O-GlcNAc Transferase Regulates Angiogenesis in Idiopathic Pulmonary Arterial Hypertension

Abstract
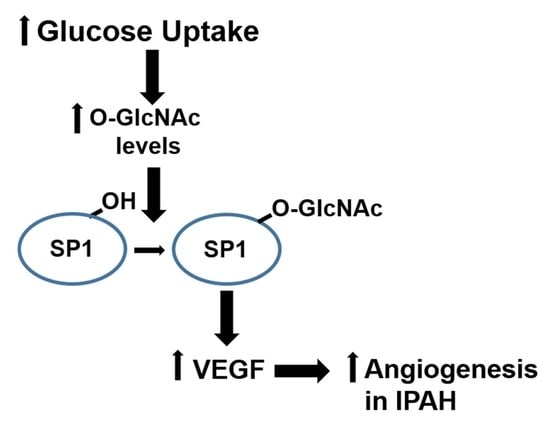
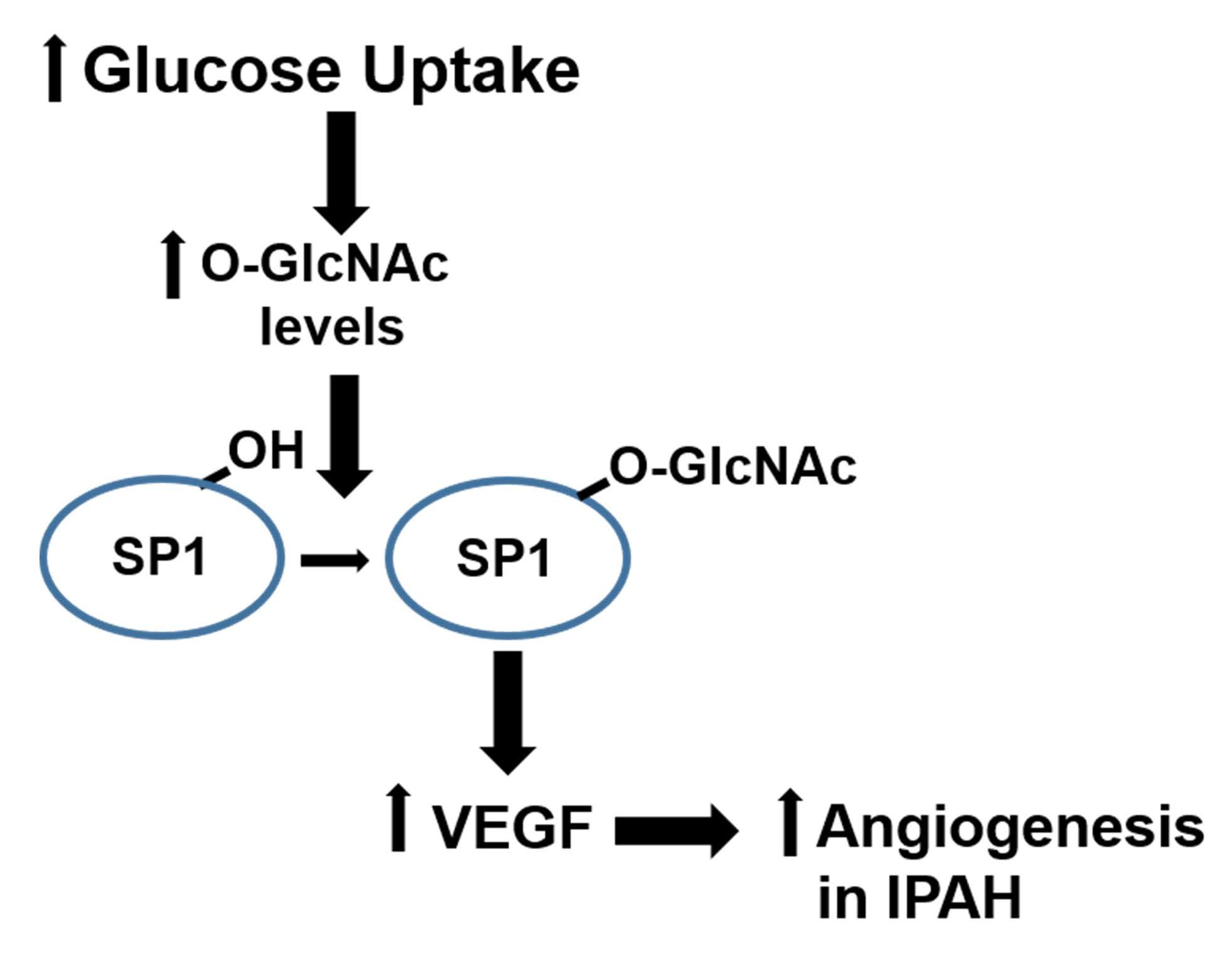
1. Introduction
2. Results
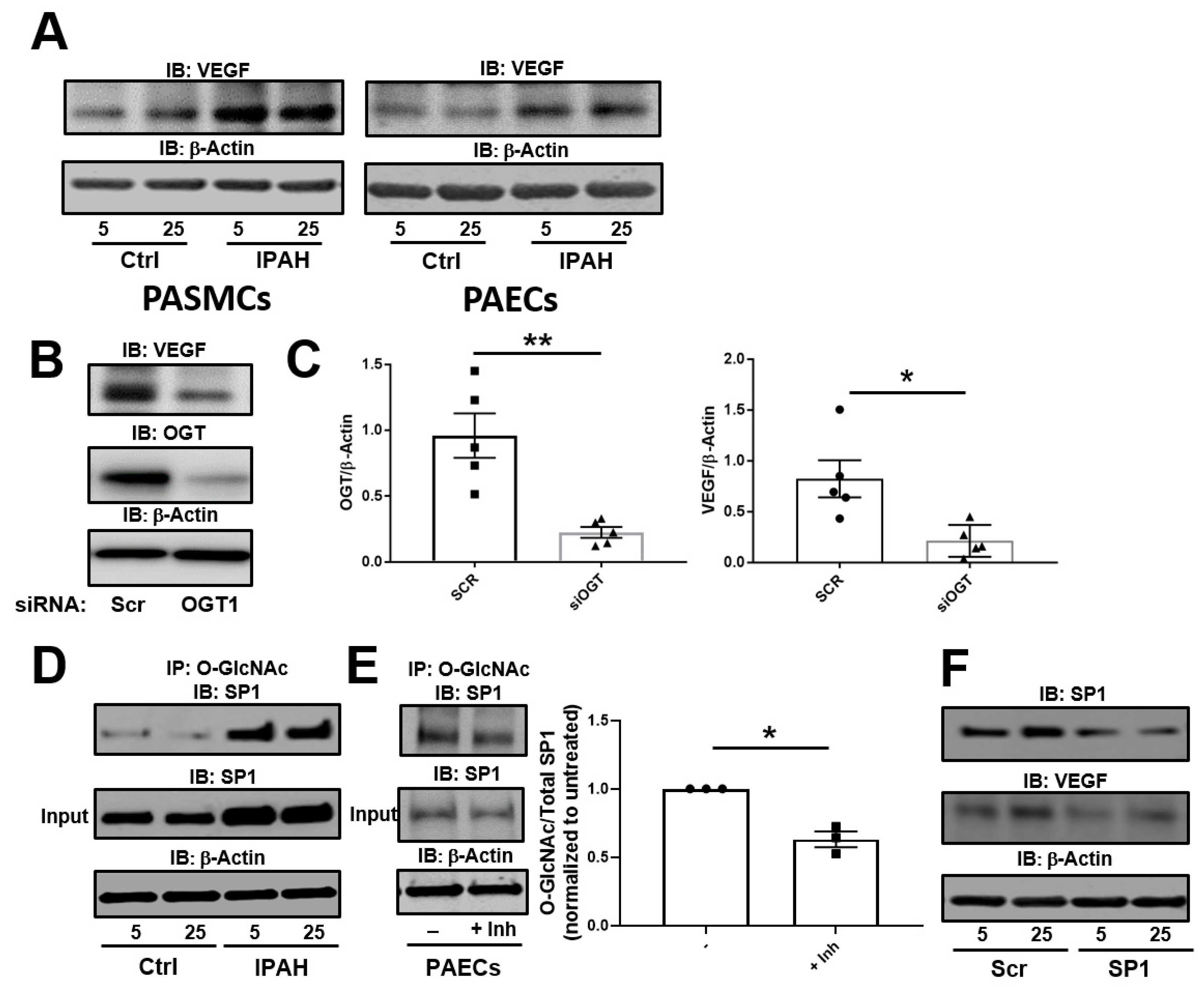
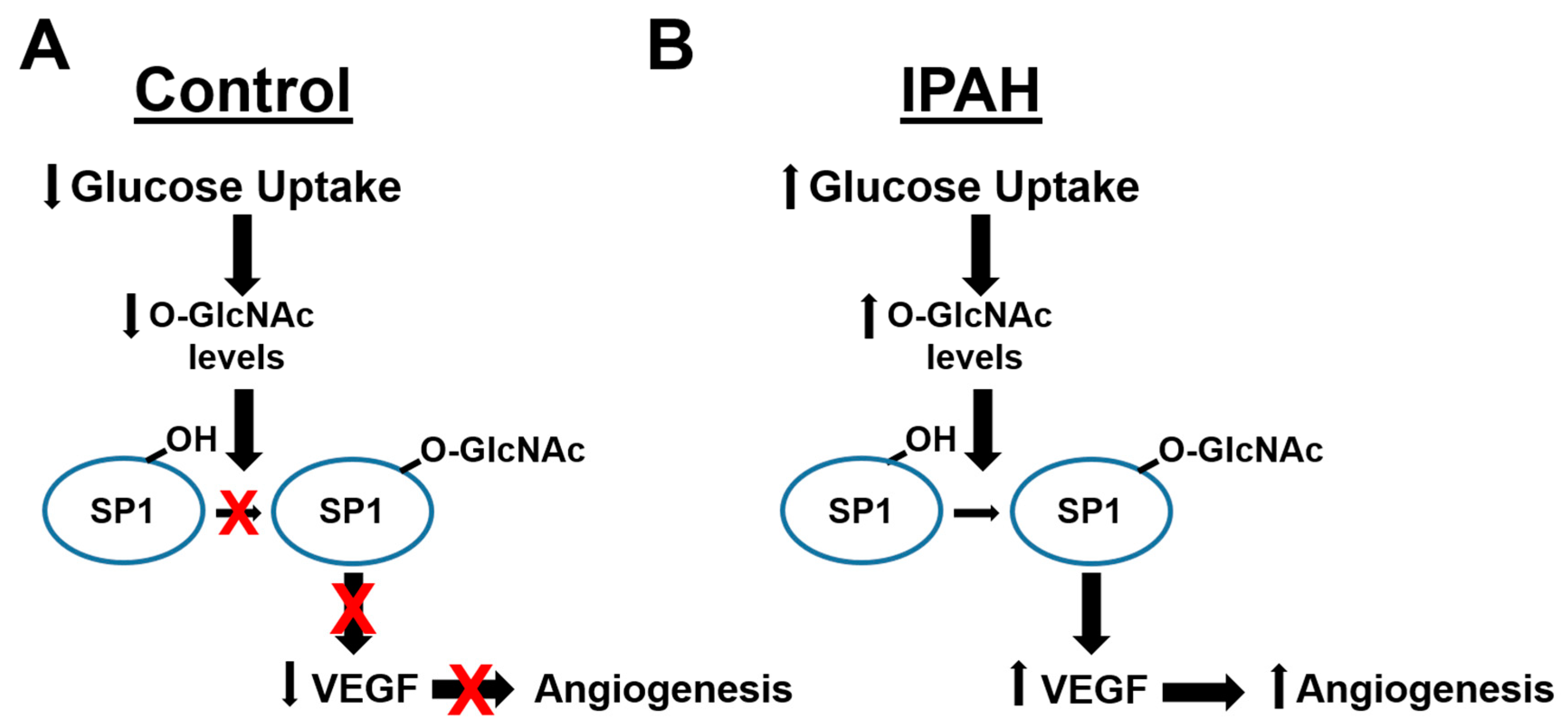
2.1. The OGT/O-GlcNAc Axis Regulates VEGF-A Expression by the O-GlcNAc Modification of SP-1
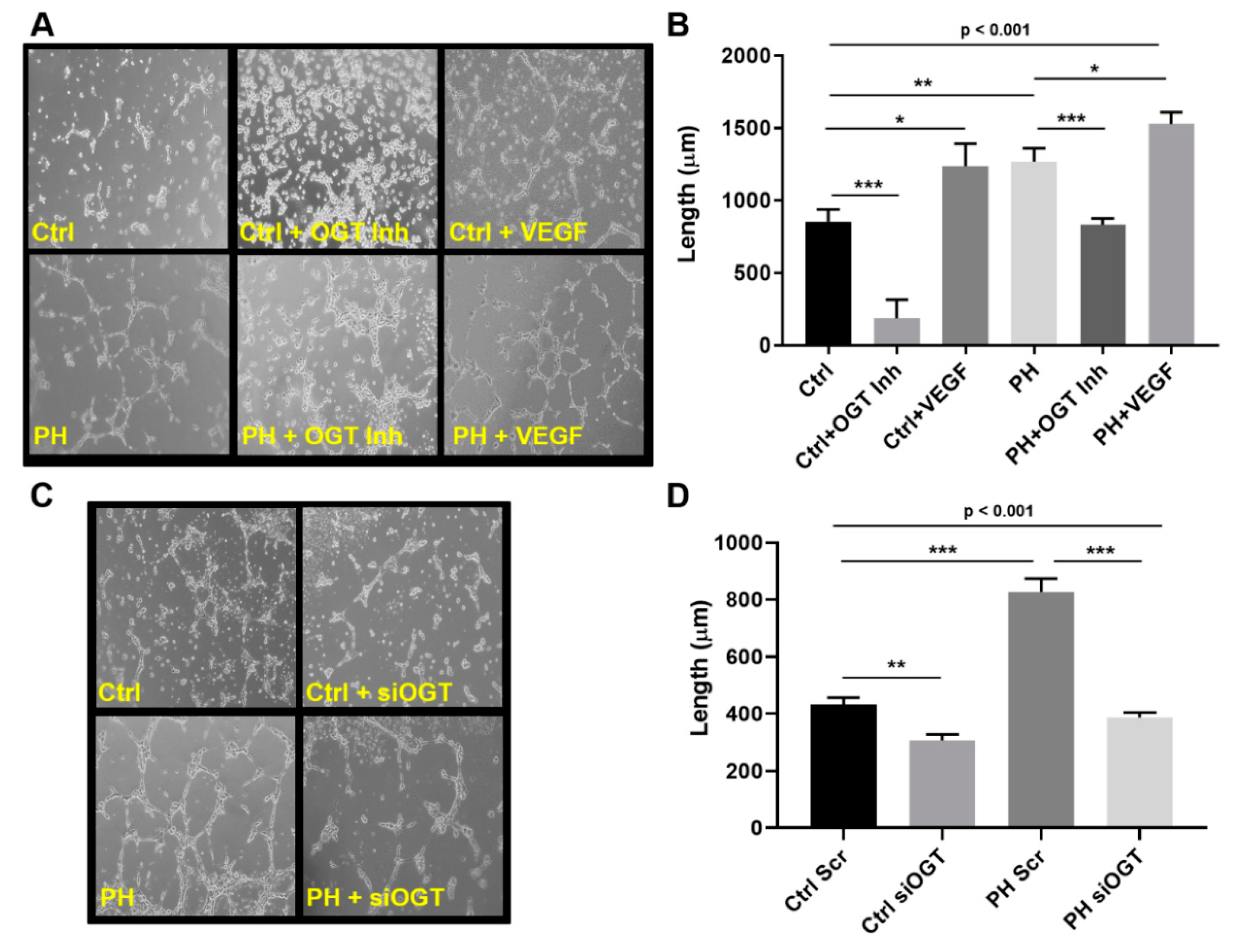
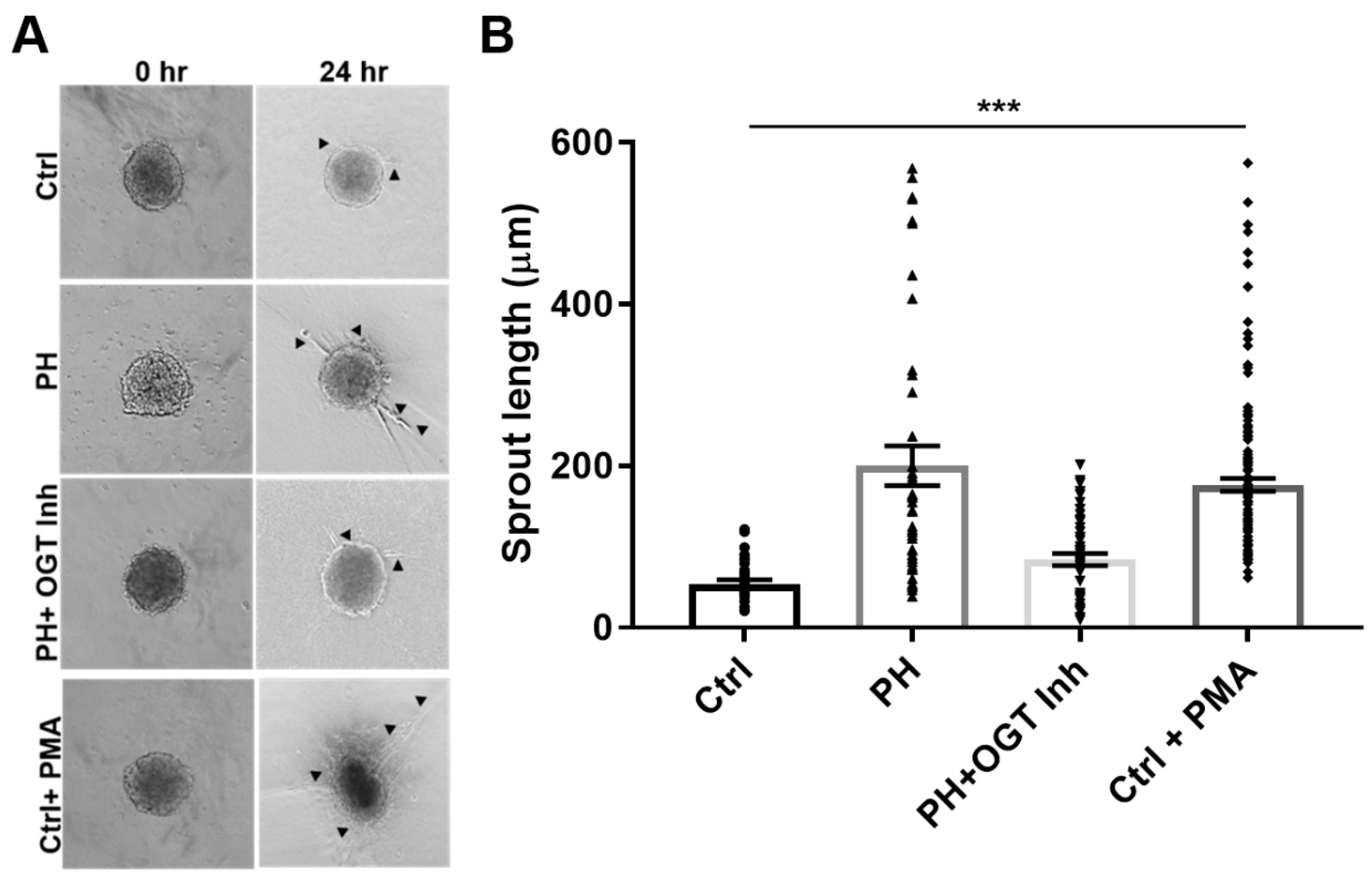
2.2. OGT Regulates Vascular Endothelial Tube Formation and Vascular Sprouting in IPAH
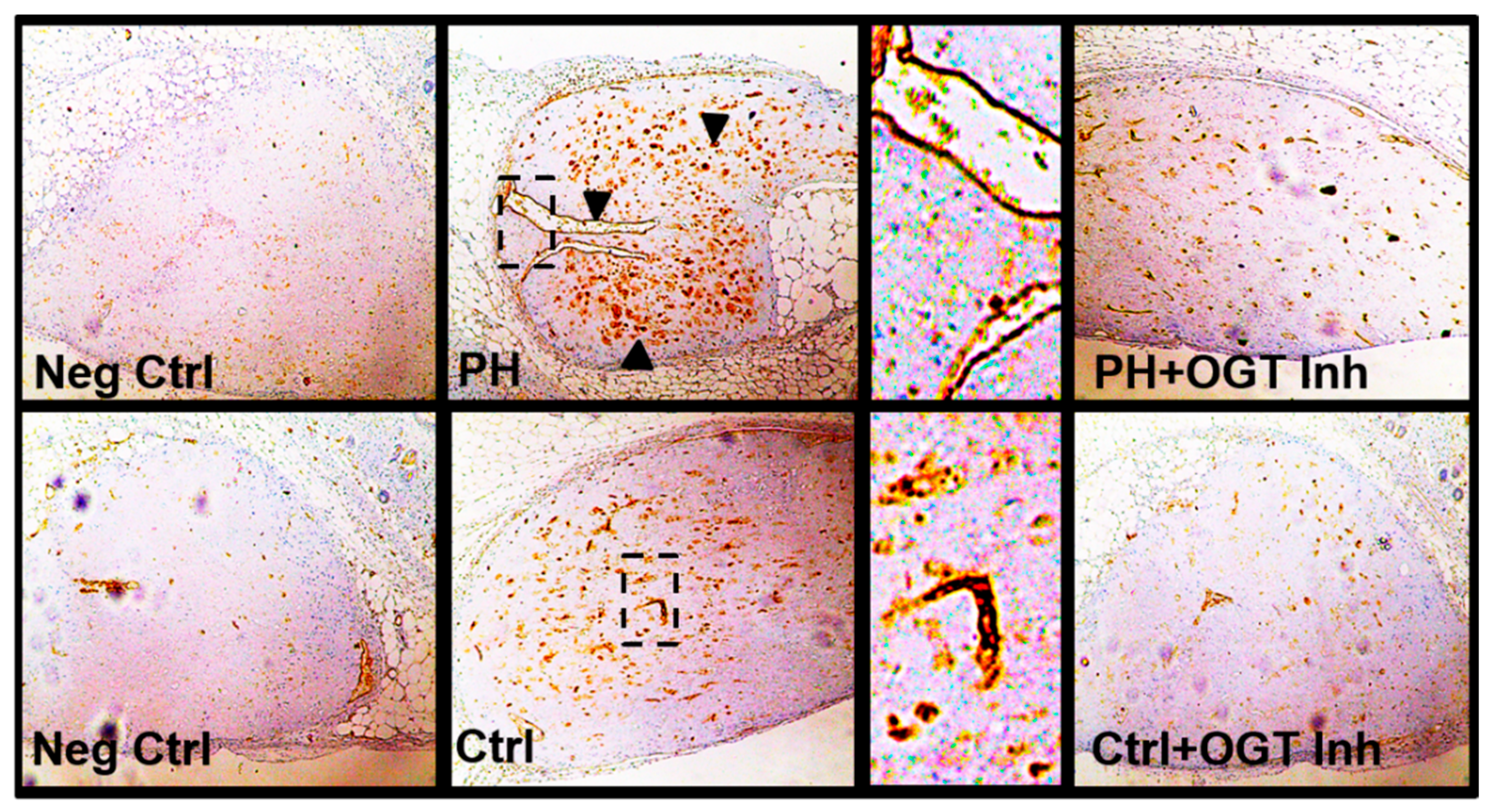
2.3. OGT Inhibition Attenuates De Novo Vascularization in a Humanized Angiogenic Mouse Model
3. Discussion
4. Materials and Methods
4.1. Pasmc and PAEC Cell Culture Conditions
4.2. Sodium Dodecyl Sulphate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Immunoblotting
4.3. Immunoprecipitation
4.4. siRNA Knockdown and Cell Culture
4.5. PAEC 2-D Tube Formation Assay
4.6. 3-D In Vitro Angiogenesis Model Using PAECs:PASMCs
4.7. De novo Vascularization in a Humanized Angiogenic Mouse Model
4.8. Statistical Analysis
5. Conclusion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dweik, R.A.; Rounds, S.; Erzurum, S.C.; Archer, S.; Fagan, K.; Hassoun, P.M.; Hill, N.S.; Humbert, M.; Kawut, S.M.; Krowka, M.; et al. An official American Thoracic Society Statement: Pulmonary hypertension phenotypes. Am. J. Respir. Crit. Care Med. 2014, 189, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Hoeper, M.M.; McLaughlin, V.V.; Dalaan, A.M.; Satoh, T.; Galie, N. Treatment of pulmonary hypertension. Lancet Respir. Med. 2016, 4, 323–336. [Google Scholar] [CrossRef]
- Seeger, W.; Adir, Y.; Barbera, J.A.; Champion, H.; Coghlan, J.G.; Cottin, V.; De Marco, T.; Galie, N.; Ghio, S.; Gibbs, S.; et al. Pulmonary hypertension in chronic lung diseases. J. Am. Coll. Cardiol. 2013, 62, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Austin, E.D.; Loyd, J.E. Heritable forms of pulmonary arterial hypertension. Semin. Respir. Crit. Care Med. 2013, 34, 568–580. [Google Scholar]
- Voelkel, N.F.; Gomez-Arroyo, J.; Abbate, A.; Bogaard, H.J.; Nicolls, M.R. Pathobiology of pulmonary arterial hypertension and right ventricular failure. Eur. Respir. J. 2012, 40, 1555–1565. [Google Scholar] [CrossRef]
- Farber, H.W.; Loscalzo, J. Pulmonary arterial hypertension. N. Engl. J. Med. 2004, 351, 1655–1665. [Google Scholar] [CrossRef]
- Rai, P.R.; Cool, C.D.; King, J.A.; Stevens, T.; Burns, N.; Winn, R.A.; Kasper, M.; Voelkel, N.F. The cancer paradigm of severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 558–564. [Google Scholar] [CrossRef]
