Identification of σE-Dependent Promoter Upstream of clpB from the Pathogenic Spirochaete Leptospira interrogans by Applying an E. coli Two-Plasmid System


Abstract
1. Introduction
2. Results and Discussion
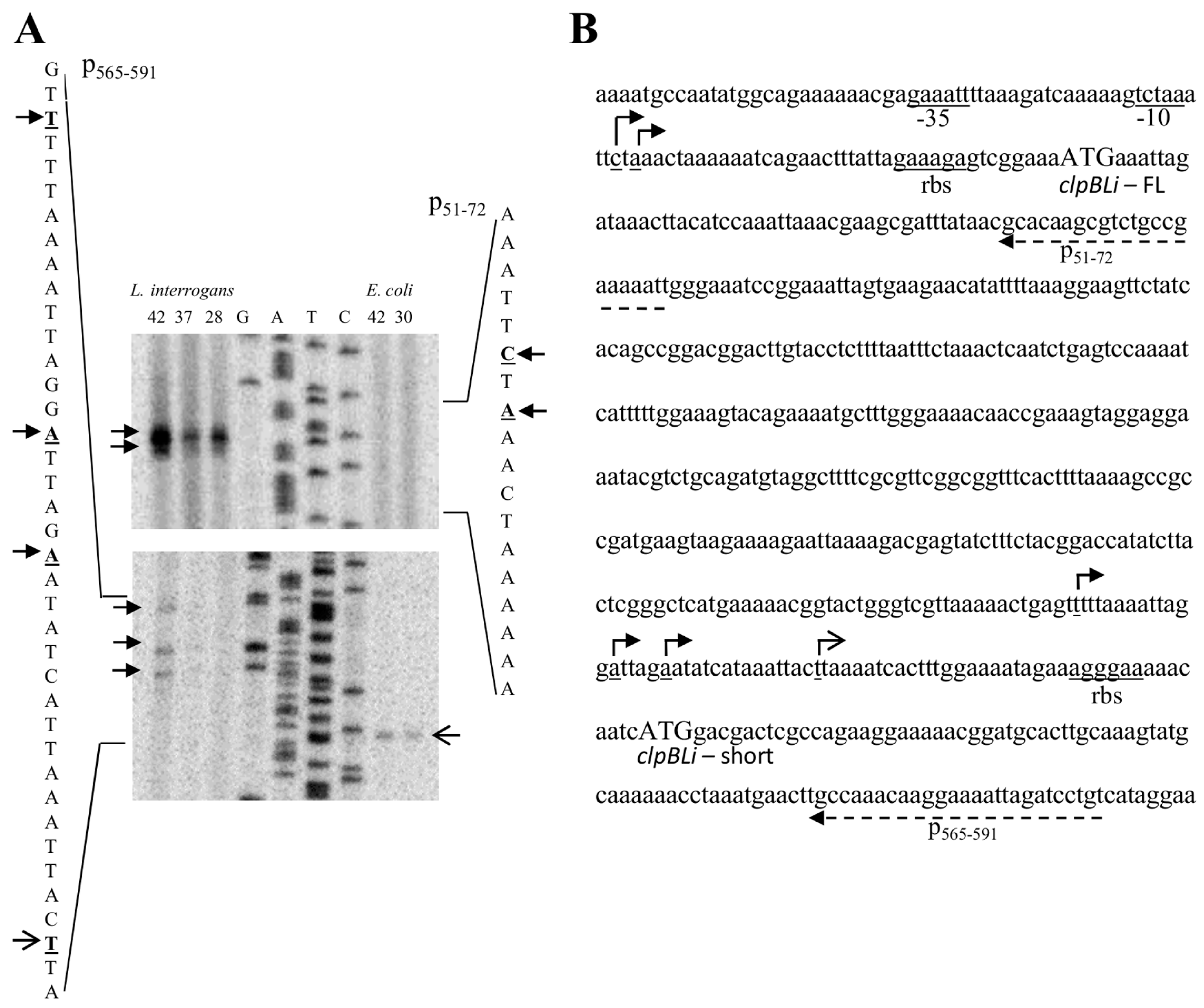
2.1. Identification of the clpB Transcriptional Start Sites by Primer Extension
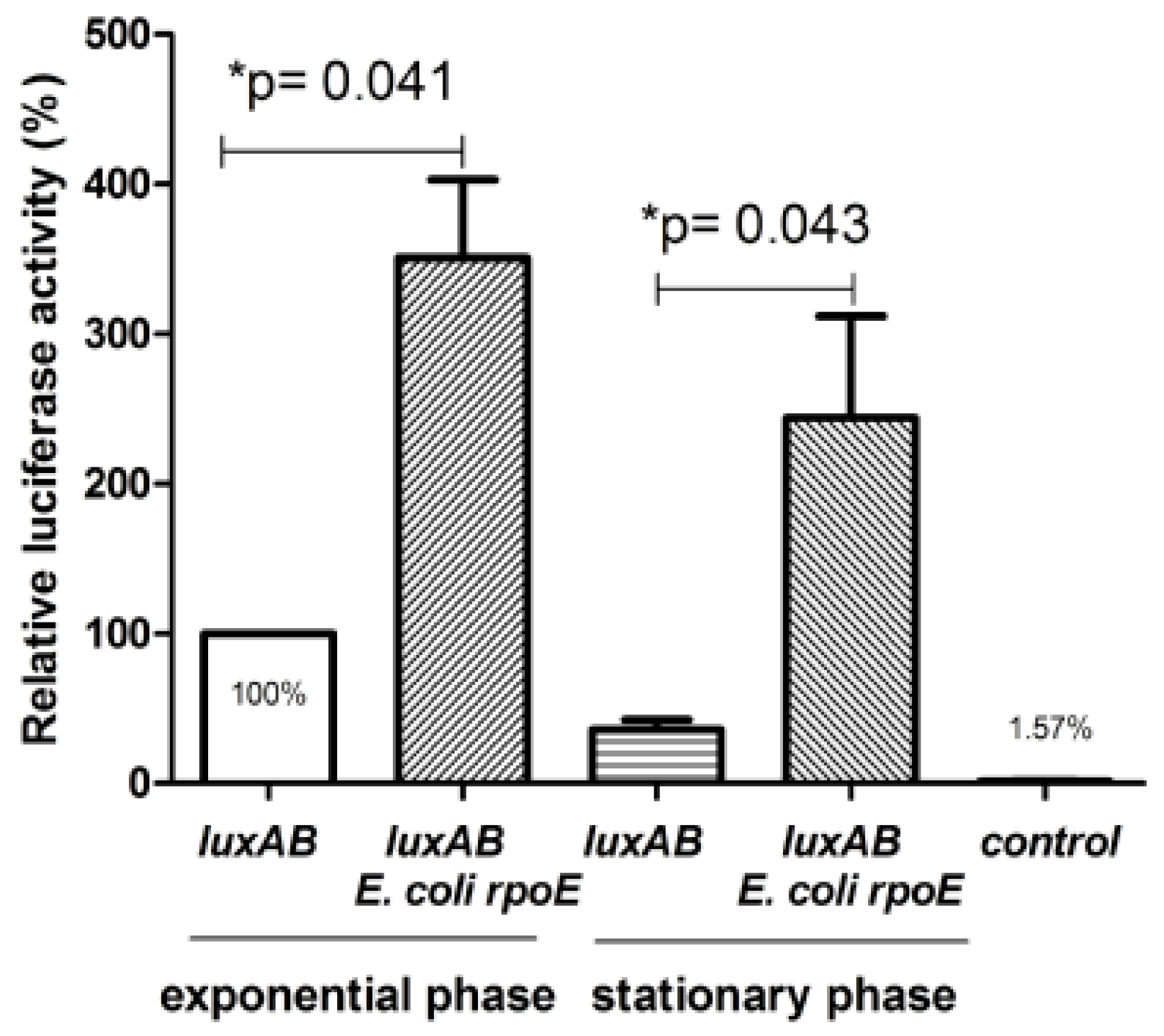
2.2. Presence of a Promoter Dependent on σE Upstream of the ClpBLi Gene and its Activity in E. coli Cells
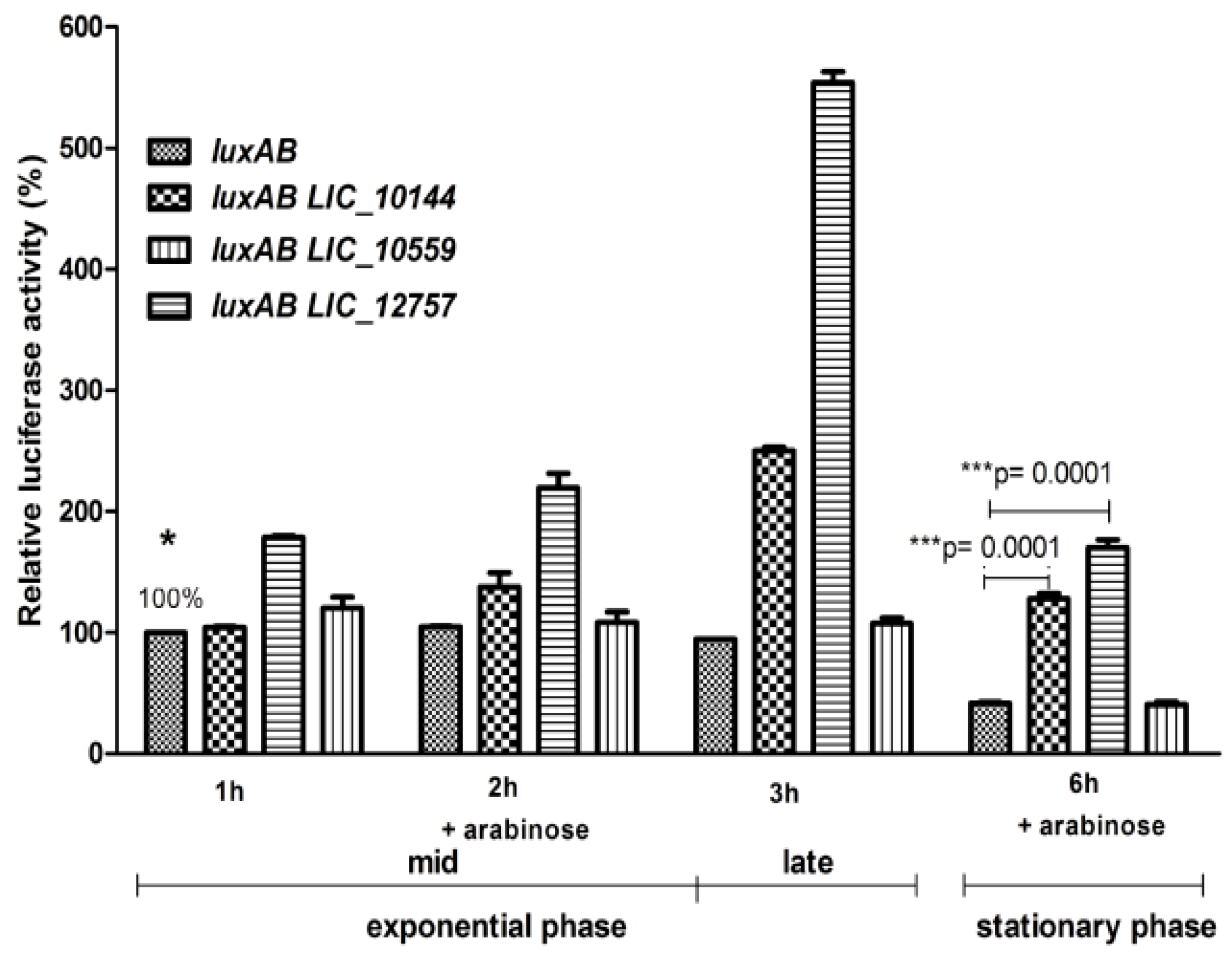
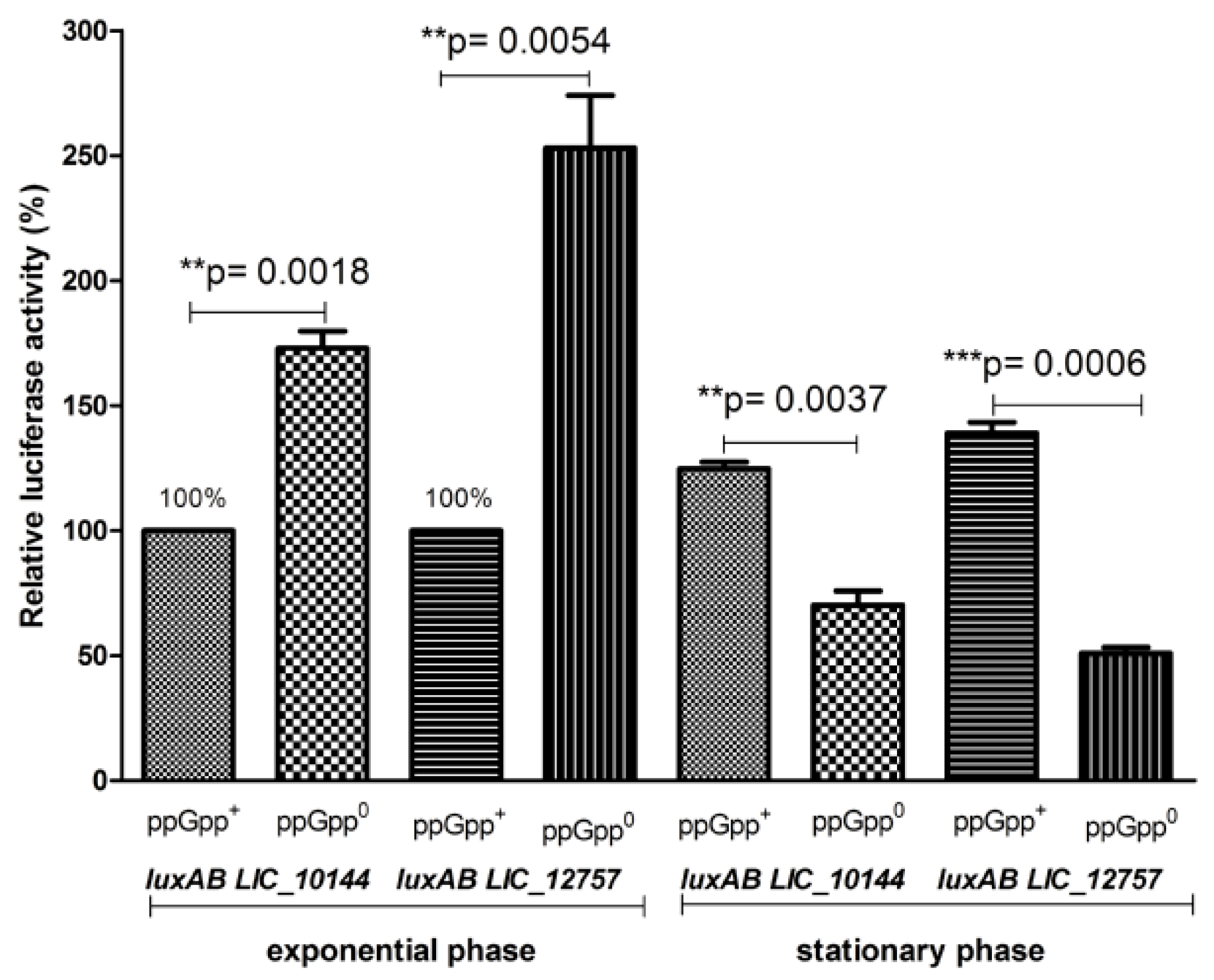
2.3. Role of Leptospiral ECFσ Factors in ClpBLi Gene Expression
3. Materials and Methods
3.1. Bacterial Strains and Plasmids
3.2. Cloning and PCR Methods
3.3. RNA Isolation and Primer Extension Assays
3.4. Luciferase Activity Assay
3.5. Detection of ClpBLi in E. coli MC4100ΔClpB Mutant Strain
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Costa, F.; Hagan, J.E.; Calcagno, J.; Kane, M.; Torgerson, P.; Martinez-Silveira, M.S.; Stein, C.; Abela-Ridder, B.; Ko, A.I. Global morbidity and mortality of leptospirosis: A systematic review. PLoS Negl. Trop. Dis. 2015, 9, e0003898. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.G.; Nola, L.; O’Grady, L.; More, S.J.; Doherty, L.M. Seroprevalence of Leptospira Hardjo in the irish suckler cattle population. Ir. Vet. J. 2012, 65, 8. [Google Scholar] [CrossRef] [PubMed]
- Truszczyński, M.; Pejsak, Z. New data concerning diagnosis and control of swine leptospirosis. Życie Weterynaryjne 2012, 87, 369–372. [Google Scholar]
- Arent, Z.; Kędzierska-Mieszkowska, S. Seroprevalence study of leptospirosis in horses in northern Poland. Vet. Rec. 2013, 172, 269. [Google Scholar] [CrossRef] [PubMed]
