular HK-2 cells and activates them via the C3a receptor. Kidney Int. 1999, 56, 1729–1736. [Google Scholar] [CrossRef]
- Lalli, P.N.; Strainic, M.G.; Yang, M.; Lin, F.; Medof, M.E.; Heeger, P.S. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood 2008, 11, 1759–1766. [Google Scholar] [CrossRef]
- Strainic, M.G.; Liu, J.; Huang, D.; An, F.; Lalli, P.N.; Muqim, N.; Shapiro, V.S.; Dubyak, G.R.; Heeger, P.S.; Medof, M.E. Locally Produced Complement Fragments C5a and C3a Provide Both Costimulatory and Survival Signals to Naive CD4 + T. Cells. Immunity 2008, 28, 425–435. [Google Scholar] [CrossRef]
- Heeger, P.S.; Lalli, P.N.; Lin, F.; Valujskikh, A.; Liu, J.; Muqim, N.; Xu, Y.; Medof, M.E. Decay-accelerating factor modulates induction of T cell immunity. J. Exp. Med. 2005, 201, 1523–1530. [Google Scholar] [CrossRef]
- Hanko, J.B.; Mullan, R.N.; O’rourke, D.M.; Mcnamee, P.T.; Maxwell, A.P.; Courtney, A.E. The changing pattern of adult primary glomerular disease. Nephrol. Dial. Transpl. 2009, 24, 3050–3054. [Google Scholar] [CrossRef]
- Nair, R.; Walker, P.D. Is IgA nephropathy the commonest primary glomerulopathy among young adults in the USA? Kidney Int. 2006, 69, 1455–1458. [Google Scholar] [CrossRef] [PubMed]
- Wada Id, Y.; Matsumoto, K.; Suzuki, T.; Saito, T.; Kanazawa, N.; Tachibanaid, S.; Iseri, K.; Sugiyama, M.; Iyoda, M.; Shibata, T. Clinical significance of serum and mesangial galactose-deficient IgA1 in patients with IgA nephropathy. PLoS ONE 2018, 13, e0206865. [Google Scholar] [CrossRef] [PubMed]
- Mestecky, J.; Raska, M.; Julian, B.A.; Gharavi, A.G.; Renfrow, M.B.; Moldoveanu, Z.; Novak, L.; Matousovic, K.; Novak, J. IgA Nephropathy: Molecular Mechanisms of the Disease. Annu. Rev. Pathol: Mech. Dis. 2012, 8, 217–240. [Google Scholar] [CrossRef] [PubMed]
- Lafayette, R.A.; Kelepouris, E.; Lafayette, R. Immunoglobulin A Nephropathy: Advances in Understanding of Pathogenesis and Treatment. Rev. Artic. Am. J. Nephrol 2018, 47, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Oortwijn, B.D.; Eijgenraam, J.W.; Rastaldi, M.P.; Roos, A.; Daha, M.R.; van Kooten, C. The Role of Secretory IgA and Complement in IgA Nephropathy. Semin. Nephrol. 2008, 28, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Roos, A.; Rastaldi, M.P.; Calvaresi, N.; Oortwijn, B.D.; Schlagwein, N.; Van Gijlswijk-Janssen, D.J.; Stahl, G.L.; Matsushita, M.; Fujita, T.; Van Kooten, C.; et al. Glomerular Activation of the Lectin Pathway of Complement in IgA Nephropathy Is Associated with More Severe Renal Disease. J. Am. Soc. Nephrol. 2006, 17, 1724–1734. [Google Scholar] [CrossRef] [PubMed]
- Thurman, J.M. Complement in Kidney Disease: Core Curriculum. Am. J. Kidney Dis. 2015, 65, 156–168. [Google Scholar] [CrossRef]
- Couser, W.G. Pathogenesis and treatment of glomerulonephritis-an update. J. Bras. Nefrol. 2016, 38, 107–122. [Google Scholar] [CrossRef]
- Gharavi, A.G.; Kiryluk, K.; Choi, M.; Li, Y.; Hou, P.; Xie, J.; Sanna-Cherchi, S.; Men, C.J.; Julian, B.A.; Wyatt, R.J.; et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat. Genet. 2011, 43, 321–329. [Google Scholar] [CrossRef]
- Xie, J.; Kiryluk, K.; Li, Y.; Mladkova, N.; Zhu, L.; Hou, P.; Ren, H.; Wang, W.; Zhang, H.; Chen, N.; et al. Fine Mapping Implicates a Deletion of CFHR1 and CFHR3 in Protection from IgA Nephropathy in Han Chinese. J. Am. Soc. Nephrol. 2016, 27, 3187–3194. [Google Scholar] [CrossRef]