- Humbert, M.; Morrell, N.W.; Archer, S.L.; Stenmark, K.R.; MacLean, M.R.; Lang, I.M.; Christman, B.W.; Weir, E.K.; Eickelberg, O.; Voelkel, N.F.; et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 13–24. [Google Scholar] [CrossRef]
- Asosingh, K.; Aldred, M.A.; Vasanji, A.; Drazba, J.; Sharp, J.; Farver, C.; Comhair, S.A.; Xu, W.; Licina, L.; Huang, L.; et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am. J. Pathol. 2008, 172, 615–627. [Google Scholar] [CrossRef]
- Guignabert, C.; Tu, L.; Le Hiress, M.; Ricard, N.; Sattler, C.; Seferian, A.; Huertas, A.; Humbert, M.; Montani, D. Pathogenesis of pulmonary arterial hypertension: Lessons from cancer. Eur. Respir. Rev. 2013, 22, 543–551. [Google Scholar] [CrossRef]
- Jonigk, D.; Golpon, H.; Bockmeyer, C.L.; Maegel, L.; Hoeper, M.M.; Gottlieb, J.; Nickel, N.; Hussein, K.; Maus, U.; Lehmann, U.; et al. Plexiform lesions in pulmonary arterial hypertension composition, architecture, and microenvironment. Am. J. Pathol. 2011, 179, 167–179. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.W.; Faley, S.; Shim, H.-N.; Prosser, J.R.; Agrawal, V.; Bellan, L.M.; West, J.D. Pulmonary Vascular Platform Models the Effects of Flow and Pressure on Endothelial Dysfunction in BMPR2 Associated Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2018, 19, 2561. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Chacon, M.; Alger, L.; Wang, J.; Taraseviciene-Stewart, L.; Kasahara, Y.; Cool, C.D.; Bishop, A.E.; Geraci, M.; Semenza, G.L.; et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: Evidence for a process of disordered angiogenesis. J. Pathol. 2001, 195, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Toba, M.; Alzoubi, A.; Ito, M.; Fagan, K.A.; Cool, C.D.; Voelkel, N.F.; McMurtry, I.F.; Oka, M. Formation of Plexiform Lesions in Experimental Severe Pulmonary Arterial Hypertension. Circulation 2010, 121, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Marecki, J.C.; Richter, A.; Fijalkowska, I.; Flores, S. Pathology of pulmonary hypertension. Clin. Chest Med. 2007, 28, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Ishida, A.; Murray, J.; Saito, Y.; Kanthou, C.; Benzakour, O.; Shibuya, M.; Wijelath, E.S. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J. Cell. Physiol. 2001, 188, 359–368. [Google Scholar] [CrossRef]
- Okuda, Y.; Tsurumaru, K.; Suzuki, S.; Miyauchi, T.; Asano, M.; Hong, Y.; Sone, H.; Fujita, R.; Mizutani, M.; Kawakami, Y.; et al. Hypoxia and endothelin-1 induce VEGF production in human vascular smooth muscle cells. Life Sci. 1998, 63, 477–484. [Google Scholar] [CrossRef]
- Morrell, N.W.; Archer, S.L.; Defelice, A.; Evans, S.; Fiszman, M.; Martin, T.; Saulnier, M.; Rabinovitch, M.; Schermuly, R.; Stewart, D.; et al. Anticipated classes of new medications and molecular targets for pulmonary arterial hypertension. Pulm. Circ. 2013, 3, 226–244. [Google Scholar] [CrossRef]