- Arent, Z.J.; Andrews, S.; Adamama-Moraitou, K.; Gilmore, C.; Pardali, D.; Ellis, W.A. Emergence of novel Leptospira serovars: A need for adjusting vaccination policies for dogs? Epidemiol. Infect. 2013, 141, 1148–1153. [Google Scholar] [CrossRef] [PubMed]
- Arent, Z.; Frizzell, C.; Gilmore, C.; Mackie, D.; Ellis, W.A. Isolation of leptospires from genital tract of sheep. Vet. Rec. 2013, 173, 582. [Google Scholar] [CrossRef] [PubMed]
- Adler, B. Pathogenesis of leptospirosis: Cellular and molecular aspects. Vet. Microbiol. 2014, 172, 353–358. [Google Scholar] [CrossRef]
- Kumar, S.; Lata, K.S.; Sharma, P.; Bhairappanavar, S.B.; Soni, S.; Das, J. Inferring pathogen-host interactions between Leptospira interrogans and Homo sapiens using network theory. Sci. Rep. 2019, 9, 1434. [Google Scholar] [CrossRef]
- Fouts, D.E.; Matthias, M.A.; Adhikarla, H.; Adler, B.; Amorim-Santos, L.; Berg, D.E.; Bulach, D.; Buschiazzo, A.; Chang, Y.-F.; Galloway, R.L.; et al. What makes a bacterial species pathogenic?: Comparative genomic analysis of the genus Leptospira. PLoS Negl. Trop. Dis. 2016, 10, e0004403. [Google Scholar] [CrossRef]
- Zhukova, A.; Fernandes, L.G.; Hugon, P.; Pappas, C.J.; Sismeiro, O.; Coppée, J.Y.; Becavin, C.; Malabat, C.; Eshghi, A.; Zhang, J.J.; et al. Genome-wide transcriptional start site mapping and sRNA identification in the pathogen Leptospira interrogans. Front. Cell. Infect. Microbiol. 2017, 7, 1–14. [Google Scholar] [CrossRef]