- Medjeral-Thomas, N.R.; Lomax-Browne, H.J.; Beckwith, H.; Willicombe, M.; McLean, A.G.; Brookes, P.; Pusey, C.D.; Falchi, M.; Cook, H.T.; Pickering, M.C. Circulating complement factor H–related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017, 92, 942–952. [Google Scholar] [CrossRef]
- Zhu, L.; Guo, W.Y.; Shi, S.F.; Liu, L.J.; Lv, J.C.; Medjeral-Thomas, N.R.; Lomax-Browne, H.J.; Pickering, M.C.; Zhang, H. Circulating complement factor H–related protein 5 levels contribute to development and progression of IgA nephropathy. Kidney Int. 2018, 94, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, S.I.; Imai, E.; Maruyama, S. Immunology of membranous nephropathy. F1000 Res. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wen, L.; Dou, Y.; Zhao, Z. Human anti-thrombospondin type 1 domain-containing 7A antibodies induce membranous nephropathy through activation of lectin complement pathway. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, C.; Jin, L.; He, F.; Li, C.; Gao, Q.; Chen, G.; He, Z.; Song, M.; Zhou, Z.; et al. IgG4 anti-phospholipase A2 receptor might activate lectin and alternative complement pathway meanwhile in idiopathic membranous nephropathy: An inspiration from a cross-sectional study. Immunol. Res. 2016, 64, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Sandor, D.G.; Beck, L.H., Jr. The role of complement inmembranous nephropathy. Semin. Nephrol. 2013, 33, 531–542. [Google Scholar] [CrossRef]
- Baker, P.J.; Ochi, R.F.; Schulze, M.; Johnson, R.J.; Campbell, C.; Couser, W.G. Depletion of C6 Prevents Development of Proteinuria in Experimental Membranous Nephropathy in Rats. Am. J. Pathol. 1989, 135, 185–194. [Google Scholar]
- Saran, A.M.; Yuan, H.; Takeuchi, E.; McLaughlin, M.; Salant, D.J. Complement mediates nephrin redistribution and actin dissociation in experimental membranous nephropathy. Kidney Int. 2003, 64, 2072–2078. [Google Scholar] [CrossRef]
- Yuan, H.; Takeuchi, E.; Taylor, G.A.; Mclaughlin, M.; Brown, D.; Salant, D.J. Nephrin Dissociates from Actin, and Its Expression Is Reduced in Early Experimental Membranous Nephropathy. J. Am. Soc. Nephrol. 2002, 13, 946–956. [Google Scholar]
- Morel-Maroger, L.; Leathem, A.; Richet, G. Glomerular abnormalities in nonsystemic diseases. Relationship between findings by light microscopy and immunofluorescence in 433 renal biopsy specimens. Am. J. Med. 1972, 53, 170–184. [Google Scholar] [CrossRef]
- Verroust, P.J.; Wilson, C.B.; Cooper, N.R.; Edgington, T.S.; Dixon, F.J. Glomerular Complement Components in Human Glomerulonephritis. J. Clin. Investig. 1974, 53, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Hisano, S.; Matsushita, M.; Fujita, T.; Takeshita, M.; Iwasaki, H. Activation of the lectin complement pathway in post-streptococcal acute glomerulonephritis. Pathol. Int. 2007, 57, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Fervenza, F.C.; Zhang, Y.; Zand, L.; Meyer, N.C.; Borsa, N.; Nasr, S.H.; Smith, R.J.H. Atypical post-infectious glomerulonephritis is associated with abnormalities in the alternative pathway of complement. Kidney Int. 2013, 83, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Ito, N.; Ohashi, R.; Nagata, M. C3 glomerulopathy and current dilemmas. Clin. Exp. Nephrol. 2017, 21, 541–551. [Google Scholar] [CrossRef]