- Lundgrin, E.L.; Park, M.M.; Sharp, J.; Tang, W.H.; Thomas, J.D.; Asosingh, K.; Comhair, S.A.; DiFilippo, F.P.; Neumann, D.R.; Davis, L.; et al. Fasting 2-deoxy-2-[18F] fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann. Am. Thorac. Soc. 2013, 10, 1–9. [Google Scholar] [CrossRef]
- Paulin, R.; Michelakis, E.D. The metabolic theory of pulmonary arterial hypertension. Circ. Res. 2014, 115, 148–164. [Google Scholar] [CrossRef]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Heresi, G.A.; Malin, S.K.; Barnes, J.W.; Tian, L.; Kirwan, J.P.; Dweik, R.A. Abnormal Glucose Metabolism and High-Energy Expenditure in Idiopathic Pulmonary Arterial Hypertension. Ann. Am. Thorac. Soc. 2017, 14, 190–199. [Google Scholar] [PubMed]
- Xu, W.; Koeck, T.; Lara, A.R.; Neumann, D.; DiFilippo, F.P.; Koo, M.; Janocha, A.J.; Masri, F.A.; Arroliga, A.C.; Jennings, C.; et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl. Acad. Sci. USA 2007, 104, 1342–1347. [Google Scholar] [CrossRef] [PubMed]
- Rehman, J.; Archer, S.L. A proposed mitochondrial-metabolic mechanism for initiation and maintenance of pulmonary arterial hypertension in fawn-hooded rats: The Warburg model of pulmonary arterial hypertension. Adv. Exp. Med. Biol. 2010, 661, 171–185. [Google Scholar] [PubMed]
- Marsboom, G.; Wietholt, C.; Haney, C.R.; Toth, P.T.; Ryan, J.J.; Morrow, E.; Thenappan, T.; Bache-Wiig, P.; Piao, L.; Paul, J.; et al. Lung (1)(8) F-fluorodeoxyglucose positron emission tomography for diagnosis and monitoring of pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 2012, 185, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.W.; Kucera, E.T.; Tian, L.; Mellor, N.E.; Dvorina, N.; Baldwin Iii, W.W.; Aldred, M.A.; Farver, C.F.; Comhair, S.A.; Aytekin, M.; et al. BMPR2 Mutation-independent Mechanisms of Disrupted BMP Signaling in IPAH. Am. J. Respir. Cell Mol. Biol. 2016. [Google Scholar] [CrossRef]
- Cikach, F.S.; Tonelli, A.R., Jr.; Barnes, J.; Paschke, K.; Newman, J.; Grove, D.; Dababneh, L.; Wang, S.; Dweik, R.A. Breath Analysis in Pulmonary Arterial Hypertension. Chest 2013, 145, 551–558. [Google Scholar] [CrossRef]
- Barnes, J.W.; Tian, L.; Heresi, G.A.; Farver, C.F.; Asosingh, K.; Comhair, S.A.; Aulak, K.S.; Dweik, R.A. O-linked beta-N-acetylglucosamine transferase directs cell proliferation in idiopathic pulmonary arterial hypertension. Circulation 2015, 131, 1260–1268. [Google Scholar] [CrossRef]
- Sage, A.T.; Walter, L.A.; Shi, Y.; Khan, M.I.; Kaneto, H.; Capretta, A.; Werstuck, G.H. Hexosamine biosynthesis pathway flux promotes endoplasmic reticulum stress, lipid accumulation, and inflammatory gene expression in hepatic cells. AJP Endocrinol. Metab. 2010, 298, 499–511. [Google Scholar] [CrossRef]