- Bervoets, I.; Van Brempt, M.; Van Nerom, K.; Van Hove, B.; Maertens, J.; De Mey, M.; Charlier, D. A sigma factor toolbox for orthogonal gene expression in Escherichia coli. Nucleic Acids Res. 2018, 46, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.L.T.O.; Verjovski-Almeida, S.; Van Sluys, M.A.; Monteiro-Vitorello, C.B.; Camargo, L.E.A.; Digiampietri, L.A.; Harstkeerl, R.A.; Ho, P.L.; Marques, M.V.; Oliveira, M.C.; et al. Genome features of Leptospira interrogans serovar Copenhageni. Braz. J. Med. Biol. Res. 2004, 37, 459–477. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Bulach, D.M.; Powell, D.R.; Haake, D.A.; Matsunaga, J.; Paustian, M.L.; Zuerner, R.L.; Adler, B. Effects of temperature on gene expression patterns in Leptospira interrogans serowar Lai as assessed by whole-genome microarrays. Infect. Immun. 2006, 74, 5848–5859. [Google Scholar] [CrossRef] [PubMed]
- Helmann, J.D. The extracytoplasmic function (ECF) sigma factors. Adv. Microb. Physiol. 2002, 46, 47–110. [Google Scholar] [PubMed]
- Lourdault, K.; Cerqueira, G.M.; Wunder, E.A., Jr.; Picardeau, M. Inactivation of clpB in the pathogen Leptospira interrogans reduces virulence and resistance to stress conditions. Infect. Immun. 2011, 79, 3711–3717. [Google Scholar] [CrossRef]
- Krajewska, J.; Arent, Z.; Więckowski, D.; Zolkiewski, M.; Kędzierska-Mieszkowska, S. Immunoreactivity of the AAA+ chaperone ClpB from Leptospira interrogans with sera from Leptospira-infected animals. BMC Microbiol. 2016, 16, 151–158. [Google Scholar] [CrossRef]
- Krajewska, J.; Arent, Z.; Zolkiewski, M.; Kędzierska-Mieszkowska, S. Isolation and identification of putative protein substrate of the AAA+ chaperone chaperone ClpB from pathogenic spirochaete Leptospira interrogans. Int. J. Mol. Sci. 2018, 19, 1234. [Google Scholar] [CrossRef]