- Masani, N.; Jhaveri, K.D.; Fishbane, S. Update on membranoproliferative GN. Clin. J. Am. Soc. Nephrol. 2014, 9, 600–608. [Google Scholar] [CrossRef]
- Noris, M.; Remuzzi, G. Glomerular Diseases Dependent on Complement Activation, Including Atypical Hemolytic Uremic Syndrome, Membranoproliferative Glomerulonephritis, and C3 Glomerulopathy: Core Curriculum 2015. Am. J. Kidney Dis. 2015, 66, 359–375. [Google Scholar] [CrossRef]
- Servais, A.; Ne Noël, L.-H.; Roumenina, L.T.; Le Quintrec, M.; Ngo, S.; -Agnès Dragon-Durey, M.; Macher, M.-A.; Zuber, J.; Karras, A.; Provot, F.; et al. Acquired and genetic complement abnormalities play a critical role in dense deposit disease and other C3 glomerulopathies. Kidney Int. 2012, 82, 454–464. [Google Scholar] [CrossRef]
- Ma, R.; Cui, Z.; Hu, S.-Y.; Jia, X.-Y.; Yang, R. The Alternative Pathway of Complement Activation May Be Involved in the Renal Damage of Human Anti-Glomerular Basement Membrane Disease. PLoS ONE 2014, 9, 91250. [Google Scholar] [CrossRef]
- Minto, A.W.; Kalluri, R.; Togawa, M.; Bergijk, E.C.; Killen, P.D.; Salant, D.J. Augmented expression of glomerular basement membrane specific type IV collagen isoforms (alpha3-alpha5) in experimental membranous nephropathy. Proc. Assoc. Am. Physicians 1998, 110, 207–217. [Google Scholar]
- Sheerin, N.S.; Springall, T.; Carroll, M.C.; Hartley, B.; Sacks, S.H. Protection against anti-glomerular basement membrane (GBM)-mediated nephritis in C3-and C4-deficient mice. Clin. Exp. Immunol. 1997, 110, 403–409. [Google Scholar] [CrossRef]
- Fischer, E.G.; Lager, D.J. Anti-glomerular basement membrane glomerulonephritis: A morphologic study of 80 cases. Am. J. Clin. Pathol. 2006, 125, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Dairaghi, D.J.; Powers, J.P.; Ertl, L.S.; Baumgart, T.; Wang, Y.; Seitz, L.C.; Penfold, M.E.; Gan, L.; Hu, P.; et al. C5a Receptor (CD88) Blockade Protects against MPO-ANCA GN Necrotizing and crescentic GN (NCGN) and vasculitis are associated with ANCA. J. Am. Soc. Nephrol. 2014, 25, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Schreiber, A.; Heeringa, P.; Falk, R.J.; Jennette, J.C. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am. J. Pathol. 2007, 170, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Jayne, D.R.W.; Bruchfeld, A.N.; Harper, L.; Schaier, M.; Venning, M.C.; Hamilton, P.; Burst, V.; Grundmann, F.; Jadoul, M.; Szombati, I.; et al. Randomized trial of C5a receptor inhibitor avacopan in ANCA-associated vasculitis. J. Am. Soc. Nephrol. 2017, 28, 2756–2767. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.S.; D’Agati, V.D. Classification of lupus nephritis. Curr. Opin. Nephrol. Hypertens. 2009, 18, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Couser, W.G. Basic and Translational Concepts of Immune-Mediated Glomerular Diseases. J. Am. Soc. Nephrol. 2012, 23, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Guo, W.Y.; Wang, F.M.; Li, Y.Z.; Song, Y.; Yu, F.; Zhao, M.H. Complement Alternative Pathway’s Activation in Patients with Lupus Nephritis. Am. J. Med. Sci. 2017, 353, 247–257. [Google Scholar] [CrossRef]
- Pickering, M.C.; Botto, M. Are anti-C1q antibodies different from other SLE autoantibodies? Nat. Publ. Gr. 2010, 6, 490–493. [Google Scholar] [CrossRef]