- Slawson, C.; Copeland, R.J.; Hart, G.W. O-GlcNAc signaling: A metabolic link between diabetes and cancer? Trends Biochem. Sci. 2010, 35, 547–555. [Google Scholar] [CrossRef]
- Vaidyanathan, K.; Durning, S.; Wells, L. Functional O-GlcNAc modifications: Implications in molecular regulation and pathophysiology. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 140–163. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.; Gao, Y.; Mahoney, J.A.; Vosseller, K.; Chen, C.; Rosen, A.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Further characterization of the nucleocytoplasmic beta-N-acetylglucosaminidase, O-GlcNAcase. J. Biol. Chem. 2002, 277, 1755–1761. [Google Scholar] [CrossRef] [PubMed]
- Hart, G.W.; Slawson, C.; Ramirez-Correa, G.; Lagerlof, O. Cross talk between O-GlcNAcylation and phosphorylation: Roles in signaling, transcription, and chronic disease. Annu. Rev. Biochem. 2011, 80, 825–858. [Google Scholar] [CrossRef] [PubMed]
- Zachara, N.E.; O’Donnell, N.; Cheung, W.D.; Mercer, J.J.; Marth, J.D.; Hart, G.W. Dynamic O-GlcNAc modification of nucleocytoplasmic proteins in response to stress. A survival response of mammalian cells. J. Biol. Chem. 2004, 279, 30133–30142. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.P.; Ferrer, C.M.; Jackson, S.R.; Shahriari, K.S.; Vosseller, K.; Reginato, M.J. Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J. Biol. Chem. 2012, 287, 11070–11081. [Google Scholar] [CrossRef]
- Jensen, R.V.; Zachara, N.E.; Nielsen, P.H.; Kimose, H.H.; Kristiansen, S.B.; Botker, H.E. Impact of O-GlcNAc on cardioprotection by remote ischaemic preconditioning in non-diabetic and diabetic patients. Cardiovasc. Res. 2013, 97, 369–378. [Google Scholar] [CrossRef]
- Lefebvre, T.; Dehennaut, V.; Guinez, C.; Olivier, S.; Drougat, L.; Mir, A.M.; Mortuaire, M.; Vercoutter-Edouart, A.S.; Michalski, J.C. Dysregulation of the nutrient/stress sensor O-GlcNAcylation is involved in the etiology of cardiovascular disorders, type-2 diabetes and Alzheimer’s disease. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 67–79. [Google Scholar] [CrossRef]
- Musicki, B.; Kramer, M.F.; Becker, R.E.; Burnett, A.L. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. Proc. Natl. Acad. Sci. USA 2005, 102, 11870–11875. [Google Scholar] [CrossRef]
- Dassanayaka, S.; Jones, S.P. O-GlcNAc and the cardiovascular system. Pharmacol. Ther. 2014, 142, 62–71. [Google Scholar] [CrossRef]
- Zachara, N.E. The roles of O-linked beta-N-acetylglucosamine in cardiovascular physiology and disease. Am. J. Physiol.-Heart Circ. Physiol. 2012, 302, 1905–1918. [Google Scholar] [CrossRef]