- Squires, C.L.; Pedersen, S.; Ross, B.M.; Squires, C. ClpB is the Escherichia coli heat shock protein F84.1. J. Bacteriol. 1991, 173, 4254–4262. [Google Scholar] [CrossRef]
- Weiner, J., III; Herrmann, R.; Browning, G.F. Transcription in Mycoplasma pneumonia. Nucleic Acids Res. 2000, 28, 4488–4496. [Google Scholar] [CrossRef]
- Park, S.K.; Kim, K.I.; Woo, K.M.; Seol, J.H.; Tanaka, K.; Ichihara, A.; Ha, D.B.; Chung, C.H. Site-directed mutagenesis of the dual translational initiation sites of the clpB gene of Escherichia coli and characterization of its gene products. J. Biol. Chem. 1993, 268, 20170–20174. [Google Scholar]
- Nagy, N.; Guenther, I.; Akoyev, V.; Barnett, M.E.; Zavodszky, M.I.; Kedzierska-Mieszkowska, S.; Zolkiewski, M. Synergistic cooperation between two ClpB isoforms in aggregate reactivation. J. Mol. Biol. 2010, 396, 697–707. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Matsunaga, J.; Haake, D.A. Cis-Acting determinant limiting expression of sphingomyelinase gene sph2 in Leptospira interrogans, identified with a gfp reporter plasmid. Appl. Environ. Microbiol. 2018, 84, e02068-1. [Google Scholar] [CrossRef] [PubMed]
- Rezuchova, B.; Kormanec, J. A two-plasmid system for identification of promoters recognized by RNA polymerase containing extracytoplasmic stress response σE in Escherichia coli. J. Microbiol. Methods 2001, 45, 103–111. [Google Scholar] [CrossRef]