- Bao, L.; Haas, M.; Quigg, R.J. Complement Factor H Deficiency Accelerates Development of Lupus Nephritis. J. Am. Soc. Nephrol. 2011, 22, 285–295. [Google Scholar] [CrossRef]
- Bao, L.; Quigg, R.J. Complement in Lupus Nephritis: The Good, the Bad, and the Unknown. Semin. Nephrol. 2007, 27, 69–80. [Google Scholar] [CrossRef]
- Tomlinson, S.; Atkinson, C.; Qiao, F.; Song, H.; Gilkeson, G.S. Mice lpr MRL/ Manifestations of Autoimmune Disease in Protects against Renal Disease and Other Low-Dose Targeted Complement Inhibition. J. Immunol. Ref. 2019, 180, 1231–1238. [Google Scholar]
- Bao, L.; Osawe, I.; Puri, T.; Lambris, J.D.; Haas, M.; Quigg, R.J. C5a promotes development of experimental lupus nephritis which can be blocked with a specific receptor antagonist. Eur. J. Immunol. 2005, 35, 2496–2506. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, Q.; MADRIt, J.A.; Rollins, S.A.; Chodera, A.; Matis, L.A.; Talmage, D.W. Amelioration of lupus-like autoimmune disease in NZB/W F1 mice after treatment with a blocking monoclonal antibody specific for complement component C5. Proc. Natl. Acad. Sci. USA 1996, 93, 8563–8568. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Colombo, B.M.; Puppo, F. Emerging biological drugs: A new therapeutic approach for Systemic Lupus Erythematosus. An update upon efficacy and adverse events. Autoimmun. Rev. 2011, 11, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, T.; Füst, G.; Farkas, H.; Jakab, L.; Temesszentandrási, G.; Nagy, G.; Kiss, E.; Gergely, P.; Zeher, M.; Griger, Z.; et al. C1-inhibitor autoantibodies in SLE. Lupus 2010, 19, 634–638. [Google Scholar]
- Al-Mayouf, S.M.; Abanomi, H.; Eldali, A. Impact of C1q deficiency on the severity and outcome of childhood systemic lupus erythematosus. Int. J. Rheum. Dis. 2011, 14, 81–85. [Google Scholar] [CrossRef]
- Heurich, M.; Martínez-Barricarte, R.; Francis, N.J.; Roberts, D.L.; Rodríguez De Córdoba, S.; Morgan, B.P.; Harris, C.L. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl. Acad. Sci. USA 2011, 108, 8761–8766. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative Role of Genetic Complement Abnormalities in Sporadic and Familial aHUS and Their Impact on Clinical Phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Manuelian, T.; Hellwage, J.; Meri, S.; Caprioli, J.; Noris, M.; Heinen, S.; Jozsi, M.; Neumann, H.P.H.; Remuzzi, G.; Zipfel, P.F. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J. Clin. Invest. 2003, 111, 1181–1190. [Google Scholar] [CrossRef]
- Caprioli, J.; Noris, M.; Brioschi, S.; Pianetti, G.; Castelletti, F.; Bettinaglio, P.; Mele, C.; Bresin, E.; Cassis, L.; Gamba, S.; et al. Genetics of HUS: The impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood 2006, 108, 1267–1279. [Google Scholar] [CrossRef]
- Sethi, S.; Haas, M.; Markowitz, G.S.; D’agati, V.D.; Rennke, H.G.; Jennette, J.C.; Bajema, I.M.; Alpers, C.E.; Chang, A.; Cornell, L.D.; et al. Mayo Clinic/Renal Pathology Society Consensus Report on Pathologic Classification, Diagnosis, and Reporting of GN. J. Am. Soc. Nephrol. 2016, 27, 1278–1287. [Google Scholar] [CrossRef] [PubMed]
- Sethi, S.; Fervenza, F.C. Pathology of Renal Diseases Associated with Dysfunction of the Alternative Pathway of Complement: C3 Glomerulopathy and Atypical Hemolytic Uremic Syndrome (aHUS). Semin. Thromb. Hemost. 2014, 40, 416–421. [Google Scholar] [PubMed]