- Hilgers, R.H.P.; Xing, D.; Gong, K.; Chen, Y.-F.; Chatham, J.C.; Oparil, S. Acute O-GlcNAcylation prevents inflammation-induced vascular dysfunction. Am. J. Physiol.-Heart Circ. Physiol. 2012, 303, 513–522. [Google Scholar] [CrossRef]
- Donovan, K.; Alekseev, O.; Qi, X.; Cho, W.; Azizkhan-Clifford, J. O-GlcNAc modification of transcription factor Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal cells. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7862–7873. [Google Scholar] [CrossRef] [PubMed]
- Fernando, K.H.N.; Yang, H.-W.; Jiang, Y.; Jeon, Y.-J.; Ryu, B. Diphlorethohydroxycarmalol Isolated from Ishige okamurae Represses High Glucose-Induced Angiogenesis In Vitro and In Vivo. Mar. Drugs 2018, 16, 375. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Ahmed, N.; Berridge, M.V. Acute Regulation of Glucose Transport After Activation of Human Peripheral Blood Neutrophils by Phorbol Myristate Acetate, fMLP, and Granulocyte-Macrophage Colony-Stimulating Factor. Blood 1998, 91, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Otake, S.; Kobayashi, M.; Narumi, K.; Sasaki, S.; Kikutani, Y.; Furugen, A.; Watanabe, M.; Takahashi, N.; Ogura, J.; Yamaguchi, H.; et al. Regulation of the Expression and Activity of Glucose and Lactic Acid Metabolism-Related Genes by Protein Kinase C in Skeletal Muscle Cells. Biol. Pharm. Bull. 2013, 36, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Prasain, N.; Meador, J.L.; Yoder, M.C. Phenotypic and Functional Characterization of Endothelial Colony Forming Cells Derived from Human Umbilical Cord Blood. J. Vis. Exp. 2012. [Google Scholar] [CrossRef]
- Yoder, M.C.; Mead, L.E.; Prater, D.; Krier, T.R.; Mroueh, K.N.; Li, F.; Krasich, R.; Temm, C.J.; Prchal, J.T.; Ingram, D.A. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 2007, 109, 1801–1809. [Google Scholar] [CrossRef]
- Mitani, Y.; Ueda, M.; Komatsu, R.; Maruyama, K.; Nagai, R.; Matsumura, M.; Sakurai, M. Vascular smooth muscle cell phenotypes in primary pulmonary hypertension. Eur. Respir. J. 2001, 17, 316–320. [Google Scholar] [CrossRef]
- Cool, C.D.; Stewart, J.S.; Werahera, P.; Miller, G.J.; Williams, R.L.; Voelkel, N.F.; Tuder, R.M. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell-specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am. J. Pathol. 1999, 155, 411–419. [Google Scholar] [CrossRef]
- Voelkel, N.F.; Bogaard, H.J. Adding complexity to plexogenic arteriopathy. Eur. Respir. J. 2016, 48, 1553–1555. [Google Scholar] [CrossRef]
- Tuder, R.M.; Davis, L.A.; Graham, B.B. Targeting Energetic Metabolism. Am. J. Respir. Crit. Care Med. 2012, 185, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Rafikova, O.; Meadows, M.L.; Kinchen, J.M.; Mohney, R.P.; Maltepe, E.; Desai, A.A.; Yuan, J.X.J.; Garcia, J.G.N.; Fineman, J.R.; Rafikov, R.; et al. Metabolic Changes Precede the Development of Pulmonary Hypertension in the Monocrotaline Exposed Rat Lung. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Erzurum, S.C. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr. Physiol. 2011, 1, 357–372. [Google Scholar] [PubMed]