- Rezuchova, B.; Miticka, H.; Homerova, D.; Roberts, M.; Kormanec, J. New members of the Escherichia coli σE regulon identified by a two-plasmid system. FEMS Microbiol. Lett. 2003, 225, 1–7. [Google Scholar] [CrossRef]
- Krajewska, J.; Modrak-Wójcik, A.; Arent, Z.; Więckowski, D.; Zolkiewski, M.; Bzowska, A.; Kędzierska-Mieszkowska, S. Characterization of the molecular chaperone ClpB from the pathogenic spirochaete Leptospira interrogans. PLoS ONE 2017, 12, e0181118. [Google Scholar] [CrossRef]
- Kazmierczak, M.J.; Wiedmann, M.; Boor, K.J. Alternative sigma factors and their roles in bacterial virulence. Microbiol. Mol. Biol. Rev. 2005, 69, 527–543. [Google Scholar] [CrossRef]
- Costanzo, A.; Ades, S.E. Growth phase-Dependent regulation of the extracytoplasmatic stress factor, σE, by guanosine 3’, 5’-bispyrophosphate (ppGpp). J. Bacteriol. 2016, 188, 4627–4634. [Google Scholar] [CrossRef][Green Version]
- Costanzo, A.; Nicoloff, H.; Barchinger, E.; Banta, A.B.; Gourse, R.L.; Ades, S.E. ppGpp and DskA likely regulate the activity of the extracytoplasmatic stress factor σE in Escherichia coli by both direct and indirect mechanisms. Mol. Microbiol. 2008, 67, 619–632. [Google Scholar] [CrossRef]
- Mutalik, V.K.; Nonaka, G.; Ades, S.E.; Rhodius, V.A.; Gross, C.A. Promoter strength properties of the complete sigma E regulon of Escherichia coli and Salmonella enterica. J. Bacteriol. 2009, 191, 7279–7287. [Google Scholar] [CrossRef]
- Potrykus, K.; Cashel, M. (p)ppGpp: Still magical? Annu. Rev. Microbiol. 2008, 62, 35–51. [Google Scholar] [CrossRef]