- Łukawska, E.; Polcyn-Adamczak, M.; Niemir, Z.I. The role of the alternative pathway of complement activation in glomerular diseases. Clin. Exp. Med. 2018, 18, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Barbour, T.D.; Pickering, M.C.; Cook, H.T. Recent insights into C3 glomerulopathy. Nephrol. Dial. Transpl. 2013, 28, 1685–1693. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schramm, E.C.; Roumenina, L.T.; Rybkine, T.; Chauvet, S.; Vieira-Martins, P.; Hue, C.; Maga, T.; Valoti, E.; Wilson, V.; Jokiranta, S.; et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood 2015, 125, 2359–2369. [Google Scholar] [CrossRef]
- Pickering, M.C.; Warren, J.; Rose, K.L.; Carlucci, F.; Wang, Y.; Walport, M.J.; Cook, H.T.; Botto, M. Prevention of C5 activation ameliorates spontaneous and experimental glomerulonephritis in factor H-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9649–9654. [Google Scholar] [CrossRef]
- Gale, D.P.; De Jorge, E.G.; Cook, H.T.; Martinez-Barricarte, R.; Hadjisavvas, A.; McLean, A.G.; Pusey, C.D.; Pierides, A.; Kyriacou, K.; Athanasiou, Y.; et al. Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 2010, 376, 794–801. [Google Scholar] [CrossRef]
- Xiao, X.; Ghossein, C.; Tortajadam, A.; Zhang, Y.; Meyer, N.; Jones, M.; Borsa, N.G.; Nester, C.M.; Thomas, C.P.; de Córdoba, S.R.; et al. Familial C3 glomerulonephritis caused by a novel CFHR5-CFHR2 fusion gene. Mol. Immunol. 2016, 77, 89–96. [Google Scholar] [CrossRef]
- Chen, Q.; Wiesener, M.; Eberhardt, H.U.; Hartmann, A.; Uzonyi, B.; Kirschfink, M.; Amann, K.; Buettner, M.; Goodship, T.; Hugo, C.; et al. Complement factor H-related hybrid protein deregulates complement in dense deposit disease. J. Clin. Investig. 2014, 124, 145–155. [Google Scholar] [CrossRef]
- Kościelska-Kasprzak, K.; Bartoszek, D.; Myszka, M.; Zabińska, M.; Klinger, M. The Complement Cascade and Renal Disease. Arch. Immunol. Ther. Exp. 2014, 62, 47–57. [Google Scholar] [CrossRef]
- Kim, M.-K.; Maeng, Y.-I.; Lee, S.-J.; Lee, I.H.; Bae, J.; Kang, Y.-N.; Park, B.-T.; Park, K.-K. Pathogenesis and significance of glomerular C4d deposition in lupus nephritis: Activation of classical and lectin pathways. Int. J. Clin. Exp. Pathol. 2013, 6, 2157–2167. [Google Scholar] [PubMed]
- Korbet, S.M. Treatment of Primary FSGS in Adults. J. Am. Soc. Nephrol. 2012, 23, 1769–1776. [Google Scholar] [CrossRef] [PubMed]
- D’Agati, V.D.; Fogo, A.B.; Bruijn, J.A.; Jennette, J.C. Pathologic Classification of Focal Segmental Glomerulosclerosis: A Working Proposal. Am. J. Kidney Dis. 2004, 43, 368–382. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gu, Q.; Huang, J.; Qu, Z.; Wang, X.; Meng, L.; Wang, F.; Liu, G.; Cui, Z.; Zhao, M. Article Clinical Significance of IgM and C3 Glomerular Deposition in Primary Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2016, 11, 1582–1589. [Google Scholar] [CrossRef]
- Strassheim, D.; Renner, B.; Panzer, S.; Fuquay, R.; Kulik, L.; Ljubanovi, D.; Holers, V.M.; Thurman, J.M. IgM Contributes to Glomerular Injury in FSGS. J. Am. Soc. Nephrol. 2013, 24, 393–406. [Google Scholar] [CrossRef]
- Sethi, S.; Fervenza, F.C.; Zhang, Y.; Smith, R.J. Secondary Focal and Segmental Glomerulosclerosis Associated with Single-Nucleotide Polymorphisms in the Genes Encoding Complement Factor H and C3 HHS Public Access. Am. J. Kidney Dis. 2012, 60, 316–321. [Google Scholar] [CrossRef]