- Caldwell, S.A.; Jackson, S.R.; Shahriari, K.S.; Lynch, T.P.; Sethi, G.; Walker, S.; Vosseller, K.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 2010, 29, 2831–2842. [Google Scholar] [CrossRef]
- Izquierdo-Garcia, J.L.; Arias, T.; Rojas, Y.; Garcia-Ruiz, V.; Santos, A.; Martin-Puig, S.; Ruiz-Cabello, J. Metabolic Reprogramming in the Heart and Lung in a Murine Model of Pulmonary Arterial Hypertension. Front. Cardiovasc. Med. 2018, 5. [Google Scholar] [CrossRef]
- Cox, E.J.; Marsh, S.A. Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc. Diabetol. 2013, 12, 101. [Google Scholar] [CrossRef]
- Hirata, Y.; Nakagawa, T.; Moriwaki, K.; Koubayashi, E.; Kakimoto, K.; Takeuchi, T.; Inoue, T.; Higuchi, K.; Asahi, M. Augmented O-GlcNAcylation alleviates inflammation-mediated colon carcinogenesis via suppression of acute inflammation. J. Clin. Biochem. Nutr. 2018, 62, 221–229. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Sodi, V.L.; Reginato, M.J. O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J. Mol. Biol. 2016, 428, 3282–3294. [Google Scholar] [CrossRef]
- Ferrer, C.M.; Lynch, T.P.; Sodi, V.L.; Falcone, J.N.; Schwab, L.P.; Peacock, D.L.; Vocadlo, D.J.; Seagroves, T.N.; Reginato, M.J. O-GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF-1 pathway. Mol. Cell 2014, 54, 820–831. [Google Scholar] [CrossRef]
- Kamigaito, T.; Okaneya, T.; Kawakubo, M.; Shimojo, H.; Nishizawa, O.; Nakayama, J. Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients. Prostate Cancer Prostatic Dis. 2014, 17, 18–22. [Google Scholar] [CrossRef]
- Ma, Z.; Vocadlo, D.J.; Vosseller, K. Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J. Biol. Chem. 2013, 288, 15121–15130. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Clark, P.M.; Mason, D.E.; Keenan, M.C.; Hill, C.; Goddard, W.A.; Peters, E.C., 3rd; Driggers, E.M.; Hsieh-Wilson, L.C. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science 2012, 337, 975–980. [Google Scholar] [PubMed]
- Da Costa, R.M.; Silva, J.F.D.; Alves, J.V.; Dias, T.B.; Rassi, D.M.; Garcia, L.V.; Lobato, N.D.S.; Tostes, R.C. Increased O-GlcNAcylation of Endothelial Nitric Oxide Synthase Compromises the Anti-contractile Properties of Perivascular Adipose Tissue in Metabolic Syndrome. Front. Physiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Federici, M.; Menghini, R.; Mauriello, A.; Hribal, M.L.; Ferrelli, F.; Lauro, D.; Sbraccia, P.; Spagnoli, L.G.; Sesti, G.; Lauro, R. Insulin-Dependent Activation of Endothelial Nitric Oxide Synthase Is Impaired by O-Linked Glycosylation Modification of Signaling Proteins in Human Coronary Endothelial Cells. Circulation 2002, 106, 466–472. [Google Scholar]