- Dalebroux, Z.D.; Svensson, S.L.; Gaynor, E.C.; Swanson, M.S. ppGpp conjures bacterial virulence. Microbiol. Mol. Biol. Rev. 2010, 74, 171–199. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Deng, C.; Liu, B.; Zeng, L.; Zhao, W.; Zhang, Y.; Jiang, X.; Guo, X.; Qin, J. Characterization of a bifunctional enzyme with (p)ppGpp-hydrolase/synthase activity in Leptospira interrogans. FEMS Microbiol. Lett. 2013, 348, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Vinella, D.; Potrykus, K.; Murphy, H.; Cashel, M. Effects on growth by changes of the balance between GreA, GreB, and DksA suggest mutual competition and functional redundancy in Escherichia coli. J. Bacteriol. 2012, 194, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Churchward, G.; Belin, D.; Nagamine, Y. A pSC101-derived plasmid which shows no sequence homology to other commonly used cloning vectors. Gene 1984, 31, 165–171. [Google Scholar] [CrossRef]
- Harlow, E.; Lane, D. Antibodies: A Laboratory Manual; CSH: Cold Spring Harbor, NY, USA, 1988. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species. | σ Factor | Function/Controlled Genes |
|---|---|---|
| L. interrogans | σ70 | housekeeping genes (>1000 genes) |
| σ28 | genes encoding components of the endoflagellum and the flagellin-specific chaperone FliS | |
| σ24/σΕ (ECF σ factor) | extracytoplasmic function (469 putative binding sites in the Leptospira genome), stress response and virulence (clpB, this study) | |
| σ54 | genes encoding putative lipoproteins and the ammonium transporter AmtB | |
| E. coli | σ70 | housekeeping genes |
| σS/σ38 | stationary phase gene expression | |
| σ32/σΗ | heat shock response | |
| σ28/σF | flagellar genes/motility genes | |