- Thurman, J.M.; Wong, M.; Renner, B.; Frazer-Abel, A.; Giclas, P.C.; Joy, M.S.; Jalal, D.; Radeva, M.K.; Gassman, J.; Gipson, D.S.; et al. Complement Activation in Patients with Focal Segmental Glomerulosclerosis. PLoS ONE 2015, 10, e0136558. [Google Scholar] [CrossRef]
- Tomlinson, S.; Thurman, J.M. Tissue-targeted complement therapeutics. Mol. Immunol. 2018, 102, 120–128. [Google Scholar] [CrossRef]
- Horiuchi, T.; Tsukamoto, H. Complement-targeted therapy: Development of C5-and C5a-targeted inhibition. Inflamm. Regen. 2016, 36, 11. [Google Scholar] [CrossRef]
- Cicardi, M.; Aberer, W.; Banerji, A.; Bas, M.; Bernstein, J.A.; Bork, K.; Caballero, T.; Farkas, H.; Grumach, A.; Kaplan, A.P.; et al. Classification, diagnosis, and approach to treatment for angioedema: Consensus report from the Hereditary Angioedema International Working Group. Allergy 2014, 69, 602–616. [Google Scholar] [CrossRef]
- Rother, R.P.; Rollins, S.A.; Mojcik, C.F.; Brodsky, R.A.; Bell, L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal hemoglobinuria. Nat. Biotechnol. 2007, 25, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Legendre, C.M.; Licht, C.; Muus, P.; Greenbaum, L.A.; Babu, S.; Bedrosian, C.; Bingham, C.; Cohen, D.J.; Delmas, Y.; Douglas, K.; et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N. Engl. J. Med. 2013, 368, 2169–2181. [Google Scholar] [CrossRef] [PubMed]
- Chatelet, V.; Frémeaux, V.; Frémeaux-Bacchi, F.; Lobbedez, T.; Ficheux, M.; Hurault De Ligny, B. Safety and Long-Term Efficacy of Eculizumab in a Renal Transplant Patient with Recurrent Atypical Hemolytic-Uremic Syndrome. Am. J. Transpl. 2009, 9, 2644–2645. [Google Scholar] [PubMed]
- Wong, E.K.S.; Goodship, T.H.J.; Kavanagh, D. Complement therapy in atypical haemolytic uraemic syndrome (aHUS). Mol. Immunol. 2013, 56, 199–212. [Google Scholar] [CrossRef]
- Kaabak, M.; Babenko, N.; Shapiro, R.; Zokoyev, A. A prospective randomized, controlled trial of eculizumab to prevent ischemia-reperfusion injury in pediatric kidney transplantation. Pediatr. Transpl. 2018, 22. [Google Scholar] [CrossRef]
- Marks, W.H.; Mamode, N.; Montgomery, R.A.; Stegall, M.D.; Ratner, L.E.; Cornell, L.D.; Rowshani, A.T.; Colvin, R.B.; Dain, B.; Boice, J.A.; et al. Safety and efficacy of eculizumab in the prevention of antibody-mediated rejection in living-donor kidney transplant recipients requiring desensitization therapy: A randomized trial. Am. J. Transpl. 2019, 1–13. [Google Scholar] [CrossRef]
- Glotz, D.; Russ, G.; Rostaing, L.; Legendre, C.; Tufveson, G.; Chadban, S.; Grinyó, J.; Mamode, N.; Rigotti, P.; Couzi, L.; et al. Safety and efficacy of eculizumab for the prevention of antibody-mediated rejection after deceased-donor kidney transplantation in patients with preformed donor-specific antibodies. Am. J. Transpl. 2019, 19, 2865–2875. [Google Scholar] [CrossRef]
- Bomback, A.S.; Smith, R.J.; Barile, G.R.; Zhang, Y.; Heher, E.C.; Herlitz, L.; Stokes, B.M.; Markowitz, G.S.; D’agati, V.D.; Canetta, P.A.; et al. Article Eculizumab for Dense Deposit Disease and C3 Glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2012, 7, 748–756. [Google Scholar] [CrossRef]