- Akimoto, Y.; Kreppel, L.K.; Hirano, H.; Hart, G.W. Hyperglycemia and the O-GlcNAc Transferase in Rat Aortic Smooth Muscle Cells: Elevated Expression and Altered Patterns of O-GlcNAcylation. Arch. Biochem. Biophys. 2001, 389, 166–175. [Google Scholar] [CrossRef]
- Lima, V.V.; Giachini, F.R.C.; Choi, H.; Carneiro, F.S.; Carneiro, Z.N.; Fortes, Z.B.; Carvalho, M.H.C.; Webb, R.C.; Tostes, R.C. Impaired Vasodilator Activity in Deoxycorticosterone Acetate-Salt Hypertension Is Associated With Increased Protein O-GlcNAcylation. Hypertension 2009, 53, 166–174. [Google Scholar]
- Luo, B.; Soesanto, Y.; McClain, D.A. Protein modification by O-linked GlcNAc reduces angiogenesis by inhibiting Akt activity in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 651–657. [Google Scholar] [CrossRef]
- Xu, C.; Liu, G.D.; Feng, L.; Zhang, C.H.; Wang, F. Identification of O-GlcNAcylation Modification in Diabetic Retinopathy and Crosstalk with Phosphorylation of STAT3 in Retina Vascular Endothelium Cells. Cell. Physiol. Biochem. 2018, 49, 1389–1402. [Google Scholar]
- Issad, T. O-GlcNAcylation of connexin 40: A sweet connection between diabetes and endothelial cell dysfunction? Focus on “O-GlcNAcase overexpression reverses coronary endothelial cell dysfunction in type 1 diabetic mice”. Am. J. Physiol. Cell Physiol. 2015, 309, 590–592. [Google Scholar] [CrossRef]
- Sodi, V.L.; Bacigalupa, Z.A.; Ferrer, C.M.; Lee, J.V.; Gocal, W.A.; Mukhopadhyay, D.; Wellen, K.E.; Ivan, M.; Reginato, M.J. Nutrient sensor O-GlcNAc transferase controls cancer lipid metabolism via SREBP-1 regulation. Oncogene 2017, 37, 924–934. [Google Scholar]
- Jackson, S.P.; Tjian, R. O-glycosylation of eukaryotic transcription factors: Implications for mechanisms of transcriptional regulation. Cell 1988, 55, 125–133. [Google Scholar] [CrossRef]
- Han, I.; Kudlow, J.E. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol. Cell. Biol. 1997, 17, 2550–2558. [Google Scholar] [CrossRef]
- Chung, S.S.; Kim, J.H.; Park, H.S.; Choi, H.H.; Lee, K.W.; Cho, Y.M.; Lee, H.K.; Park, K.S. Activation of PPARγ negatively regulates O-GlcNAcylation of Sp1. Biochem. Biophys. Res. Commun. 2008, 372, 713–718. [Google Scholar] [CrossRef]
- Majumdar, G.; Harrington, A.; Hungerford, J.; Martinez-Hernandez, A.; Gerling, I.C.; Raghow, R.; Solomon, S. Insulin Dynamically Regulates Calmodulin Gene Expression by Sequential O-Glycosylation and Phosphorylation of Sp1 and Its Subcellular Compartmentalization in Liver Cells. J. Biol. Chem. 2006, 281, 3642–3650. [Google Scholar] [CrossRef] [PubMed]