| σ24/σΕ (ECF σ factor) | extracytoplasmic function, cell surface stress response, resistance to heat shock and other environmental stresses | |
| σ19/σFecI (ECF σ factor) | extracytoplasmic function | |
| σ54/σΝ | nitrogen metabolism genes |
| Gene ID a | Protein Accession Number | Number of Amino Acids | Identity/Similarity b (%) |
|---|---|---|---|
| LIC_10144 | AAS68777 | 174 | 26.0/50.3 |
| LIC_10225 | AAS68853 | 301 | 20.8/51.6 |
| LIC_10386 | AAS69009 | 182 | 20.1/55.6 |
| LIC_10559 | AAS69180 | 181 | 28.3/58.7 |
| LIC_10644 | AAS69265 | 174 | 26.8/61 |
| LIC_11817 | AAS70405 | 184 | 26.1/55.7.9 |
| LIC_12490 | AAS71055 | 206 | 32.7/70.8 |
| LIC_12757 | AAS71314 | 180 | 24.0/55.0 |
| LIC_13266 | AAS71810 | 192 | 31.1/63.3 |
| LIC_13285 | AAS71829 | 169 | 27.6/62.9 |
| Oligonucleotide | Sequence (5’ to 3’) | Purpose |
|---|---|---|
| p51–72 | ATTTTTCGGCAGACGCTTGTGC | primer extension |
| p290–321 | AAGATACTCGTCTTTTAATTCTTTTCTTACTT | primer extension |
| p565–591 | ACAGGATCTAATTTTCCTTGTTTGGC | primer extension |
| luxANdeI | CATATGAAATTTGGAAACTTCCTTCTC | cloning of luciferase reporter genes |
| luxBHindIII | CGACCAAAGCTTACAGTGGTATTTGACGATG | cloning of luciferase reporter genes |
| SDLiLuxAXmaI | CCCGGGAACTTTATTAGAAAGAGTC | cloning of luciferase reporter genes |
| clpBLiNdeI | CATATGAAATTAGATAAACTTACATCCAAATT | cloning of clpBLi |
| clpBLiHindIII | AAGCTTTTAAACTACAACAACTACCTTTCCCT | cloning of clpBLi |
| prLiXmaI | CCCGGGATAAAATTTCCGAGTCCGATT | cloning of clpBLi promoter region |
| prLIC_10144NdeI | CATATGGTTCAATCTGATTCTGC | cloning of LIC_10144 |
| prLIC_10144HindIII | AAGCTTAGAATTGAAATCCTTGTAG | cloning of LIC_10144 |