- Radhakrishnan, S.; Lunn, A.; Kirschfink, M.; Thorner, P.; Hebert, D.; Langlois, V.; Pluthero, F.; Licht, C. Eculizumab and refractory membranoproliferative glomerulonephritis. N. Engl. J. Med. 2012, 366, 1165–1166. [Google Scholar] [CrossRef]
- Mccaughan, J.A.; O’rourke, D.M.; Courtney, A.E. Recurrent Dense Deposit Disease After Renal Transplantation: An Emerging Role for Complementary Therapies. Am. J. Transpl. 2012, 12, 1046–1051. [Google Scholar] [CrossRef]
- Thurman, J.M.; Le Quintrec, M. Targeting the complement cascade: Novel treatments coming down the pike. Kidney Int. 2016, 90, 746–752. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Name. | Class | Pharmacodinamics | Disease | Status | Additional Info |
|---|---|---|---|---|---|
| Eculizumab | Humanized monoclonal antibody | Binds C5 preventing MAC generation | aHUS, DDD, C3GN | Available for use in PHN and aHUS | First USA FDA-approved among anti-complement drugs |
| Ravulizumab ALXN1210 | Humanized monoclonal antibody | Binds C5 preventing MAC generation | aHUS | Phase III for PHN | Induces prolonged decease of C5 plasmatic levels allowing longer dosing intervals compared to Eculizumab |
| Coversin | Small dimension recombinant protein | Prevents cleavage of C5 into C5a/C5b by C5 convertase | aHUS | Phase II for PHN | Valid alternative for patients bearer of C5 molecule polymorphisms which interferes with correct binding of Eculizumab |
| Avacopan CCX168 | Small dimension anti-inflammatory molecule | Inhibits selectively C5aR | aHUS ANCA-vasculitides | Phanse III for ANCA vasculitides. Phase II for aHUS | Effective replacing high-dose glucocorticoids in treating vasculitis |
| CDX-1135 | C1R-based molecule | Inhibits CR1 | DDD | Phase I for DDD | |
| Mirococept APT070 | CR1-based molecule | Inhibits CR1 | IRI in Tx | Phase I for DDD, C3GN | |
| Cinryze | C1 estarase | Inhibits CR1 | Antibody-mediated rejection in renal transplant | Available for use in HAE. Phase III for prevention of DGF in cadaveric allograft | FDA approved for hereditary angioedema |
| Narsoplimab OMS721 | Humanized monoclonal antibody | Binds the mannan-binding lectin-associated serinprotease-2 | aHUS, TTP IgAN | Phase II for aHUS, IgA, LES, MN, C3G | Multi-dose administration is needed |
| ACH-4471 | Small dimension molecule | Inhibits factor D | aHUS | Phase II for IC-MPGN, DDD, C3GN | Oral assumption with delivery advantage over intravenously infused agents |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrighetto, S.; Leventhal, J.; Zaza, G.; Cravedi, P. Complement and Complement Targeting Therapies in Glomerular Diseases. Int. J. Mol. Sci. 2019, 20, 6336. https://doi.org/10.3390/ijms20246336
Andrighetto S, Leventhal J, Zaza G, Cravedi P. Complement and Complement Targeting Therapies in Glomerular Diseases. International Journal of Molecular Sciences. 2019; 20(24):6336. https://doi.org/10.3390/ijms20246336
Chicago/Turabian StyleAndrighetto, Sofia, Jeremy Leventhal, Gianluigi Zaza, and Paolo Cravedi. 2019. "Complement and Complement Targeting Therapies in Glomerular Diseases" International Journal of Molecular Sciences 20, no. 24: 6336. https://doi.org/10.3390/ijms20246336
APA StyleAndrighetto, S., Leventhal, J., Zaza, G., & Cravedi, P. (2019). Complement and Complement Targeting Therapies in Glomerular Diseases. International Journal of Molecular Sciences, 20(24), 6336. https://doi.org/10.3390/ijms20246336