- Samson, S.; Wong, N. Role of Sp1 in insulin regulation of gene expression. J. Mol. Endocrinol. 2002, 29, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Comhair, S.A.; Xu, W.; Mavrakis, L.; Aldred, M.A.; Asosingh, K.; Erzurum, S.C. Human primary lung endothelial cells in culture. Am. J. Respir. Cell Mol. Biol. 2012, 46, 723–730. [Google Scholar] [CrossRef]
- Abramoff, M.D.; Magelhaes, P.J.; Ram, S.J. Image Processing with ImageJ. Biophotonics Int. 2004, 11, 36–42. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Korff, T.; Kimmina, S.; Martiny-Baron, G.; Augustin, H.G. Blood vessel maturation in a 3-dimensional spheroidal coculture model: Direct contact with smooth muscle cells regulates endothelial cell quiescence and abrogates VEGF responsiveness. FASEB J. 2001, 15, 447–457. [Google Scholar] [CrossRef]
- Pfisterer, L.; Korff, T. Spheroid-Based In Vitro Angiogenesis Model. In Angiogenesis Protocols; Martin, S.G., Hewett, P.W., Eds.; Springer: New York, NY, USA, 2016; pp. 167–177. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IPAH PASMCs (n) | 4 |
| Age, years (MIN–MAX) | 41.0 (26–49) |
| Female | 3 (75.0%) |
| PAH category: | |
| Idiopathic PAH | 3 (75.0%) |
| Heritable PAH (BMPR2 Mutation) | 1 (25.0%) |
| IPAH PAECs (n) | 4 |
| Age, years | 34.7 (17–49) |
| Female | 4 (100.0%) |
| PAH category: | |
| Idiopathic PAH | 3 (75.0%) |
| Heritable PAH (BMPR2 Mutation) | 1 (25.0%) |
| Control PASMCs (n) | 4 |
| Age, years | 53.5 (43–58) |
| Female | 2 (67.0%) |
| Control PAECS (n) | 4 |
| Age, years | 45.6 (21–49) |
| Female | 2 (50.0%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barnes, J.W.; Tian, L.; Krick, S.; Helton, E.S.; Denson, R.S.; Comhair, S.A.A.; Dweik, R.A. O-GlcNAc Transferase Regulates Angiogenesis in Idiopathic Pulmonary Arterial Hypertension. Int. J. Mol. Sci. 2019, 20, 6299. https://doi.org/10.3390/ijms20246299
Barnes JW, Tian L, Krick S, Helton ES, Denson RS, Comhair SAA, Dweik RA. O-GlcNAc Transferase Regulates Angiogenesis in Idiopathic Pulmonary Arterial Hypertension. International Journal of Molecular Sciences. 2019; 20(24):6299. https://doi.org/10.3390/ijms20246299
Chicago/Turabian StyleBarnes, Jarrod W., Liping Tian, Stefanie Krick, E. Scott Helton, Rebecca S. Denson, Suzy A. A. Comhair, and Raed A. Dweik. 2019. "O-GlcNAc Transferase Regulates Angiogenesis in Idiopathic Pulmonary Arterial Hypertension" International Journal of Molecular Sciences 20, no. 24: 6299. https://doi.org/10.3390/ijms20246299
APA StyleBarnes, J. W., Tian, L., Krick, S., Helton, E. S., Denson, R. S., Comhair, S. A. A., & Dweik, R. A. (2019). O-GlcNAc Transferase Regulates Angiogenesis in Idiopathic Pulmonary Arterial Hypertension. International Journal of Molecular Sciences, 20(24), 6299. https://doi.org/10.3390/ijms20246299