| prLIC_10559NdeI | CATATGATGCTGAATCCGAATTGC | cloning of LIC_10559 |
| prLIC_10559HindIII | AAGCTTTCATTCTTCATAAAATTTCTCC | cloning of LIC_10559 |
| prLIC_12757NdeI | CATATGAGCCAAAATTCCGAAAC | cloning of LIC_12757 |
| prLIC_12757HindIII | AAGCTTCTATATACTCTCAAAGTCG | cloning of LIC_12757 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kędzierska-Mieszkowska, S.; Potrykus, K.; Arent, Z.; Krajewska, J. Identification of σE-Dependent Promoter Upstream of clpB from the Pathogenic Spirochaete Leptospira interrogans by Applying an E. coli Two-Plasmid System. Int. J. Mol. Sci. 2019, 20, 6325. https://doi.org/10.3390/ijms20246325
Kędzierska-Mieszkowska S, Potrykus K, Arent Z, Krajewska J. Identification of σE-Dependent Promoter Upstream of clpB from the Pathogenic Spirochaete Leptospira interrogans by Applying an E. coli Two-Plasmid System. International Journal of Molecular Sciences. 2019; 20(24):6325. https://doi.org/10.3390/ijms20246325
Chicago/Turabian StyleKędzierska-Mieszkowska, Sabina, Katarzyna Potrykus, Zbigniew Arent, and Joanna Krajewska. 2019. "Identification of σE-Dependent Promoter Upstream of clpB from the Pathogenic Spirochaete Leptospira interrogans by Applying an E. coli Two-Plasmid System" International Journal of Molecular Sciences 20, no. 24: 6325. https://doi.org/10.3390/ijms20246325
APA StyleKędzierska-Mieszkowska, S., Potrykus, K., Arent, Z., & Krajewska, J. (2019). Identification of σE-Dependent Promoter Upstream of clpB from the Pathogenic Spirochaete Leptospira interrogans by Applying an E. coli Two-Plasmid System. International Journal of Molecular Sciences, 20(24), 6325. https://doi.org/10.3390/ijms20246325